

Abstract
Little information is available on peripheral levels of Hsp72, Hsp60, and anti-Hsp60 antibodies in patients with left ventricular (LV) dysfunction due to non-atherosclerotic cardiac disease. In this study, serum Hsp72, Hsp60 and anti-Hsp60 antibodies, IL-6, and C-reactive protein (CRP) were measured in 44 healthy controls and in 82 patients with angiographically normal coronary arteries (LV ejection fraction [EF] ≥ 50%, n = 22; ≥35% to <50%, n = 32; <35%, n = 28). Patients with more severe disease (more depressed myocardial blood flow at rest and during dipyridamole, indicative of coronary microvascular impairment) showed more elevated circulating Hsp60 and auto-antibodies, Hsp72, and CRP levels. IL-6 was increased progressively as a function of severity of LV dysfunction. Anti-Hsp60 antibodies, Hsp72, and IL-6 were significantly correlated with brain natriuretic peptide (BNP) levels and LV end-diastolic dimensions (LVEDD) values. IL-6 tended to be related with Hsp72 in particular in patients with more severe disease (r = 0.45, P = 0.021). Hsp60 and Hsp72 activation and inflammatory markers were correlated with the extent of cardiac and microvascular dysfunction in patients with angiographycally normal coronary arteries. These results suggest a pathogenic role of infective-metabolic insult and inflammatory reaction in the development of vascular and myocardial damage in patients with heart failure even in the absence of overt coronary artery disease.
INTRODUCTION
Heat shock proteins (Hsp) are a family of intracellular proteins with well-known cytoprotective functions (Morimoto 1993). They are considered as molecular chaperones essential for cell survival both in physiological and stress conditions (Hightower 1991; Hartl 1996). However, although they are evidence of cellular insult, they contribute at the same time to the inflammatory reaction. In fact, following stress, Hsps can be presented on the cell surface or released to the surroundings, thereby activating the immune system (Srivastava 2002) and mediating the production of proinflammatory cytokines (Asea et al 2000).
Hsps of the 60-kD family and the stress-inducible Hsp72 of the 70-kD family recently have been linked to the atherosclerotic process (Xu 2002) and to ischemic myocardial damage (Dybdahl et al 2005). In particular, Hsp60 is expressed on the endothelial cell surface and in the myocyte in response to biochemical and/or infective insults (Knowlton et al 1998; Kanwar et al 2001; Xu 2002). It is thought to participate in inflammatory processes causing early atherogenesis and destabilization of the atherosclerotic plaque (Xu 2002; Mandal et al 2004) by activation of the autoimmune response. Both vascular and myocardial expressed Hsp60 also may elicit an autoimmune reaction able to cause further vascular and myocardial damage. On the other hand, Hsp72 is an inducible myocyte protein that has a specific role in myocardial protection from “chronic” ischemia (Martin et al 1997; Knowlton et al 1998; Biasucci et al 2003). It is expressed in the myocyte in response to brief periods of ischemia or mechanical stretch (Knowlton et al 1991a, 1991b) and participates in myocardial adaptive processes to chronic or repetitive ischemia known as “hibernation” (viable but dysfunctional myocardium) (Fallavollita et al 1999; Depré et al 2004). Thus, high levels of circulating Hsp60 and high titers of anti-Hsp60 auto-antibodies have been reported in patients with acute or chronic coronary artery disease (Portig et al 1997; Latif et al 1999; Xu 2002; Biasucci et al 2003; Genth-Zotz et al 2004) and high tissue levels of Hsp72 have been documented in myocardial hibernation (Depré et al 2004).
Little is known about the possible involvement of the Hsp60 and Hsp72 systems in ventricular dysfunction associated with non-atherosclerotic cardiac diseases. It has been reported that in response to a metabolic/infective insult Hsps may mediate coronary endothelial dysfunction and produce microvascular damage (Kuhl et al 2005; Sampietro et al 2005). Coronary microvascular impairment is a common feature in patients with idiopathic left ventricular dysfunction and may contribute to myocardial damage (Van den Heuvel et al 2000; Neglia et al 2002). This mechanism may in turn elicit myocardial Hsps and an inflammatory response in these patients (Depré et al 2004).
We thus hypothesized that Hsps could be increased, in association with systemic markers of inflammation, in patients with idiopathic left ventricular dysfunction, a clinical model that, by definition, excludes the presenceof coronary artery disease. To test this hypothesiswe evaluated Hsp60, Hsp72, anti-Hsp60 auto-antibodies, and inflammatory markers in the peripheral circulation of selected patients with normal coronary angiography, variable severity of ventricular dysfunction and, in a subgroup, characterization of coronary microvascular function by positron emission tomography (PET).
METHODS
Patients
Among 597 patients admitted to the cardiological ward of the Consiglio Nazionale delle Ricerche (CNR) Institute of Clinical Physiology from June 2001 to June 2005 and angiographically documented normal coronary arteries, we enrolled 60 consecutive patients (age 59 ± 12 years) with idiopathic LV systolic dysfunction. All patients had come to medical attention because of dyspnea on effort, ventricular arrhythmias, chest pain, and/or conduction disturbances, with evidence of regional or global LV dysfunction at 2-dimensional–(2D) echocardiographic evaluation leading to coronary angiography. Inclusion criteria were: (1) LV ejection fraction (EF) <50% at 2-D echocardiography; (2) functional New York Heart Association (NYHA) class I–III; (3) angiographically normal coronary arteries; (4) exclusion of vasospastic angina, congenital, or valvular heart disease, hypertrophic cardiomyopathy, myocarditis, pericarditis, lung disease, and primary thyroid disease; (5) presence of sinus rhythm; (6) exclusion of alcohol abuse or other systemic diseases.
Patients were subdivided into 2 groups according to the presence of mild (LVEF ≥35% to <50%, 32 patients, group I) or moderate–severe (LVEF <35%, 28 patients, group II) LV systolic dysfunction. Twenty-two additional patients (age 51 ± 12 years) submitted to coronary angiography because of the same clinical presentation but with normal LV systolic function at 2D echo evaluation and normal coronary angiography, also were enrolled (group N).
A local ethics review committee approved the study, and the investigation conformed to the principles outlined in the Declaration of Helsinki.
All patients gave their written informed consent to be involved in the PET study.
LV function, coronary microvascular, and biohumoral profiles
LVEF, LV end-diastolic dimensions (LVEDD, mm), and LV mass index were obtained by 2D–echocardiographic evaluation according to international standards (Lang et al 2005). In 38 patients coronary microvascular function also was assessed by rest/stress PET myocardial blood flow (MBF) study. Absolute MBF was measured, using 13N-Ammonia as a flow tracer, at rest and during i.v. dipyridamole (0.56 mg/Kg in 4 min) or adenosine (140 μg/ Kg/min for 6 min). Coronary resistance was computed in each condition as the ratio of mean aortic pressure over MBF value. Coronary reserves of flow and resistance were obtained as the ratio of stress/rest MBF or rest/stress resistance, respectively. Both the PET protocol and the data analysis have been described elsewhere (Neglia et al 1995). Brain natriuretic peptide (BNP) was obtained with 2-site immunoradiometric assays ([IRMA]; Shionogi, Osaka, Japan), as previously described (Del Ry et al 2000). Lipid, glucose, and insulin profiles were obtained by standard measurements.
Hsps and inflammatory profile
To determine heat shock proteins levels, auto-antibodies, and inflammatory markers, blood samples were withdrawn in all patients at 8 AM following an overnight fast. Blood samples also were obtained in the same conditions in 44 age-matched healthy subjects (group C) included as control group for these measurements. All healthy subjects were not obese, had normal arterial blood pressure, and were free from acute disease, as determined by an interview with a clinician. All of them had normal values for the main plasma parameters and normal leukocyte count. Furthermore, they denied the use of any drug during the 4 weeks before the study.
Serum samples were used for determination of Hsps, auto-antibody anti-Hsps, and C-reactive protein (CRP). For the cytokine assay, blood samples were collected into ice-chilled polypropylene tubes in the presence of ethylenediamine-tetraacetic acid ([EDTA] 1 mg/mL) in an ice bath and the plasma was prepared within 1 hour of the withdrawal, by centrifugation at 1500 × g at +4°C and stored frozen at −80°C until assay.
Hsp60 and Hsp72 were measured by commercially available specific enzyme-linked immunosorbent assay (ELISA) methods (StressGen Biotechnologies, Victoria, British Columbia, Canada). The lower detection limit was 1.3 ± 0.14 and 0.33 ± 0.07 ng/mL for Hsp60 and Hsp72, respectively. In our conditions, the working range (analyte determination with an imprecision less than 10%) resulted 10–100 ng/mL and 1–25 ng/mL, for Hsp60 and Hsp72, respectively, as evaluated by the imprecision profile determined for each dose-response curve (Pilo et al 1982).
Similarly, anti-Hsp60 auto-antibody levels in serum samples were determined by a commercially available ELISA kit (StressGen Biotechnologies). This method allows us to determine the concentration instead of the titer of anti-Hsp60 auto-antibodies in human serum. The working range resulted 11–250 ng/mL and sensitivity was 2.0 ± 0.20 ng/mL, mean ± standard error of mean (SEM); the between- and the within-assay variability were 18.7 ± 0.46 (7.3%, n = 9) and 18.3 ± 0.20 μg/mL (2.2%), respectively. A parallelism of the response from 1 to 0.05 μL of serum and a quantitative recovery of the anti-Hsp60 antibody standard preparation added to a 1: 500 diluted sample ruled out any matrix effect. Usually 1:500 dilution of unknown samples was suitable for the assay.
Plasma level of IL-6 was measured by a high-sensitivity ELISA technique (Diaclone Research, Besancon, France). Sensitivity level was 0.33 ± 0.04 pg/mL; the working range was 2.2–50 pg/mL; between-assay variability resulted 23.5 ± 0.48 pg/mL (CV% 8.5, n = 17) and 0.50 ± 0.06 pg/mL (31.6%, n = 6).
C-reactive protein (CRP) was measured in human serum by a high-sensitivity ELISA method (Diagnostics Biochem Canada Inc, London, ON, Canada); lower detection limit was about 0.001 mg/dL and the working range 0.005–1.0 mg/dL.
Statistical analysis
All the results are reported as mean ± SEM. All the values of sample concentration and the quality control of the immunometric assays of Hsp60, Hsp72, and anti-Hsp60 antibodies, as well as of IL-6 and CRP, were calculated by using a 4-parameter logistic function to interpolate dose-response curve (Pilo et al 1982).
For multiple comparisons, analysis of variance (ANOVA) followed by Fisher's test was used for continous variables after a logarithmic transformation of the original variables not normally distributed. The frequency of events was compared by Chi-square test for parametric variables. Simple regression analysis was performed to correlate different variables. A P value <0.05 was considered significant.
RESULTS
Clinical, functional, microvascular, and metabolic profiles
Clinical, functional, and microvascular data are represented in Table 1 and compared among the 3 groups of patients with normal or moderately or severely depressed LV function (groups N, I, and II, respectively). Patients in group II were older, had significantly higher BNP values, larger ventricle, and lower systemic pressure values. Although all patient groups showed abnormal values of MBF, as compared with previously published values in normals (Neglia et al 2002), patients in group II had a more severe depression of coronary microvascular function than the other groups, as expressed by significantly higher coronary resistance during dipyridamol and lower coronary resistance reserve.
Table 1.
Clinical, functional, and positron emission tomography (PET) characteristics at enrollment

The 3 groups did not show any significant difference in risk factors as shown in Table 2. They differed for treatment regimens as follows: ace-inhibitors were assumed by 64%, 97%, and 93% of patients in groups N, I, and II, respectively (P = 0.001); beta-blockers in 32%, 56%, and 75% (P = 0.0094); digitalis in 23%, 25%, and 50%; diuretics in 36%, 59%, and 89%; and amiodarone in 18% of all patients.
Table 2.
Metabolic risk factors at enrollment

Hsps, inflammatory profile, and LV function
Data obtained in healthy controls (group C) and in patients of groups N, I, and II are shown in Table 3.
Table 3.
Circulating levels of Hsps, auto-antibodies to Hsps and inflammatory markers in patients and in healthy controls

Anti-Hsp60 antibodies and Hsp60 protein concentration were significantly elevated only in patients with more severe LV dysfunction (group II). However, Hsp60 protein frequently was undetectable (in 72% of all subjects) and, interestingly, was more undetectable in patients with LV dysfunction (90% in group I and 80% in group II) than in patients of group N (68%) and in healthy controls (58%) (P = 0.0161).
Hsp72 protein concentration was significantly elevated in patients with more severe disease (group II) and only slightly elevated in the other groups of patients as compared with controls (Fig 1).
Fig 1.
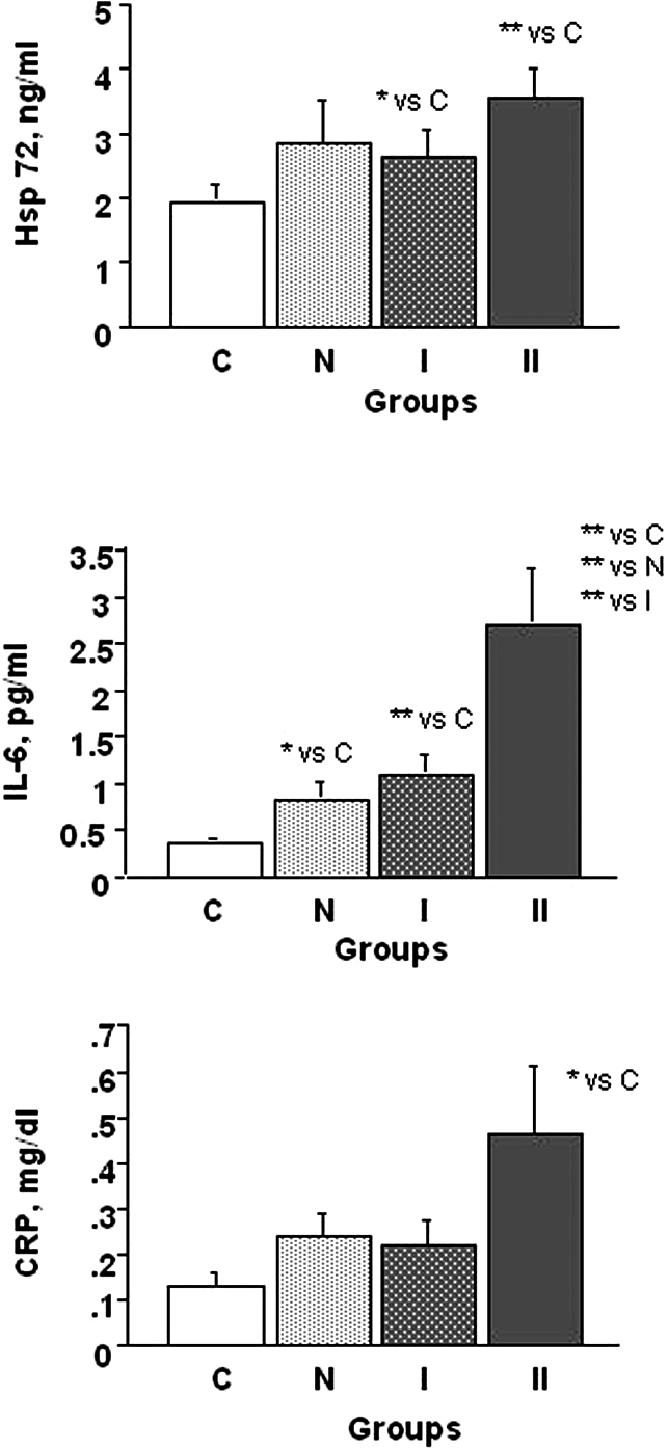
Serum levels of Hsp72, IL-6, and C-reactive protein (CRP) as a function of severity of left ventricle (LV) systolic dysfunction: group C (healthy controls), group N (LV ejection fraction [EF] ≥ 50%), group I (LVEF ≥35% to <50%), and group II (LVEF <35%). Hsp72: (**)P < 0.001 group II vs group C; (*)P < 0.05 group I vs group C. IL-6: (**)P < 0.001 group II and group I vs group C; P < 0.001 group II vs group N; P < 0.005 group II vs group I; (*)P < 0.05 group N vs group C. CRP: (*)P < 0.05 Group II vs Group C
Patients in all groups showed a systemic activation of the cytokine system as documented by significantly elevated values of IL-6, as compared with healthy controls. Among patients, IL-6 progressively increased with the severity of LV dysfunction (Fig 1). At variance with IL-6, CRP was significantly increased only in group II patients.
In the whole patient population there was a significant direct correlation of Hsp72, anti-Hsp60 autoantibodies, and IL-6 with BNP levels (Fig 2) or LVEDD values (Fig 3). Moreover, IL-6 tended to be related to Hsp72 (r = 0.24, P = 0.056), particularly in patients with more severe disease (group II, r = 0.45, P= 0.021). When patients were subdivided according to NYHA functional classes, all the parameters previously described, but not CRP, tended to be related to symptoms of heart failure reproducing the same pattern described for LV function (Fig 4).
Fig 2.

Correlation of Hsp72, anti-Hsp60 auto-antibodies and IL-6 with brain natriuretic peptide (BNP) levels in the whole population of patients: group C (healthy controls), group N (left ventricular ejection fraction [LVEF] ≥50%), group I (LVEF ≥35% to <50%) and group II (LVEF <35%)
Fig 3.
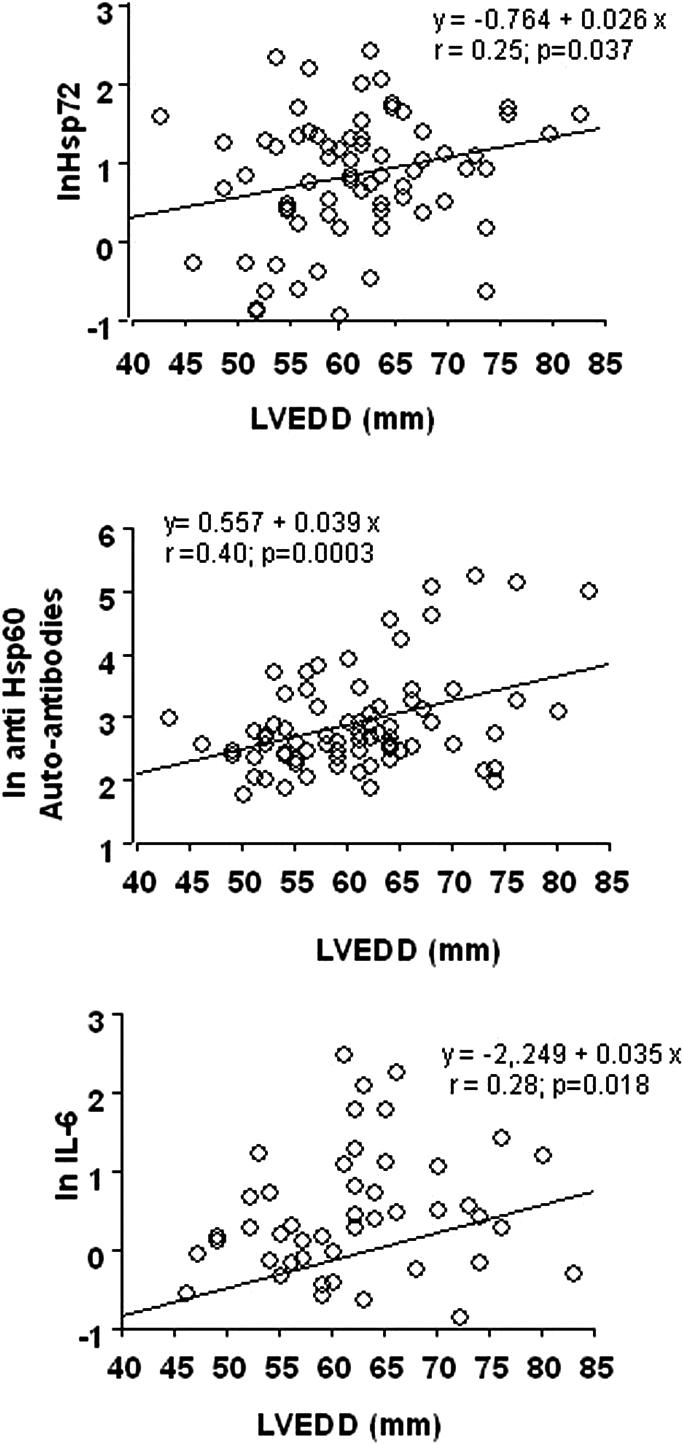
Correlation of Hsp72, anti-Hsp60 auto-antibodies, and IL-6 with left ventricular end-diastolic dimensions (LVEDD) values in the whole population of patients: group C (healthy controls), group N (LV ejection fraction [EF] ≥50%), group I (LVEF ≥35% to <50%) and group II (LVEF <35%)
Fig 4.
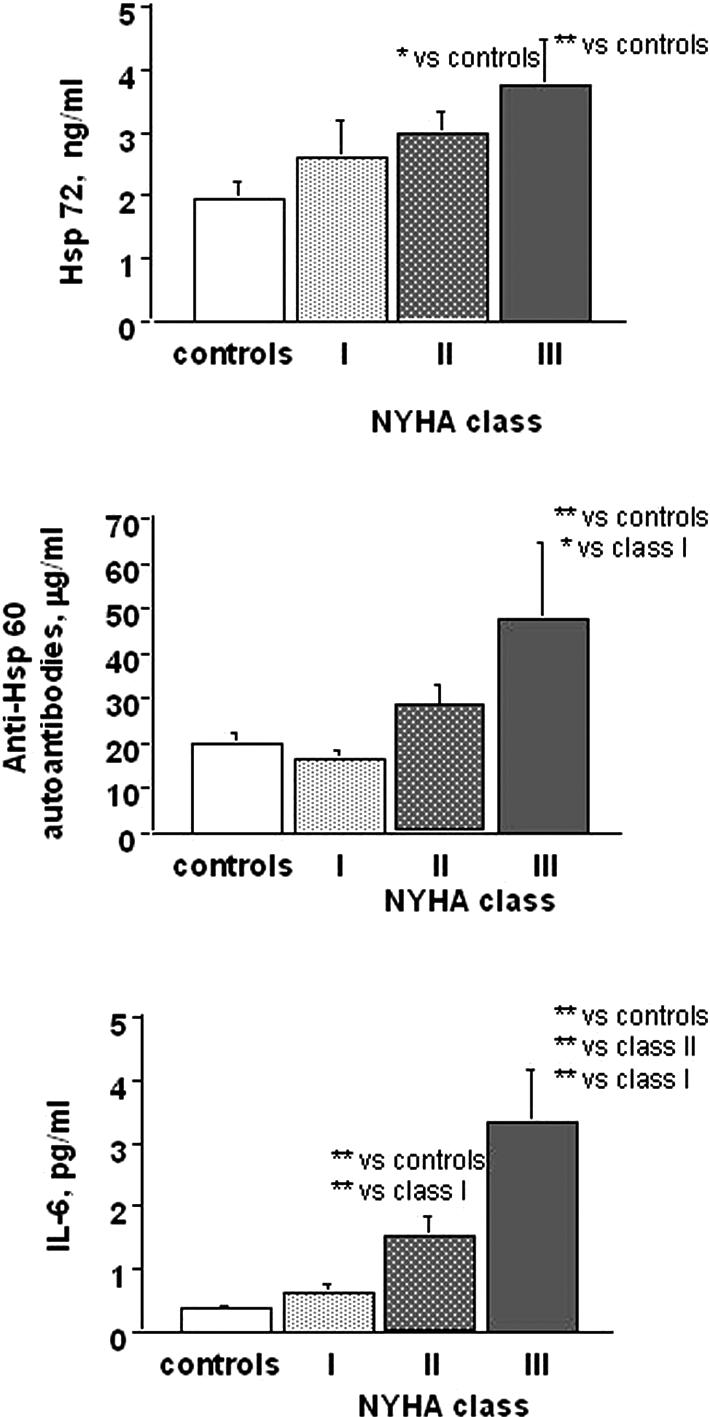
Serum levels of Hsp72, IL-6, anti-Hsp60 auto-antibodies as a function of NYHA class. Hsp72: (**)P < 0.005 NYHA class II and III vs controls. Anti-Hsp60 auto-antibodies: (*)P < 0.01 NYHA class III vs controls; (*)P < 0.05 NYHA class I vs III. IL-6: (**)P < 0.0001 NYHA class II and III vs controls; (**)P < 0.005 NYHA class I vs II; (**)P < 0.0001 NYHA class I vs III; (**)P < 0.005 NYHA class II vs III
Hsps, inflammatory profile, and coronary microvascular function
In the subgroup of 34 patients who had undergone PET study, Hsp and inflammatory profiles were analyzed according to the extent of coronary microvascular dysfunction as expressed by minimal coronary resistance during dipyridamole (Fig 5). Patients with dipyridamole resistance higher than the median value in the patients' population (High R) had significantly higher Hsp72 protein expression and tended to show higher Hsp60 protein, anti-Hsp60 auto-antibodies, and IL-6 levels than patients with lower dipiridamole coronary resistance (Low R). Patients with more or less severe coronary microvascular function differed for LV functional parameters, as expected, and did not differ for any other clinical and metabolic variable (Table 4). CRP values were similar in the 2 groups of patients. Dipyridamole coronary resistance was correlated inversely with Hsp72 protein levels (r = −0.388; P = 0.034) but with no other Hsps or inflammatory parameter.
Fig 5.

Hsp72, IL-6, Hsp60, and anti-Hsp60 auto-antibodies in patients with minimal coronary resistance, respectively, higher (High R) and lower (Low R) than the median value in the patient population. Patients with more severely impaired coronary microvascular function (High R) had significantly increased circulating Hsp72 protein and IL-6 levels
Table 4.
Clinical, functional, and metabolic parameters as a function of microvascular dysfunction

DISCUSSION
The present data demonstrate that, in patients with idiopathic LV dysfunction, the Hsp60 and 70 systems are activated. Circulating Hsp60 protein and, in particular, the related auto-antibodies, as well as Hsp70 protein are particularly elevated in patients with more severe disease who show both more pronounced LV dysfunction and more evident coronary microvascular impairment. Because the activation of these Hsp systems appears related both with biomarkers of systemic inflammation (in particular cytokine levels) and the extent of cardiac and microvascular dysfunction, it might be speculated that, as in coronary artery disease, Hsps constitute a link between an infective-metabolic insult, the consequent inflammatory reaction, and the microvascular and myocardial damage.
Relationships between Hsp60–70 activation, endothelial/vascular dysfunction, and myocardial damage in ischemic heart disease
The constitutively expressed Hsps of the 60-kD family recently have been associated with the pathogenesis of ischemic heart disease (Xu 2002; Dybdahl et al 2005). In response to an infective or metabolic insult, members of the Hsp60 family may act as auto-antigens, inducing vascular inflammatory responses and hence contributing to acute and chronic dysfunction of the endothelium with development or destabilization of coronary atherosclerosis (Xu 2002; Mandal et al 2004). Moreover, Hsp60, a constitutive myocyte protein with cytoprotective functions, is increased in the myocardium of patients with heart failure secondary to ischemic heart disease (Knowlton et al 1998). Accordingly, it is not surprising that high levels of Hsp60 and high titers of anti-Hsp60 auto-antibodies have been demonstrated in blood samples from patients with acute coronary syndromes and with chronic heart failure of atherosclerotic origin (Xu 2002; Biasucci et al 2003).
In patients with heart failure due to ischemic or idiopathic LV dysfunction (Genth-Zotz et al 2004), as well as in patients with unstable angina or after acute myocardial infarction (Dybdahl et al 2005, Valen et al 2000), increased levels of circulating Hsp72 have been documented. In particular, different experimental studies have demonstrated that Hsp72 is expressed rapidly in the myocardium in response to brief periods of ischemia and mechanical stretch (Knowlton et al 1991a, 1991b) and is a relevant part of the myocardial adaptive processes to chronic or repetitive ischemia known as myocardial hibernation (Fallavollita et al 1999; Depré et al 2004). It is believed that Hsp72 exerts a protective effect against ischemia (Mestril et al 1994; Martin et al 1997), but also a proinflammatory effect by inducing IL-6 production (Asea et al 2000; Campisi et al 2003).
Hsp60–70 activation and idiopathic LV dysfunction
Few data are available on circulating levels of Hsps and their related auto-antibodies in non-atherosclerotic cardiac diseases. Previous clinical reports (Portig et al 1997; Latif et al 1999) mainly refer to populations of patients with mixed etiology, prevalently of ischemic origin, and advanced heart failure where many different systems, including Hsps, are known to be secondarily activated. None of previous studies was able to document a direct relationship between circulating levels of Hsps or related antibodies from one site and inflammatory markers or indexes of LV dysfunction from the other, specifically in patients with idiopathic LV dysfunction with or without overt heart failure. In patients with idiopathic LV dysfunction, a frequent impairment of coronary microvascular function has been documented, even before the occurrence of heart failure, likely due to endothelial damage possibly causing repetitive and chronic myocardial ischemia (Van den Heuvel et al 2000; Neglia et al 2002). We hypothesized that, similarly to patients with ischemic heart disease, patients with non-atherosclerotic LV impairment and coronary microvascular dysfunction also could show an early activation of the Hsp60 and Hsp70 systems as part of the disease process.
The results of the present study confirm the hypothesis. We carefully selected a population of patients without coronary artery disease, with different degrees of idiopathic LV dysfunction and, in the large majority, with mild heart failure symptoms (NYHA I–II in 83% of the population). Anti-Hsp60 antibodies were elevated abnormally in these patients, in particular in those with more severe dysfunction, without correlation either with inflammatory markers or with microvascular function. Hsp60 was more frequently undetectable in patients as compared with controls (possibly due to the binding with the specific auto-antibodies) and, when detectable, it reached very high levels in patients with severe dysfunction. Whether the activation of Hsp60 and anti-Hsp60 auto-antibodies could depend on a previous or recurrent bacterial–viral infection and could be involved in the generation of myocardial damage in these patients cannot be stated from the present study. As a matter of fact, a similar increase of anti-chlamydial pneumoniae-Hsp60 auto-antibodies has been demonstrated in acute coronary syndromes persisting to some extent later on, but also in these case no correlation with inflammatory markers has been reported (Biasucci et al 2003). It is suggestive that Hsp60 system activation could be linked to vascular endothelial activation both in patients with or without coronary artery disease.
A relevant result of the present study was the clear demonstration of increased circulating levels of Hsp72 in patients with idiopathic LV dysfunction. Hsp72 was related with the degree of LV dysfunction, with IL-6 levels and with the degree of coronary microvascular impairment. No such relationship had been described previously in heart failure, probably due to the confounding effects of systemic neurohumoral activation in advanced disease and to mixed etiology of cardiac damage. We hypothesize that Hsp72 might be expressed in the myocardium of patients with idiopathic LV dysfunction in response to myocardial “microvascular” ischemia and might induce overexpression of cytokines, therefore mediating a secondary inflammatory damage. As a matter of fact, high levels of Hsp72 were documented in the myocardium of patients with dilated cardiomyopathy (Knowlton et al 1998) as well as in experimental models of progressive ischemic LV dysfunction (Fallavollita et al 1999). High levels of Hsp72 also induce expression of IL-6 (Asea et al 2000).
In our study, activation of Hsp systems and cytokines were also proportional to the severity of heart failure as indicated by the NYHA class. Possibly due to the prevalence of patients with no or mild heart failure, there was no relationship between Hsps and aspecific markers of neurohumoral activation such as norepinephrine levels, plasma renin activity, or aldosterone (data not shown). Conversely, a significant relationship was found with BNP levels, which more directly reflects LV myocardial impairment.
Hsp60, anti-Hsp60 auto-antibodies, and Hsp72 levels were not correlated with CRP levels otherwise increased in this population as compared with controls. CRP levels were related with IL-6 but not with LV or microvascular dysfunction. These data most likely express a systemic activation of inflammatory mechanisms in patients with LV dysfunction. However, although the Hsp and cytokine systems are known to be related reciprocally in the response of vascular and myocardial tissues to stress, CRP levels are influenced by many factors and show higher interindividual variations.
CONCLUSION
In patients with idiopathic LV dysfunction Hsp60 and Hsp70 systems appear to be activated systemically as they are in patients with coronary artery disease and ischemic LV dysfunction. Because the particular population of this study did not show the systemic derangement characteristic of advanced heart failure, the Hsp activation most probably reflect early pathophysiologic mechanisms involving microvascular and myocardial dysfunctions. The increasing attention to the role of impaired coronary microcirculation and subclinical atherosclerosis in causing adverse prognosis and progressive LV dysfunction stresses the importance of detecting systemic markers of early vascular and myocardial damage, which also could be responsive to aggressive pharmacological treatment. Hsp72, anti-Hsp60 auto-antibodies, and IL-6 in association with signs of LV dysfunction and/or increased BNP levels might constitute a biohumoral alert of an ongoing vascular-myocardial damage process to be actively and aggressively treated.
REFERENCES
- Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. Hsp70 stimulates cytokine production through a CD-14–dependent pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–442. doi: 10.1038/74697.1078-8956(2000)006[0435:HSCPTA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Biasucci LM, Liuzzo G, and Ciervo A. et al. 2003 Antibody response to chlamydial heat shock protein 60 is strongly associated with acute coronary syndromes. Circulation. 107:3015–3017. [DOI] [PubMed] [Google Scholar]
- Campisi J, Leem TH, Fleshner M. Stress-induced extracellular Hsp72 is a functionally significant danger signal to the immune system. Cell Stress Chaperones. 2003;8:272–286. doi: 10.1379/1466-1268(2003)008<0272:sehiaf>2.0.co;2.1466-1268(2003)008[0272:SEHIAF]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Ry S, Clerico A, and Giannessi D. et al. 2000 Measurement of brain natriuretic peptide in plasma samples and cardiac tissue extracts by means of an immunoradiometric assay method. Scand J Clin Lab Invest. 60:81–90.10817394 [Google Scholar]
- Depré C, Kim SJ, and John AS. et al. 2004 Program of cell survival underlying human and experimental hibernating myocardium. Circ Res. 95:433–440. [DOI] [PubMed] [Google Scholar]
- Dybdahl B, Slordahl SA, Waage A, Kierulf P, Espevik T, Sundan A. Myocardial ischaemia and the inflammatory response: release of heat shock protein 70 after myocardial infarction. Heart. 2005;91:299–304. doi: 10.1136/hrt.2003.028092.1355-6037(2005)091[0299:MIATIR]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallavollita JA, Jacob S, Young RF, Canty JM Jr.. Regional alterations in SR Ca2+−ATPase, phospholamban, and Hsp70 expression in chronic hibernating myocardium. Am J Physiol Heart Circ Physiol. 1999;277:H1418–1428. doi: 10.1152/ajpheart.1999.277.4.H1418.0363-6135(1999)277[H1418:RAISCP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Genth-Zotz S, Bolger AP, Kalra PR, von Haehling S, Doehner W, Coats AJS, Volk HD, Anker SD. Heat shock protein 70 in patients with chronic heart failure: relation to disease severity and survival. Int J Cardiol. 2004;96:397–401. doi: 10.1016/j.ijcard.2003.08.008.0167-5273(2004)096[0397:HSPIPW]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Hartl F. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0.1476-4687(1996)381[0571:MCICPF]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Hightower LE. Heat shock, stress proteins, chaperones, and proteotoxicity. Cell. 1991;166:191–197. doi: 10.1016/0092-8674(91)90611-2.0092-8674(1991)166[0191:HSSPCA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Kanwar RK, Kanwar JR, Wang D, Ormrod DJ, Krissansen GW. Temporal expression of heat shock proteins 60 and 70 at lesion-prone sites during atherogenesis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:1991–1997. doi: 10.1161/hq1201.100263.1079-5642(2001)021[1991:TEOHSP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Knowlton AA, Brecher P, Apstein CS. Rapid expression of heat shock proteins in the rabbit after brief cardiac ischemia. J Clin Invest. 1991a;87:139–147. doi: 10.1172/JCI114963.0021-9738(1991)087[0139:REOHSP]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton AA, Eberli FR, Brecher P, Romo GM, Owen A, Apstein CS. A single myocardial stretch or decreased systolic fiber shortening stimulates the expression of heat shock protein 70 in the isolated, erythrocyte-perfused rabbit heart. J Clin Invest. 1991b;88:2018–2025. doi: 10.1172/JCI115529.0021-9738(1991)088[2018:ASMSOD]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton AA, Kapadia S, Torre-Amione G, Durand JB, Bies R, Young J, Mann DL. Differential expression of heat shock proteins in normal and failing human hearts. J Mol Cell Cardiol. 1998;30:811–818. doi: 10.1006/jmcc.1998.0646.0022-2828(1998)030[0811:DEOHSP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Kuhl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W, Pauschinger M. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation. 2005;112:1965–1970. doi: 10.1161/CIRCULATIONAHA.105.548156.0009-7322(2005)112[1965:VPITMI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Lang RM, Bierig M, and Devereux RB. et al. 2005 Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 18:1440–1463. [DOI] [PubMed] [Google Scholar]
- Latif N, Taylor PM, Khan MA, Yacoub MH, Dunn MJ. The expression of heat shock protein 60 in patients with dilated cardiomyopathy. Basic Res Cardiol. 1999;94:112–119. doi: 10.1007/s003950050133.0300-8428(1999)094[0112:TEOHSP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Mandal K, Jahangiri M, Xu Q. Autoimmunity to heat shock proteins in atherosclerosis. Autoimmunity Reviews. 2004;3:31–37. doi: 10.1016/S1568-9972(03)00088-0.1568-9972(2004)003[0031:ATHSPI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Martin JL, Mestril R, Hilal-Dandan R, Brunton LL, Dillmann WH. Small heat shock proteins and protection against ischemic injury in cardiac myocytes. Circulation. 1997;96:4343–4348. doi: 10.1161/01.cir.96.12.4343.0009-7322(1997)096[4343:SHSPAP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Mestril R, Chi S, Sayen R, O'Reilly K, Dillman WH. Expression of inducible stress protein 70 in rat heart myogenic cells confers protection against stimulated ischemia-induced injury. J Clin Invest. 1994;93:759–767. doi: 10.1172/JCI117030.0021-9738(1994)093[0759:EOISPI]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Science. 1993;259:1409–1410. doi: 10.1126/science.8451637.0193-4511(1993)259[1409:CISTAO]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Neglia D, Michelassi C, and Trivieri MG. et al. 2002 Prognostic role of myocardial blood flow impairment in idiopathic left ventricular dysfunction. Circulation. 105:186–193. [DOI] [PubMed] [Google Scholar]
- Neglia D, Parodi O, and Gallopin M. et al. 1995 Myocardial blood flow response to pacing tachycardia and to dipyridamole infusion in patients with dilated cardiomyopathy without overt heart failure. Circulation. 92:796–804. [DOI] [PubMed] [Google Scholar]
- Neglia D, Sambuceti G, Iozzo P, L'Abbate A, Strauss HW. Myocardial metabolic and receptor imaging in idiopathic dilated cardiomyopathy. Eur J Nucl Med Mol Imaging. 2002;29:1403–1413. doi: 10.1007/s00259-002-0898-y.1619-7089(2002)029[1403:MMARII]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Pilo A, Zucchelli GC, Malvano R, Masini S. Main features of computer algorithms for RIA data reduction. Comparison of same different approaches for the interpolation of dose-response curve. J Nucl Med All Sci. 1982;26:235–248.0392-0208(1982)026[0235:MFOCAF]2.0.CO;2 [PubMed] [Google Scholar]
- Portig I, Pankuweit S, Maisch B. Antibodies against stress proteins in sera of patients with dilated cardiomyopathy. J Mol Cell Cardiol. 1997;29:2245–2251. doi: 10.1006/jmcc.1997.0463.0022-2828(1997)029[2245:AASPIS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Sampietro T, Neglia D, and Bionda A. et al. 2005 Inflammatory markers and serum lipids in idiopathic dilated cardiomyopathy. Am J Cardiol. 96:1718–1720. [DOI] [PubMed] [Google Scholar]
- Srivastava P. Roles of heat shock proteins in innate and adaptative immunity. Nature Rev Immunol. 2002;2:185–194. doi: 10.1038/nri749.1474-1733(2002)002[0185:ROHSPI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Valen G, Hansson GK, Dumitrescu A, Vaage J. Unstable angina activates myocardial heat shock protein 72, endothelial nitric oxide synthase, and transcription factor NF-kb and AP-1. Cardiovasc Res. 2000;47:49–56. doi: 10.1016/s0008-6363(00)00071-7.0008-6363(2000)047[0049:UAAMHS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Van den Heuvel AF, Van Veldhuisen DJ, Van der Wall EE, Blanksma PK, Siebelink HM, Vaalburg WM, van Gilst WH, Crijns HJ. Regional myocardial blood flow reserve impairment and metabolic changes suggesting myocardial ischemia in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2000;35:19–28. doi: 10.1016/s0735-1097(99)00499-4.0735-1097(2000)035[0019:RMBFRI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Xu Q. Role of heat shock proteins in atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22:1547–1559. doi: 10.1161/01.atv.0000029720.59649.50.1079-5642(2002)022[1547:ROHSPI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]