

Abstract
Traumatic brain injury (TBI) remains a major public health problem globally. Presently, there is no way to restore cognitive deficits caused by TBI. In this study, we seek to evaluate the effect of statins (simvastatin and atorvastatin) on the spatial learning and neurogenesis in rats subjected to controlled cortical impact. Rats were treated with atorvastatin and simvastatin 1 day after TBI and daily for 14 days. Morris water maze tests were performed during weeks 2 and 5 after TBI. Bromodeoxyuridine (BrdU; 50 mg/kg) was intraperitoneally injected 1 day after TBI and daily for 14 days. Brain tissue was processed for immunohistochemical staining to identify newly generated cells and vessels. Our data show that (1) treatment of TBI with statins improves spatial learning on days 31–35 after onset of TBI; (2) in the non-neurogenic region of the hippocampal CA3 region, statin treatment reduces the neuronal loss after TBI, demonstrating the neuroprotective effect of statins; (3) in the neurogenic region of the dentate gyrus, treatment of TBI with statins enhances neurogenesis; (4) statin treatment augments TBI-induced angiogenesis; and (5) treatment with simvastatin at the same dose provides a therapeutic effect superior to treatment with atorvastatin. These results suggest that statins may be candidates for treatment of TBI.
Keywords: neurogenesis, rat, spatial learning, statins
INTRODUCTION
Traumatic brain injury (TBI) remains a major public health problem globally. Despite considerable investigation using models of TBI, no therapy has been successfully translated from the bench to the bedside. One reason for this failure may be that most therapies tested treat a single event or pathology, such as intracellular overload of calcium (Maeda et al., 2005). However, multiple pathological mechanisms are involved in brain injury after TBI, including ischemia resulting from secondary thrombosis and vascular damage, and hematoma and delayed cell death related to apoptosis (Narayan et al., 2002). Therefore, an effective treatment should target the particular interactive events that will prevent or reduce secondary injury and amplify endogenous neuroplasticity (Lu et al., 2004a). Therapies targeting a single pathological mechanism after TBI may not be sufficient to obtain significant improvement of clinical outcome. Therefore, employing a drug cocktail or an agent (e.g., statins) that can act on many different pathological mechanisms is likely to be superior to targeting a single approach to treat TBI.
Our preliminary data, and complementary data from others, demonstrate that secondary injury attributed to delayed thrombosis is present in the controlled cortical impact (CCI) model of TBI, and prevention or inhibition of delayed thrombosis by statin treatment reduces delayed neuronal death in the hippocampal CA3 region after TBI (Lu et al., 2004b,c; Stein and Smith, 2004). Injury-induced neuroplasticity is also triggered at the onset of brain trauma, including neurogenesis, synaptogenesis, and angiogenesis (Davidson et al., 2004). Whether statins enhance these aspects of neuroplasticity after TBI remains unknown.
3-Hydroxy-3-methyglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) have been developed as lipid-lowering drugs that reduce morbidity and mortality from coronary artery and cerebral vascular diseases (Essig et al., 1998). In addition to lowering lipids, statins reduce vascular inflammation (Maeda et al., 2003), decrease platelet aggregation and thrombosis (Laufs, 2003), reduce volume of hematoma (Delanty et al., 2001), and increase endothelium-derived nitric oxide production (Eto et al., 2002). These pluripotent effects of statins act to reduce the secondary vascular injury, thrombosis, and lesion volume after TBI (Lu et al., 2004b,c). We have also shown that delayed (i.e., 24 h) treatment of middle cerebral artery occlusion in the rat with statins enhances neurogenesis, synaptogenesis, and angiogenesis, and significantly improves neurological outcome after stroke (Chen et al., 2003). Therefore, statins may be highly suitable candidates for the treatment of TBI, and statins provide a strategy of targeting multiple events of neuroprotection and neurorestoration after brain injury.
Many statins are commercially available in the U.S. market. They can be categorized into different groups, such as natural, synthesized, and semi-synthesized (based on their generation), and lipophilic (hydrophobic) or lipophobic (hydrophilic) (based on their molecular characteristics) (McKenney, 2003; Stein, 2003). In this study, we restrict our efforts to two statins, simvastatin (i.e., natural, lipophilic, and capable of crossing the blood–brain barrier [BBB]) and atorvastatin (i.e., semi-synthesized, lipophilic, and unable to cross the BBB). Many studies have shown the overall consistent effects of statins on vascular hemostasis and cholesterol (McKenney, 2003; Stein, 2003); whether there are differences in the effects of these two statins on neuronal loss, neurogenesis and angiogenesis, and functional outcome (i.e., spatial learning), has not been investigated.
METHODS
All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Henry Ford Hospital.
Animal Models
A CCI model of TBI in the rat was utilized for the present study (Dixon et al., 1991; Mahmood et al., 2001). Male Wistar rats (300–400 g) were anesthetized intraperitoneally with chloral hydrate, 350 mg/kg/body weight. Rectal temperature was controlled at 37°C with a feedback-regulated water heating pad. A controlled cortical impact device was used to induce the injury. Rats were placed in a stereotactic frame. Two 10-mm-diameter craniotomies were performed adjacent to the central suture, midway between lambda and bregma. The second craniotomy allowed for movement of cortical tissue laterally. The dura was kept intact over the cortex. Injury was induced by impacting the left cortex (ipsilateral cortex) with a pneumatic piston containing a 6-mm-diameter tip at a rate of 4 m/sec and 2.5 mm of compression. Velocity was measured with a linear velocity displacement transducer.
Experimental Groups
Based on our earlier studies (Lu et al., 2004a-c; Chen et al., 2003), we designed the following studies.
Experiment 1
Thirty male Wistar rats were randomly divided into three groups with 10 rats per group: (1) sham control, (2) TBI + saline, and (3) TBI + atorvastatin. Rats in the sham control group received scalp surgery without craniotomy, and rats in the TBI + saline group were exposed to TBI and given saline orally. Rats in the TBI + atorvastatin group were subjected to TBI, and 1 day later, atorvastatin was administered orally daily at a dose of 1 mg/kg/day until the day before sacrifice. All rats from the above groups were injected BrdU (100 mg/kg) intraperitoneally 1 day after TBI and daily until the day before sacrifice. These rats were also tested on the Morris water maze (MWM) during the last 5 days before the rats were sacrificed at 15 days after TBI.
Experiment 2
Forty rats were randomly divided into four groups with 10 rats per group, including sham control, TBI + saline, TBI + atorvastatin, and TBI + simvastatin. All rats received the same intervention as the rats in Experiment 1, except that rats were sacrificed 35 days after TBI, and the MWM test was performed during the last 5 days before sacrifice (days 31–35). Rats in the TBI + simvastatin group were treated orally with simvastatin (1 mg/kg) 1 day after TBI and daily for 14 days, as in the regimen for atorvastatin.
Statin Administration by Oral Gavage
The animals were firmly restrained (the animal was grasped by the loose skin of the neck and back) to immobilize the head and maintained in an upright (vertical) position. The gavage needle was passed through the side of the mouth, followed the roof of the mouth, and advanced into the esophagus and toward the stomach. After the needle was passed to the correct length, the statins (0.01% statin in saline) were injected. The dose for both atorvastatin and simvastatin was 1 mg/kg.
Morris Water Maze Tests
The testing procedure was a modification of that described previously (Whalen et al., 1999a,b; Whalen 2000; Day and Schallert, 1996). Data collection was automated by the HVS Image 2020 Plus Tracking System (U.S. HVS Image, San Diego, CA). For descriptive data collection, the pool was subdivided into four equal quadrants formed by imaging lines. All animals were tested during the last 5 days before sacrifice. At the start of a trial, the rat was placed randomly at one of four fixed starting points, randomly facing either toward the wall or the center (designated North, South, East, and West) and allowed to swim for 90 sec or until they found the platform. The platform was located in a randomly changing position within the NE quadrant throughout the test period (e.g., sometimes equidistant from the center and edge of the pool, against the wall, near the center of the pool, and at the edges of the NE quadrant). The percentage of time traveled within the NE (correct) quadrant was calculated relative to the total amount of time spent swimming before reaching the platform, and was employed for statistical analysis.
Tissue Preparation
Rats were anesthetized intraperitoneally with ketamine (80 mg/kg) and xylazine (4 mg/kg), and perfused transcardially with saline solution containing heparin, followed by 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS), pH 7.4. Their brains were removed, post-fixed in 10% formalin for 1–2 days at room temperature, and then processed for paraffin sectioning. A series of 6-μm-thick sections were cut with a microtome through each of seven standard sections.
Immunoperoxidase Staining
To identify the newly generated cells, brain sections, after being deparaffinized, were incubated in 2% bovine serum albumin (BSA)–PBS at room temperature for 30 min, and were subsequently treated with mouse anti–bromodeoxyuridine (BrdU) (Dako, Carpinteria, CA) antibody diluted at 1:200 in PBS at 4°C overnight. Following sequential incubation with biotin-conjugated anti-mouse immunoglobulin G (IgG) (dilution 1:100; Dakopatts, CA), the sections were treated with an avidinbiotin-peroxidase system (ABC kit; Vector Laboratories, Inc., Burlingame, CA). Diaminobenzidine (DAB) was then used as a sensitive chromogen for light microscopy.
Immunofluorescence
After dehydration, sections were boiled in 1% citric acid buffer (pH 6.0) in a microwave oven for 10 min, cooled down to room temperature, and incubated in 1% sapornin for 3 h. Subsequently, the sections were incubated in 1% BSA to block the non-specific signals. Using the same buffer solution, the sections were incubated overnight at 4°C in primary antibodies (monoclonal mouse anti-MAP-2; Chemicon, Temecula, CA), followed by 2 h at room temperature in corresponding fluorochrome-conjugated goat secondary antibodies (anti-mouse fluorescein isothiocyanate) [FITC]; Jackson ImmunoResearch, West Grove, PA). The sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Each of the above steps was followed by four 5-min rinses in PBS. The sections were mounted with ProLong antifade medium (Molecular Probes, Eugene, OR). Double immunofluorescent staining was performed as described for identification of the newly generated vessels, which contained BrdU-labeled cells and new neurons. Briefly, BrdU labeling was performed after staining for vWF (the endothelial cell marker) and neuronal nuclei (NeuN; a neuronal marker). After dehydration, sections were incubated for 1 h in 0.1 M PBS containing 1% goat serum and 0.3% Triton X-100. Using the same buffer solution, the sections were incubated overnight at 4°C in primary antibodies [monoclonal mouse anti-vWF (1:200) and anti-NeuN (1:400); Dako], respectively, and followed by 2 h at room temperature in corresponding fluorochrome-conjugated goat secondary antibodies (anti-mouse FITC). The sections were incubated with monoclonal mouse anti-BrdU (Dako) at room temperature for 2 h and subsequently with anti-mouse Cy5 at room temperature for 2 h. All secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). Each of the above steps was followed by four 5-min rinses in PBS. The sections were mounted with ProLong antifade medium (Molecular Probes, Eugene, OR). Sections were observed under a fluorescent microscope.
Cell Counting
To evaluate whether orally administered atorvastatin reduces neuronal damage after TBI, cell counts were performed by observers blinded to the individual treatment status of the animals. MAP-2/DAPI-positive cells were defined as the survival neurons and counted in the CA3 region of the hippocampal formation as previously described (Becher et al., 1999). Five sections with 50-μm interval through the dorsal DG were analyzed with a fluorescent microscope at ×400 magnification (at the interaural 5.20-mm levels). The number of MAP-2/DAPI-positive cells was counted in the CA3 region both in the ipsilateral and contralateral hippocampus using an MCID image analyzer (MCID, St. Catherine’s, Ontario, Canada). The percentage of MAP-2/DAPI-positive cells of CA3 regions in the ipsilateral hippocampus compared to those in the contralateral hippocampus was estimated and used as a parameter to evaluate histological changes. Although just an estimate of the cell number, this method permits a meaningful comparison of differences between groups. DAPI-labeled nuclei were also counted in the boundary zone and the CA3 region on the same sections as used above to determine cell densities in these regions.
Analysis of Neurogenesis in the Ipsilateral Dentate Gyrus after TBI in Rat
Additional sections used in the above studies were used to evaluate neurogenesis in the dentate gyrus (DG) by calculating the density of BrdU-labeled cells and BrdU/NeuN-colabeled cells (cells/mm). We focused mainly on the ipsilateral DG and its subregions, including the subgranular zone (SGZ), granular cell layer (GCL), and the molecular layer. First, we detected the number of BrdU-labeled cells in these three regions by using single-fluorescent staining and then performed double-fluorescent staining for identification of mature neurons (NeuN). Proliferation and differentiation of newly generated cells were evaluated by counting the colabeled-positive cells.
Measurement of Vascular Density
Five sections with 50-μm intervals through the dorsal DG were stained for vWF, and the images were digitized with a light microscope at ×400 magnification (at the interaural 5.20-mm levels). The vWF-positive vessels were counted in the boundary zone of the lesion and the CA3 region of the hippocampus, using the MCID system. The vascular density in both regions was determined by dividing the immunoreactive vessels by the corresponding area (Lu et al., 2004a) and was used as a parameter of angiogenesis. To evaluate angiogenesis and the effect of statin treatment, an additional five rats from each group were perfused with fluorescence-conjugated dextran 1 min before sacrifice, and the brain tissues were prepared for vibratome sectioning (Lu et al., 2004b). Every fifth section was used to measure the density of vessels in the ipsilateral DG, and one section adjacent to each of the above sections was used for double-fluorescent staining with BrdU and NeuN. Vessels containing the BrdU-labeled endothelial cells were digitized with a fluorescent microscope at ×400 magnification (at the interaural 5.20-mm levels) and counted in the DG using the MCID system. The vascular density was determined by dividing the immunoreactive vessels by the corresponding area and used as a parameter of angiogenesis.
Statistical Analysis
Data were analyzed by analysis of variance (ANOVA) for multiple comparisons of functional test data (Mahmood et al., 2001). Paired t-test was used to test the difference of cell counts and vascular density in the ipsilateral hemisphere between the atorvastatin-treated group and the control group, as well as the simvastatin treated-group. All measurements were performed by observers blinded to individual treatments.
RESULTS
Functional Response of TBI Rats after Treatment with Statins
To test whether statin treatment promotes spatial learning in the early phase (within 15 days) after TBI, rats were tested on the MWM during the last 5 days (days 11–15 after TBI) before sacrifice. For the sham control, the percentage of the mean time spent in the correct quadrant (containing the platform) was increased from approximately ~25% to ~55% within the 5-day daily training session (Fig. 1). However, the maximum percentage of time within the correct quadrant was 36.2 ± 2.3% for the TBI + saline group, and 39 ± 2.5% for the TBI + atorvastatin group, with no statistical significance between these groups (p > 0.05). These data demonstrate that treatment with atorvastatin does not improve spatial learning in the early phase after TBI.
FIG. 1.
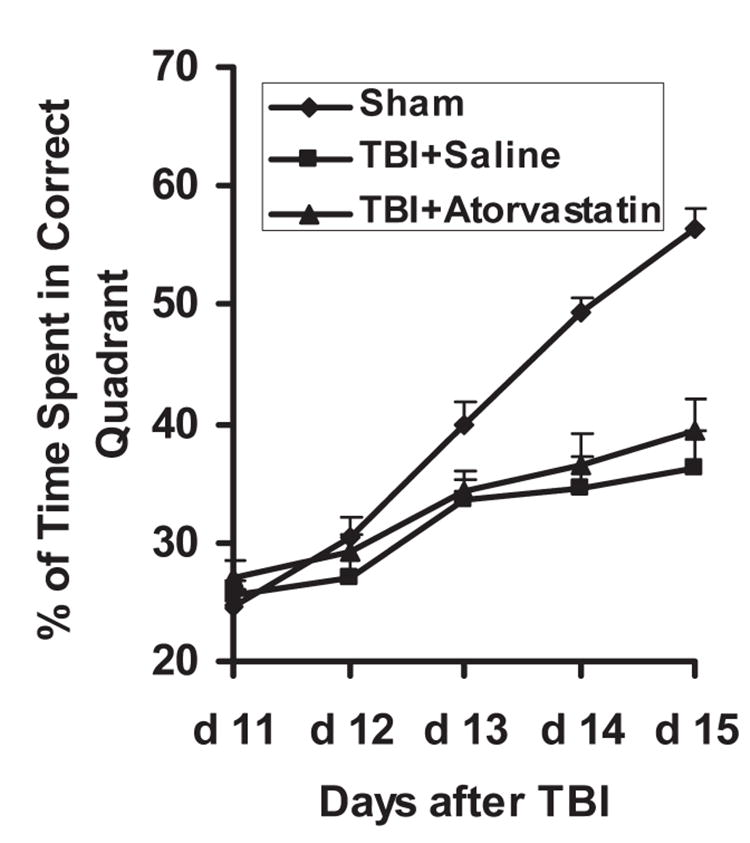
The plot shows the spatial learning deficits of rats on days 11–15 after traumatic brain injury (TBI). Rats were trained on the Morris water maze test starting at day 11 after TBI, and daily for 5 days (before sacrifice). Rats in the sham group spent a longer time in the correct quadrant searching for the platform (~55%) than rats from the saline- and atorvastatin-treated groups, with no significant difference between the saline-treated group and atorvastatin-treated group.
To test whether statin treatment promotes spatial learning in the relative late phase (i.e., approximately 1 month) after TBI, rats from control, TBI + saline, and TBI + atorvastatin groups received the same procedures and treatment as described previously, and were tested on the MWM during the last 5 days (days 31–35 after TBI) before sacrifice. For the sham control, the percentage of the mean time spent in the correct quadrant (containing the platform) was increased from approximately ~25% to ~55% within the five daily training sessions, as shown in the plots (Fig. 2). However, the mean percentages of time spent in the correct quadrant were significantly higher in the atorvastatin-treated group than the saline-treated group at days 34 (36.7 ± 1.1% vs. 31.8 ± 1.6%) and 35 (42.2 ± 2.1% vs. 34.4 ± 2.4%) after TBI (p < 0.05). However, treatment with simvastatin significantly improved spatial learning as early as 33 days (35.5 ± 2.5%) after TBI and the improvement continued at days 34 (42.5 ± 1.5%) and 35 (49.5 ± 2.9%), and the percentage was significantly higher than that of the atorvastatin-treated group (p < 0.05). These data demonstrate that both atorvastatin and simvastatin promote restoration of spatial learning in the relatively late phase after TBI. However, simvastatin appears to be more effective in the overall improvement of spatial learning therapy than atorvastatin, which only improves late phase spatial learning.
FIG. 2.
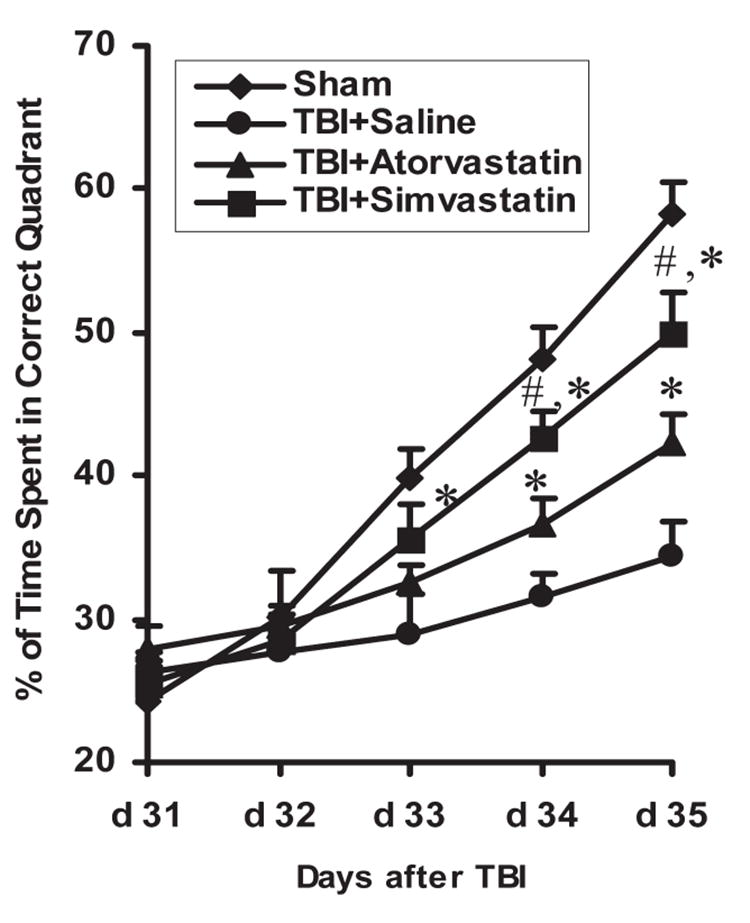
The plot shows the spatial learning deficits of rats on days 31–35 after traumatic brain injury (TBI). Rats were trained on the Morris water maze test on days 31–35 after TBI. Rats in the statin-treated groups showed significant increases in time spent in the correct quadrant on days 33–35 after TBI when compared to the saline-treated group (*p < 0.05). Simvastatin-treated rats showed significant increases of time spent in the correct quadrant at days 34 and 35 when compared to the atorvastatin-treated group (#p < 0.05).
Neuroprotective Effect of Statins on the Hippocampal CA3 Region
From rats sacrificed at days 15 and 35 after TBI, sections with a 100-μm interval from block E (containing the DG and the dorsal hippocampus) were selected for fluorescent immunohistochemical staining for MAP-2 to identify neuronal cells in the hippocampal CA3 region and counterstained with propidium iodide (PI). The MAP-2-positive cells in the ipsilateral hemisphere were counted under a ×20 objective using the MCID system, and then divided by the corresponding length (mm) to calculate the density of surviving neuronal cells. Figure 3 and Table 1 show that damage to the hippocampal formation in the TBI model causes significant loss of neurons in the hippocampal CA3 region in the saline-treated group (42 ± 6 neurons/mm) compared to sham (92 ± 12 neurons/mm, p < 0.05) at 15 days after TBI. However, the atorvastatin-treated group demonstrated a significant reduction in the loss of neurons in the hippocampal CA3 region (68 ± 7 neurons/mm) compared with the loss seen in the saline-treated group (42 ± 6 neurons/mm, p < 0.05). The number of neurons in the hippocampal CA3 region in the saline-treated group continued to decrease 35 days after TBI compared to 15 days after TBI (31 ± 7 neurons/mm vs. 42 ± 6 neurons/mm). Additionally, treatment with atorvastatin and simvastatin reduced the loss of neurons (i.e., 57 ± 8 neurons/mm and 61 ± 11 neurons/mm), which were similar to the 15-day level (68 ± 7 neurons/mm) after atorvastatin treatment. The reduction of percentage loss of neurons (Table 1) also further demonstrates the neuroprotective effect of atorvastatin treatment in that 16% of neurons were lost at 35 days after TBI in the TBI + atorvastatin group, versus 26% lost in the TBI + saline group (p < 0.05). The atorvastatin- and simvastatin-treated groups showed no significant differences in the density of neurons in the hippocampal CA3 region at day 35 after TBI (p > 0.05).
FIG. 3.
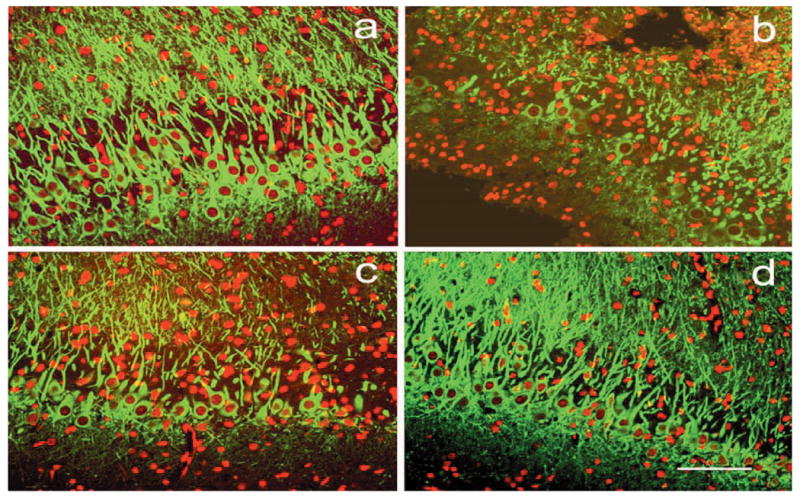
Fluorescent staining for MAP-2 (green) counterstained with PI (red) shows the density of neurons in the ipsilateral hippocampal CA3 region in the sham (a), saline-treated (b), atorvastatin-treated (c), and simvastatin-treated (d) animals at 35 days after traumatic brain injury (TBI). Scale bar = 50 μm (d).
TABLE 1.
Number of Neurons in the Hippocampal CA3 Region in the Animals Sacrificed 15 and 35 Days after TBI
| Sham | TBI + saline | TBI + Atorvastatin | TBI + Simvastatin | |
|---|---|---|---|---|
| Day 15 after TBI | 92 ± 12 | 42 ± 6 | 68 ± 7a | |
| Day 35 after TBI | 31 ± 7b | 57 ± 8a | 61 ± 11a | |
| Loss of neurons (%) | 26 ± 4 | 16 ± 3a |
p < 0.05, compared with TBI ± saline group.
p < 0.05, compared with day 15 after TBI.
TBI, traumatic brain injury.
These data demonstrate that, in the non-neurogenic region of the hippocampal CA3 region, TBI causes loss of the pyramidal cells, and this loss is a continuing process. The data also show that statin treatment not only reduces the early neuronal loss (within 15 days after TBI) but also reduces the relatively late neuronal loss shown at 35 days after TBI, demonstrating the neuroprotective effect of statins in the injured CA3 region.
Analysis of Neurogenesis in the Ipsilateral DG after TBI in Rat
We focused mainly on the ipsilateral DG and its subregions, including SGZ, GCL, and the molecular layer. First, we detected the number of BrdU-labeled cells in these three regions by using single staining (Fig. 4) and then performed double-fluorescent staining for identification of mature neurons (NeuN) (Fig. 5). Proliferation and differentiation of newly generated cells was evaluated by counting the colabeled-positive cells. Table 2 shows the numbers of BrdU-labeled cells and the BrdU/NeuN-colabeled cells in the DG at days 15 and 35 after TBI and the effect of statin treatment on them. TBI significantly increases the number of BrdU-positive cells when compared to sham control at days 15 (136 ± 22 cells/mm vs. 42 ± 13 cells/mm) and 35 (32 ± 3 cells/mm vs. 5 ± 2 cells/mm) after TBI.
FIG. 4.
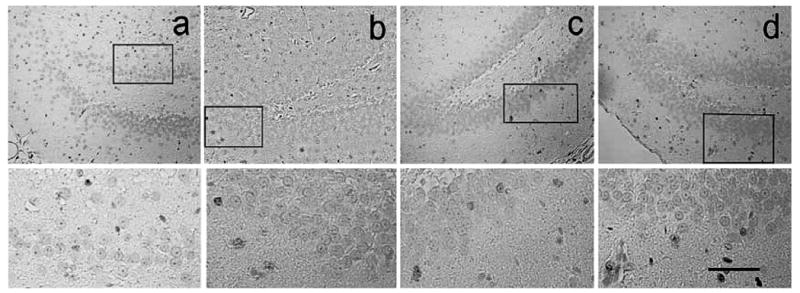
Immunohistochemical staining with diaminobenzidine (DAB) for bromodeoxyuridine (BrdU; counterstained with hematoxylin shows the density of BrdU-labeled cells in the ipsilateral dentate gyrus in the sham (a), saline-treated (b), atorvastatin-treated (c), and simvastatin-treated (d) animals at 35 days after traumatic brain injury (TBI). The lower panel is the inset of the upper panel. Scale bar = 100 μm.
FIG. 5.
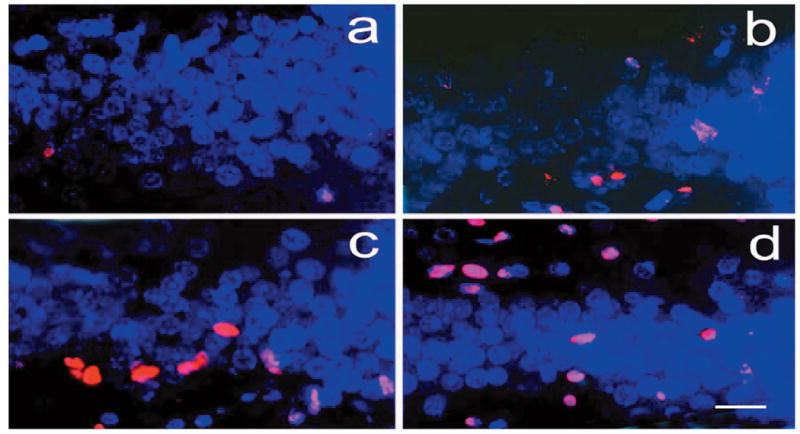
Double-fluorescent staining for bromodeoxyuridine (BrdU; red) and neuronal nuclei (NeuN; blue) shows the BrdU-labeled (red only) and BrdU/NeuN-colabeled cells (pink) in the ipsilateral dentate gyrus in the sham (a), saline-treated (b), atorvastatin-treated (c), and simvastatin-treated (d) animals at 35 days after traumatic brain injury (TBI). Scale bar = 25μm (d).
TABLE 2.
Number of BrdU-Labeled and BrdU/NeuN-Colabeled Cells in the Ipsilateral Dentate Gyrus (Mean ± SD)
| Day 15 after TBI | Day 35 after TBI | |
|---|---|---|
| BrdU+ cells (/mm) | ||
| Sham | 42 ± 13 | 5 ± 2 |
| TBI + saline | 136 ± 22 | 32 ± 3 |
| TBI + Atorvastatin | 186 ± 34a | 74 ± 13a |
| TBI + Simvastatin | 103 ± 16a,b | |
| BrdU+/NeuN+ cells (/mm) | ||
| Sham | 6 ± 2 (14%) | 2 ± 0.4 |
| TBI + saline | 26 ± 4 (19%) | 12 ± 3 (37%) |
| TBI + Atorvastatin | 48 ± 8a (25%) | 32 ± 9a (43%) |
| TBI + Simvastatin | 58 ± 7a,b (56%) |
p < 0.05 vs. TBI + Saline group.
p < 0.05 vs. TBI + Atorvastatin group.
Percentages in parentheses indicate the proportion of BrdU/NeuN-colabeled cells to BrdU-labeled cells.
BrdU, bromodeoxyuridine; NeuN, neuronal nuclei; TBI, traumatic brain injury.
Approximately 19% of the BrdU-labeled cells express NeuN (26 ± 4 neurons/mm) at day 15, and 37% at day 35 (12 ± 3 neurons/mm) after TBI. These data demonstrate that (1) TBI induces cell proliferation in the DG and some of the newly generated cells become neuron-like, suggestive of TBI-induced neurogenesis; and (2) a reduced proportion of the BrdU-labeled cells detected at day 15 after TBI survive at day 35 (23%, 32/136). Approximately 46% (12/26) of BrdU/NeuN-colabeled cells counted at day 15 after TBI survive at day 35, indicating that the majority of the newly generated TBI-induced cells die in the DG. The number of BrdU-labeled cells is significantly greater in the atorvastatin-treated group not only at day 15 (186 ± 34 cells/mm), but also at day 35 after TBI (74 ± 13 cells/mm) compared to the saline-treated group (TBI + saline). Differentiation of the newly generated cells into neurons was also augmented compared to the saline-treated group at days 15 (25% vs. 19%) and 35 (43% vs. 37%).
Approximately 66% of the newly formed neurons at day 15 after TBI (48) survive at day 35 (32). These data demonstrate that atorvastatin treatment 1 day after TBI increases the number of newly generated cells, augments differentiation of the newly formed cells into neurons, and reduces the death of the new neurons. When comparing the effects of atorvastatin and simvastatin at 35 days after TBI, simvastatin was found to significantly increase the number of BrdU-labeled cells (103 ± 16 cells/mm vs. 74 ± 13 cells/mm) and BrdU/NeuN-cola-beled cells (58 ± 7 cells/mm vs. 32 ± 9 cells/mm) compared to atorvastatin. This demonstrates that simvastatin is more effective than atorvastatin at the same dose of 1 mg/kg/day.
Analysis of Angiogenesis in the Ipsilateral DG after TBI in Rat
Figure 6 shows the density of fluorescence (FITC)–dextran perfused vessels in the upper blade of the ipsilateral DG at 35 days after TBI. A lower density of the vessels was present in the saline-treated rats than in rats treated with statins. Treatment with atorvastatin significantly enhanced TBI-induced angiogenesis at 15 days (12.5 ± 1.4 vessels/mm2) and 35 days (16.5 ± 1.9 vessels/mm2) after TBI compared with the saline-treated group (p < 0.05) (Fig. 7). However, simvastatin is more effective than atorvastatin in inducing angiogenesis at 35 days after TBI (p < 0.05). These data suggest that TBI induces angiogenesis in the DG and that statin treatment augments TBI-induced angiogenesis after TBI.
FIG. 6.
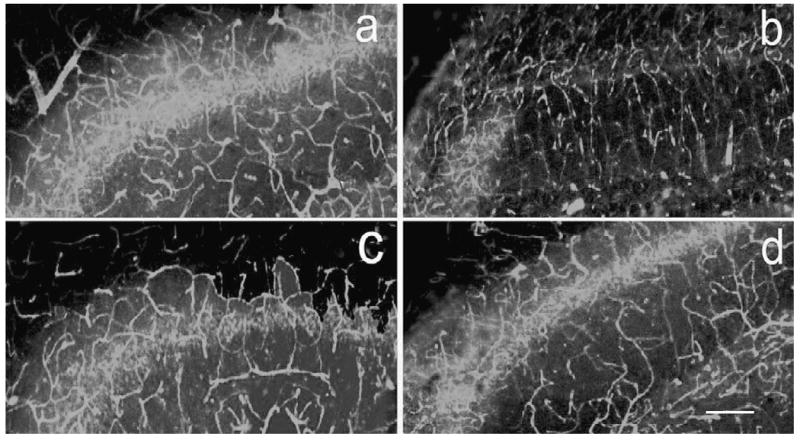
Fluorescein isothiocyanate (FITC)–conjugated dextran perfused vessels were captured by laser confocal microscopy in the ipsilateral dentate gyrus in the sham (a), saline-treated (b), atorvastatin-treated (c), and simvastatin-treated (d) animals at 35 days after traumatic brain injury (TBI). Scale bar = 100 μm (d).
FIG. 7.
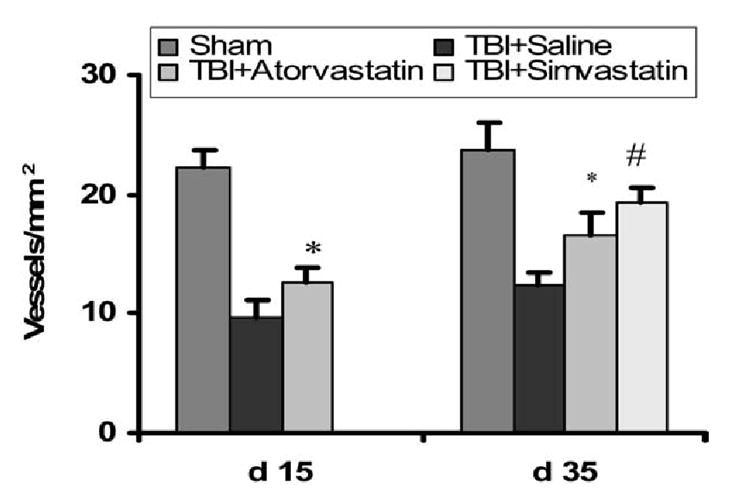
Bar graph shows that vessels containing bromodeoxyuridine (BrdU)–labeled cells were detected in the saline-treated rats at 15 days after traumatic brain injury (TBI) and the density of vessels increased at 35 days after TBI, demonstrating the presence of TBI-induced angiogenesis in the dentate gyrus, consistent with TBI-induced neurogenesis in the dentate gyrus. Treatment with atorvastatin significantly enhanced TBI-induced angiogenesis at 15 and 35 days after TBI compared with the saline-treated group (*p < 0.05). However, simvastatin is more effective than atorvastatin in the induction of angiogenesis at 35 days after TBI (#p < 0.05).
DISCUSSION
Our primary findings in this study are as follows: (1) treatment of TBI with statins improves spatial learning during week 5 (days 31–35) after the onset of TBI, but atorvastatin does not during week 2 (days 11–15), and improvement of spatial learning is temporally associated with the enhanced neurogenesis; (2) in the non-neurogenic region of the hippocampal CA3 region, TBI causes loss of the pyramidal cells, and this loss is a continuing process, and statin treatment not only reduces the early neuronal loss (within 15 days after TBI) but also reduces the relatively late neuronal loss shown at 35 days after TBI, which demonstrates the neuroprotective effect of statins in the injured CA3 region; (3) in the neurogenic region of the DG, TBI also induces neurogenesis compared to sham control, and statin treatment appears to enhance TBI-induced neurogenesis in the DG compared to the saline-treated group at both days 15 and 35 after TBI, suggesting that statin enhances neurogenesis in the region; and (4) statin treatment augments TBI-induced angiogenesis in the DG.
The most prevalent and debilitating features in survivors of brain trauma are cognitive deficits and motor dysfunction. The most common cognitive impairment among severely head-injured patients is memory loss and learning disability (Narayan et al., 2002; Fujimoto et al., 2004). Natural recovery from cognitive deficits after TBI is greatest within the first 6 months after the injury and is more gradual after that, but outcome varies with different types of brain injury (Whalen et al., 2004; Kline et al., 2002). To date, there is no effective treatment to promote recovery of memory and learning except routine medical intervention and care (Consensus Conference, 1999). The CCI model of TBI has many clinically relevant features, such as deficits of spatial learning and memory that have been evaluated by the MWM test (Royo et al., 2003; Fujimoto et al., 2004). This model has served as a principal tool for elucidating the pathophysiologic sequelae of brain trauma and for developing novel therapeutic approaches to treat TBI (Whalen et al., 2004). Rats suffering from CCI are deficient in spatial learning and memory, which has been attributed to damage to the trisynaptic pathway (Kline et al., 2002; Fox and Faden, 1998; Fox et al., 1998). The trisynaptic pathway from the entorhinal cortex to the hippocampus has long been regarded as the major route of information transfer underlying learning and memory consolidation (Day et al., 1999). Selective destruction of any portion of this pathway results in spatial learning disability, and intervention that reduces the injury to the trisynaptic pathway has shown improvement of the spatial learning (Day et al., 1999). In the CCI model of TBI, damage occurs selectively in the CA3 (non-neurogenic) region and in the DG (neurogenic), but not in the CA1 region, which provides an excellent model to study neuroprotection (on the CA3 region) and neurogenesis (on the DG) in a relative narrow structure.
Our data show that spatial learning was affected (indicated by decreased percent of time spent in the correct quadrant) not only during the second week after onset of injury but also during the fifth week in the control group (TBI + saline), demonstrating the successful duplication of spatial learning deficits which was targeted in this study. Following treatment with atorvastatin, the percentage of time spent in the correct quadrant does not show significant changes when compared with the saline-treated group during the second week after the onset of injury. However, a significantly increased percentage of time spent in the correct quadrant was observed during the fifth week in both atorvastatin- and simvastatintreated groups. This finding raises the following questions: (1) Why is the benefit of statin treatment on spatial learning recovery manifest during the fifth week but not during the second week? (2) Is the improvement of spatial learning associated with the neuroprotective effect of statins or with their pro-neurogenic effect, or both?
Our data show that statin treatment has a neuroprotective effect on neurons in the CA3 region. However, significant improvement of spatial learning was detected in the fifth week but not in the second week after TBI, demonstrating that the neuroprotective effect of statins may be insufficient to contribute to the improvement of spatial learning at an early time after TBI. Neurogenesis in the DG enhanced by statin treatment was also detected at 15 and 35 days after TBI, which parallels the neuroprotective effect in the CA3 region. The difference is that the number of pyramidal cells in the CA3 region continues to decrease in both the saline- and statin-treated groups, with a significantly smaller decrease in the latter group. However, the number of BrdU-labeled cells in the DG significantly increased in both statin-treated groups, but not in the saline-treated group, indicating that statins induce new cell generation (neurogenesis). The temporal match of the spatial learning recovery with neurogenesis suggests that neurogenesis may contribute to the recovery of spatial learning. The newly generated cells in the DG must take four important steps to become functional neurons, including differentiation, maturation, migration, and formation of new synapses (Kempermann et al., 2004a,b; Kempermann and Gage, 2002). The newly generated cells in the DG need four weeks to perform these four steps (Kempermann et al., 2004b). Therefore, the recovery of spatial learning function occurring in the fifth week after TBI in this study is consistent with established findings (Kempermann and Gage, 2002).
Statin treatment significantly increases the numbers of NeuN/BrdU-colabeled cells compared to the TBI + saline group, indicating that statin treatment enhances differentiation of newly generated cells into mature neurons. Based on the electrophysiological studies (Van Praag et al., 2002), the newly generated cells migrating into the granular zone may function as mature neurons. However, measurement of direct electrophysiological activity of the newly generated neurons in the DG has not been performed in this study, and future studies should include such measurements.
In the adult hippocampus, proliferating cell clusters are found in close proximity to blood vessels, implicating that vasculature- or blood-derived factors are regulators of neurogenesis (Palmer et al., 1999). Therefore, in this study we measured the parameters for angiogenesis, including the density of newly formed vessels. Our data indicate that statin treatment enhances angiogenesis. In addition, a large body of literature has demonstrated that statins promote angiogenesis (Dimmerler et al., 2001).
One important finding in this study is that simvastatin is superior to atorvastatin at the same dose of 1 mg/kg/day in improving spatial learning and neurogenesis. One possible reason for the differential therapeutic effect is that simvastatin crosses the BBB, while atorvastatin does not (Joukhadar et al., 2001), indicating that simvastatin may have a broader effect than atorvastatin on brain tissue. Our data demonstrate that treatment of TBI with statins reduces neuronal loss, increases neurogenesis and angiogenesis in the ipsilateral hippocampus, and improves spatial learning. In addition, at the same dose, simvastatin provides a greater restorative effect than atorvastatin.
Based on our work in stroke, the therapeutic benefit of statins, both neurorestorative and neuroprotective, may in part be attributed to their effect on the cerebral microvasculature (Zhang et al., 2005). We have also shown that statins have direct effects on neurons (Chen et al., 2005). Thus, it is likely that the combination of vascular and parenchymal response to statins provides the therapeutic benefit. However, further studies are needed to elucidate the underlying mechanisms.
In conclusion, statin treatment after TBI induces neurogenesis and angiogenesis in the DG and promotes improvement in spatial learning.
Acknowledgments
This work was supported by the NIH (grants RO1 NS52280 and PO1 42345).
References
- Becher A, Drenckhahn A, Pahner I, Ahnert-Hilger G. The synaptophysin–synaptobrevin complex is developmentally upregulated in cultivated neurons but is absent in neuroendocrine cells. Eur J Cell Biol. 1999;78:650–656. doi: 10.1016/S0171-9335(99)80050-8. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang ZG, Li Y, et al. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol. 2003;53:743–751. doi: 10.1002/ana.10555. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang C, Jiang H, et al. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J Cereb Blood Flow Metab. 2005;25:281–290. doi: 10.1038/sj.jcbfm.9600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consensus Conference. Rehabilitation of persons with traumatic brain injury. NIH Consensus Development Panel on Rehabilitation of Persons with Traumatic Brain Injury. JAMA. 1999;282:974–983. [PubMed] [Google Scholar]
- Davidson MH, Ballantyne CM, Kerzner B, et al. Efficacy and safety of ezetimibe coadministered with statins: randomised, placebo-controlled, blinded experience in 2382 patients with primary hypercholesterolemia. Int J Clin Pract. 2004;58:746–755. doi: 10.1111/j.1368-5031.2004.00289.x. [DOI] [PubMed] [Google Scholar]
- Day LB, Crews D, Wilczynski W. Spatial and reversal learning in congeneric lizards with different foraging strategies. Anim Behav. 1999;57:393–407. doi: 10.1006/anbe.1998.1007. [DOI] [PubMed] [Google Scholar]
- Day LB, Schallert T. Anticholinergic effects on acquisition of place learning in the Morris water task: spatial mapping deficit or inability to inhibit nonplace strategies? Behav Neurosci. 1996;110:998–1005. doi: 10.1037//0735-7044.110.5.998. [DOI] [PubMed] [Google Scholar]
- Delanty N, Vaughan CJ, Sheehy N. Statins and neuroprotection. Expert Opin Invest Drugs. 2001;10:1847–1853. doi: 10.1517/13543784.10.10.1847. [DOI] [PubMed] [Google Scholar]
- Dimmerler S, Aicher A, Vasa M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–397. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- Essig M, Vrtovsnik F, Nguyen G, Sraer JD, Friedlander G. Lovastatin modulates in vivo and in vitro the plasminogen activator/plasmin system of rat proximal tubular cells: role of geranylgeranylation and Rho proteins. J Am Soc Nephrol. 1998;9:1377–1388. doi: 10.1681/ASN.V981377. [DOI] [PubMed] [Google Scholar]
- Eto M, Kozai T, Cosentino F, Joch H, Luscher TF. Statin prevents tissue factor expression in human endothelial cells: role of Rho/Rho-kinase and Akt pathways. Circulation. 2002;105:1756–1759. doi: 10.1161/01.cir.0000015465.73933.3b. [DOI] [PubMed] [Google Scholar]
- Fox GB, Faden AI. Traumatic brain injury causes delayed motor and cognitive impairment in a mutant mouse strain known to exhibit delayed Wallerian degeneration. J Neurosci Res. 1998;53:718–727. doi: 10.1002/(SICI)1097-4547(19980915)53:6<718::AID-JNR9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Fox GB, Fan L, Levasseur RA, Faden AI. Effect of traumatic brain injury on mouse spatial and nonspatial learning in the Barnes circular maze. J Neurotrauma. 1998;15:1037–1046. doi: 10.1089/neu.1998.15.1037. [DOI] [PubMed] [Google Scholar]
- Fujimoto ST, Longhi L, Saatman KE, Mcintosh TK. Motor and cognitive function evaluation following experimental traumatic brain injury. Neurosci Biobehav Rev. 2004;28:365–378. doi: 10.1016/j.neubiorev.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukhadar C, Klein N, Prinz M, et al. Similar effects of atorvastatin, simvastatin and pravastatin on thrombogenic and inflammatory parameters in patients with hypercholesterolemia. Thromb Haemost. 2001;85:47–51. [PubMed] [Google Scholar]
- Kempermann G, Gage FH. Genetic determinants of adult hippocampal neurogenesis correlate with acquisition, but not probe trial performance, in the water maze task. Eur J Neurosci. 2002;16:129–136. doi: 10.1046/j.1460-9568.2002.02042.x. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Jessberger S, Steiner B, Kronenberg G. Milestones of neuronal development in the adult hippocampus. Trends Neurosci. 2004a;27:447–452. doi: 10.1016/j.tins.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Wiskott L, Gage FH. Functional significance of adult neurogenesis. Curr Opin Neurobiol. 2004b;14:186–191. doi: 10.1016/j.conb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Kline AE, Massucci JL, Marion DW, Dixon CE. Attenuation of working memory and spatial acquisition deficits after a delayed and chronic bromocriptine treatment regimen in rats subjected to traumatic brain injury by controlled cortical impact. J Neurotrauma. 2002;19:415–425. doi: 10.1089/08977150252932370. [DOI] [PubMed] [Google Scholar]
- Laufs U. Beyond lipid-lowering: effects of statins on endothelial nitric oxide. Eur J Clin Pharmacol. 2003;58:719–731. doi: 10.1007/s00228-002-0556-0. [DOI] [PubMed] [Google Scholar]
- Lu D, Goussev A, Chen J, et al. Atorvastatin reduces neurological deficit and increases synaptogenesis, angiogenesis and neuronal survival in rats subjected to traumatic brain injury. J Neurotrauma. 2004a;21:21–32. doi: 10.1089/089771504772695913. [DOI] [PubMed] [Google Scholar]
- Lu D, Mahmood A, Goussev A, et al. Atorvastatin reduces intravascular thrombosis, increases cerebral microvascular patency and integrity, and enhances spatial learning in rats subjected to traumatic brain injury. J Neurosurg. 2004b;101:819–827. doi: 10.3171/jns.2004.101.5.0813. [DOI] [PubMed] [Google Scholar]
- Lu D, Mahmood A, Goussev A, Qu C, Zhang ZG, Chopp M. Delayed thrombosis after traumatic brain injury in rats. J Neurotrauma. 2004c;21:1756–1766. doi: 10.1089/neu.2004.21.1756. [DOI] [PubMed] [Google Scholar]
- Maeda T, Lee SM, Hovda DA. Restoration of cerebral vasoreactivity by an L-type calcium channel blocker following fluid percussion brain injury. J Neurotrauma. 2005;22:763–771. doi: 10.1089/neu.2005.22.763. [DOI] [PubMed] [Google Scholar]
- Maeda T, Kawane T, Horiuchi N. Statins augment vascular endothelial growth factor expression in osteoblastic cells via inhibition of protein prenylation. Endocrinology. 2003;144:681–692. doi: 10.1210/en.2002-220682. [DOI] [PubMed] [Google Scholar]
- Mahmood A, Lu D, Yi L, Chen JL, Chopp M. Intracranial bone marrow transplantation after traumatic brain injury improving functional outcome in adult rats. J Neurosurg. 2001;94:589–595. doi: 10.3171/jns.2001.94.4.0589. [DOI] [PubMed] [Google Scholar]
- Mckenney JM. Pharmacologic characteristics of statins. Clin Cardiol. 2003;26:11132–11138. doi: 10.1002/clc.4960261507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan RK, Michel ME, Ansell B, et al. Clinical trials in head injury. J Neurotrauma. 2002;19:503–557. doi: 10.1089/089771502753754037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH. Fibroblast growth factors activate a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci. 1999;19:8487–8497. doi: 10.1523/JNEUROSCI.19-19-08487.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royo NC, Shimizu S, Schouten JW, Stover JF, Mcintosh TK. Pharmacology of traumatic brain injury. Curr Opin Pharmacol. 2003;3:27–32. doi: 10.1016/s1471-4892(02)00006-1. Review. [DOI] [PubMed] [Google Scholar]
- Stein EA. The power of statins: aggressive lipid lowering. Clin Cardiol. 2003;26:11125–11131. doi: 10.1002/clc.4960261506. [DOI] [PubMed] [Google Scholar]
- Stein SC, Smith DH. Coagulopathy in traumatic brain injury. Neurocrit Care. 2004;1:479–488. doi: 10.1385/NCC:1:4:479. [DOI] [PubMed] [Google Scholar]
- Van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Dixon CE, et al. Effect of traumatic brain injury in mice deficient in intercellular adhesion molecule–I: assessment of histopathologic and functional outcome. J Neurotrauma. 1999a;16:299–309. doi: 10.1089/neu.1999.16.299. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Clark RSB, Dixon CE, et al. Reduction of cognitive and motor deficits after traumatic brain injury in mice deficient in poly(ADP-ribose) polymerase. J Cereb Blood Flow Metab. 1999b;19:835–842. doi: 10.1097/00004647-199908000-00002. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Dixon CE, et al. Reduced brain edema after traumatic brain injury in mice deficient in P-selectin and intercellular adhesion molecule-I. J Leukoc Biol. 2000;67:160–168. doi: 10.1002/jlb.67.2.160. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Dixon CE, et al. Effect of traumatic brain injury in mice deficient in intercellular adhesion molecule–1: assessment of histopathologic and functional outcome. J Neurotrauma. 2004;16:299–309. doi: 10.1089/neu.1999.16.299. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Jin K, Mao XO, Greenberg DA. Vascular endothelial growth factor promotes proliferation of cortical neuron precursors by regulating E2F expression. FASEB J. 2003;17:186–193. doi: 10.1096/fj.02-0515com. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhang ZG, Ding GL, et al. Multitargeted effects of statin-enhanced thrombolytic therapy for stroke with recombinant human tissue-type plasminogen activator in the rat. Circulation. 2005;112:3486–3494. doi: 10.1161/CIRCULATIONAHA.104.516757. [DOI] [PubMed] [Google Scholar]