

Abstract
The gastrin-releasing peptide receptor (GRP-R) is one of three members of the mammalian bombesin subfamily of seven-transmembrane G protein-coupled receptors that mediate diverse biological responses including secretion, neuromodulation, chemotaxis, and growth. The X chromosome-linked GRP-R gene is expressed widely during embryonic development and predominantly in gastrointestinal, neuronal, and neuroendocrine systems in the adult. Surprisingly, gene-targeted mice lacking a functional GRP-R gene develop and reproduce normally and show no gross phenotypic abnormalities. However, peripheral administration of bombesin at dosages up to 32 nmol/kg to such mice had no effect on the suppression of glucose intake, whereas normal mice showed a dose-dependent suppression of glucose intake. These data suggest that selective agonists for the GRP-R may be useful in inducing satiety.
Bombesin (BN) is an amidated tetradecapeptide originally purified from the skin of the European frog Bombina bombina (1). The two known mammalian BN-like peptides are gastrin-releasing peptide (GRP) (2) and neuromedin B (NMB) (3). Exogenous introduction of these peptides into various organ systems elicits a wide range of responses including secretion of gastrointestinal, adrenal, and pituitary hormones and gastric acid, pancreatic enzyme, and mucous; regulation of smooth muscle contraction; and modulation of neuronal firing rate (for review, see ref. 4). In the central nervous system, these peptides are postulated to play a role in thermoregulation, metabolism, behavior, and satiety (for review, see refs. 4 and 5). Mammalian BN-like peptides can also act as mitogens, as shown for Swiss 3T3 cells in vitro (6) and for human small cell lung carcinoma in vivo. In some of these cells, BN-like peptides appear to function as autocrine growth factors (7). As with many gastrointestinal hormones and peptides, which of these diverse responses are pharmacological and which are physiological remain unclear, primarily due to lack of highly selective agonists or antagonists with useful pharmacokinetic properties.
Molecular cloning studies have revealed the identity of three mammalian BN receptors: the GRP receptor (GRP-R) (8, 9), the NMB receptor (NMB-R) (10), and BN receptor subtype 3 (BRS-3) (11, 12). These three receptors share about 50% amino acid sequence identity. Although structurally similar, the three receptors are pharmacologically distinct. The GRP-R binds GRP with higher affinity than NMB, and the NMB-R binds NMB with higher affinity than GRP (13). In contrast, BRS-3 binds neither GRP nor NMB with an affinity higher than in the micromolar range, suggesting that the natural ligand for this receptor remains to be discovered. The GRP-R gene has been mapped at 70 map units to the telomeric region of the mouse X chromosome, which is homologous with human Xp22.2–p22.13 (Mouse Genome Database, August 1997. Mouse Genome Informatics, The Jackson Laboratory, Bar Harbor, MA. http://www.informatics.jax.org/).
Several studies of BN receptor expression provide potential clues about the function of these receptors in mammals. In adult animals, GRP-R and NMB-R are widely expressed in the central nervous system (10) and gastrointestinal tract (14, 15), whereas BRS-3 shows limited expression in the hypothalamus but is expressed in secondary spermatocytes (12). In the central nervous system, GRP-R expression is most prominent in several hypothalamic nuclei that are associated with feeding behavior, thermoregulation, and glucose regulation, whereas NMB-R is absent from these regions (16). Studies examining these effects after introduction of BN into specific hypothalamic nuclei suggest that GRP-R and/or BRS-3 are the BN receptors mediating these responses.
In the periphery, GRP-R is localized to many regions of the gastrointestinal tract, with particularly high densities in the pancreas (17) and stomach (18). With the exception of the esophagus, NMB-R in the gastrointestinal tract is more sparsely represented. Peripheral administration of BN produces a variety of effects directly or indirectly linked to activation of receptors for BN-like peptides in the gastrointestinal tract, including exocrine pancreatic secretion, gastrointestinal peptide hormone release, smooth muscle contraction, and reduction of food intake. As in the central nervous system, the existence of GRP-R in regions of the gastrointestinal tract involved in these responses suggests that they are mediated by this BN receptor subtype. Although GRP-R and NMB-R antisense oligonucleotides have been successfully used to examine receptor subtype contribution to BN-induced colonic smooth muscle contraction in vitro (19), their utility in peripheral in vivo studies is limited.
During development, expression of GRP-R occurs earlier and is more widespread than expression of NMB-R. In addition, GRP-R shows a transient expression in the posterior pituitary, with peak levels of expression occurring around embryonic day 20 followed by a rapid decline during the postnatal period (20). These studies suggest that GRP-R may be involved in posterior pituitary development. Analysis of a rhesus monkey model system suggested that GRP secreted from neuroendocrine cells in the lung stimulated airway development by binding to GRP-R expressed in airway epithelial cells (21). Thus, animals lacking a functional GRP-R gene might show abnormalities in lung development. We have now created a line of mice with a disrupted GRP-R gene to determine whether the GRP-R plays an essential role in mediating one or more of these varied biological responses to BN.
MATERIALS AND METHODS
Targeting Vector Construction and Selection of Targeted Embryonic Stem (ES) Cells.
The mouse GRP-R gene was isolated by screening a 129/SvJ mouse genomic library constructed from liver genomic DNA partially digested with MboI and cloned into the bacteriophage substitution vector Lambda GEM-11 (Promega) by using standard methodology (22). A 10-kb genomic clone that hybridized to a mouse GRP-R cDNA fragment was found to contain 3 kb of 5′ upstream region, exon 1, and part of intron 1 of the GRP-R gene. A replacement-type targeting vector was constructed from the plasmid vector pPNT [provided by V. Tybulewicz (23)] that contains a neomycin-resistance (neo) gene and a herpes simplex virus thymidine kinase (HSVtk) gene cassette, each driven by a phosphoglycerate kinase (PGK-1) promoter. A 1-kb NotI–SmaI fragment containing the 5′ upstream region and part of exon 1 of GRP-R was inserted 5′ of the neo cassette, and a 6-kb SmaI–BamHI fragment containing the remaining part of GRP-R exon 1 and part of intron 1 was inserted between the neo and the HSVtk cassettes. An additional BamHI site was incorporated into the construct to facilitate screening. The construct was linearized at a unique NotI site prior to electroporation into the 129/SvJ ES cell line R4 (Genome Systems, St. Louis). ES clones were selected with Geneticin (G418) and 1-[2-deoxy-2-fluoro-β-d-arabinofuranosyl]-5-iodouracil.
Generation of GRP-R-Deficient Mice.
G418/1-[2-deoxy-2-fluoro-β-d-arabinofuranosyl]-5-iodouracil-resistant clones were screened by Southern blot hybridization as indicated in the text and an ES cell clone that had undergone homologous recombination with the targeting vector was used to create blastocyst chimeras. From the offspring, an ES/C57BL/6J chimeric male was selected and backcrossed to C57BL/6J females. Germ-line transmission was confirmed by Southern blot hybridization with tail DNA. All further backcrosses were to the C57BL/6J strain and genotypes of offspring were subsequently determined by PCR. PCR was performed in duplicate on tail DNA by using two independent pairs of oligonucleotides specific for the neo and GRP-R genes that amplify a product only in mice that contain the targeted GRP-R allele.
Pathology and Histology.
A pathological and histological examination of the animals was performed by SAIC Frederick (Science Applications International, Frederick, MD).
Preparation of Dispersed Pancreatic Acini and Amylase Release Assay.
Dispersed mouse pancreatic acini were prepared as described (24, 25) with the modification that one instead of three mouse pancreata were used to prepare an acini suspension per experiment. This yielded a lower quantity of acini, but the procedures were otherwise identical. Acini from one mouse were resuspended in 3–4 ml of incubation buffer. Of this suspension, 1 ml was diluted in 30 ml incubation buffer for determination of amylase release, 1 ml was used directly for binding of 125I-labeled [Tyr4]BN, 1 ml was used directly for binding of 125I-labeled BH-CCK-8 (where CCK-8 is carboxyl-terminal octapeptide of cholecystokinin) and the remainder was saved for protein determination. Amylase release was measured as described (24, 25).
Binding of 125I-labeled [Tyr4]BN and 125I-labeled BH-CCK-8.
125I-labeled [Tyr4]BN was prepared as described (26) and binding was performed as described (25, 26). Briefly, 125I-labeled [Tyr4]BN (0.5 μCi/ml; 1 Ci = 37 GBq) was added to concentrated mouse acini (1-ml suspension) with nonradioactive BN to give a final concentration of 1 nM BN. Five hundred microliters was then incubated at 37°C for 60 min alone (total binding) or with 1 μM nonradioactive BN (nonsaturable binding). At the end of the incubation, the acini were resuspended and 100 μl was layered over ice-cold incubation buffer containing 4% (wt/vol) BSA. The suspensions were centrifuged 15 sec at 10,000 × g, the supernatants were aspirated, and the pellets were rinsed twice with 4% (wt/vol) BSA. The bound radioactivity was then quantitated with a γ counter along with an aliquot of uncentrifuged acini (to determine the total added counts). Binding was expressed as saturable binding of 125I-labeled [Tyr4]BN per milligram of acinar protein (fmol/mg of protein). The binding of 125I-labeled BH-CCK-8 was performed identically to the binding of 125I-labeled [Tyr4]BN except either 125I-labeled BH-CCK-8 or CCK-8 was substituted. Binding was expressed as saturable binding of 125I-labeled BH-CCK-8 per mg of acini protein (fmol/mg of protein).
Protein Determination.
Protein was determined in the acini suspensions by using the Bio-Rad protein assay according to the manufacturer’s specifications. Before protein determination, the acinar suspensions were dissolved by vortex mixing in 4 vol of 0.1% Triton X-100.
Glucose Intake After BN Administration.
Adult male GRP-R-targeted (n = 7) and wild-type (n = 6) mice with a starting weight of 28–36 g were the subjects for this experiment. They were individually housed in a temperature-controlled room on a 12-hr light/12-hr dark cycle. Rodent chow and tap water were available ad libitum unless specified otherwise. All behavioral testing was conducted during the light phase of the light/dark cycle at 1400 hr.
Mice were familiarized with the experimental procedure before behavioral testing. Each mouse was administered an intraperitoneal injection of 0.9% saline [1 ml/100 g (body weight)], placed in an 8 inch × 8 inch Plexiglas test chamber (1 inch = 2.54 cm), and given immediate access to a glucose solution (0.5 kcal/ml; 1 cal = 4.184 J) for 60 min. This procedure was repeated five times before testing began to stabilize glucose intake.
GRP-R-targeted and wild-type mice were then evaluated for their ability to suppress glucose intake in response to intraperitoneal administration of BN at 0 (0.9% saline), 1.0, 3.2, 10, and 32 nmol/kg delivered in a volume of 1 ml/100 g (body weight). Doses of BN were given in randomized order in a counterbalanced design and separated by at least 48 hr. Each dose of BN was preceded by a day in which saline was administered. Glucose intake was recorded to the nearest 0.1 ml at 15-min intervals for 60 min.
Glucose consumption after intraperitoneal saline administration was averaged across days for each mouse (baseline intake) and compared with intake after BN administration. Data were analyzed separately for each group by using repeated measures ANOVA followed by planned t test comparisons at each time point.
RESULTS
Generation of GRP-R-Deficient Mice.
To generate a GRP-R mutation, a replacement-type targeting vector was prepared in which exon 1 of the GRP-R was interrupted by a neo gene cassette and whose 3′ end is flanked by a HSVtk cassette, thus allowing positive–negative selection (Fig. 1A). The linearized vector was electroporated into a 129/SvJ ES line and clones were selected in the presence of G418 and 1-[2-deoxy-2-fluoro-β-d-arabinofuranosyl]-5-iodouracil. They were identified by Southern blot analysis using genomic DNA digested with BamHI and a 5′ genomic probe not contained within the targeting vector (Fig. 1A). Correct targeting in an ES cell clone was indicated by the loss of the wild-type 10-kb BamHI fragment normally residing on the single X chromosome of the male ES cells, and the generation of a novel 5.2-kb BamHI fragment resulting from the insertion of a BamHI site downstream of the neo gene (Fig. 1A and data not shown). This clone was injected into C57BL/6J blastocysts, yielding a male chimeric mouse that transmitted the targeted allele through the germ line, as shown for a heterozygous female offspring (Fig. 1B). This female was crossed with a C57BL/6J male to produce hemizygous males carrying the targeted allele (−/Y), and all subsequent backcrosses were to the C57BL/6J strain. In addition, heterozygous (+/−) females were crossed with hemizygous (−/Y) males to produce homozygous (−/−) females.
Figure 1.
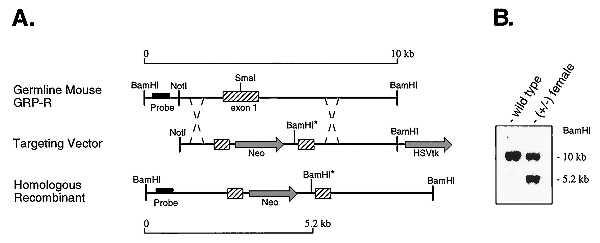
Targeted disruption of the mouse GRP-R gene. (A) Diagram of the GRP-R genomic structure, targeting vector construct, and gene structure after homologous recombination. Hatched boxes, GRP-R exon 1 sequence; shaded arrows, neo and HSVtk cassettes. The neo cassette is inserted into the SmaI site of the GRP-R exon 1 along with an additional BamHI site (noted by ∗). The location of a 5′ probe sequence is indicated by a solid bar. Homologous recombination after electroporation of ES cells with this construct yields a GRP-R mutant allele in which the 10-kb wild-type BamHI genomic fragment is replaced with a 5.2-kb BamHI fragment. (B) Southern blot of BamHI-digested genomic DNA from a wild-type (+/Y) male and a heterozygous (+/−) female. Hybridization was to a 32P-labeled 600-bp 5′ fragment designated as “probe” in A. The molecular sizes of the genomic DNA fragments hybridizing to the probe are indicated.
The Targeted GRP-R Allele Expresses no Functional GRP-R.
BN, cholecystokinin, and carbachol stimulate amylase release when applied to dispersed rat pancreatic acinar cells, each by interacting with a distinct receptor (27). Because the BN-stimulated amylase release is mediated through the GRP-R, we predict that this response would be absent in GRP-R-targeted mice but the cholecystokinin- and carbachol-induced amylase release would be intact. Wild-type (+/Y) and GRP-R-targeted (−/Y) male mice were indistinguishable when CCK-8 (Fig. 2 Left) and carbachol (Fig. 2 Right) were used to elicit amylase release, but the BN-induced amylase release was robust in the wild-type mice but absent in the GRP-R-targeted mice (Fig. 2 Middle). Additional binding studies confirmed the complete absence of a high-affinity binding site for BN on the GRP-R-targeted male mice, in contrast to their wild-type male littermates, but the presence of normal binding sites for the cholecystokinin a receptor (data not shown).
Figure 2.
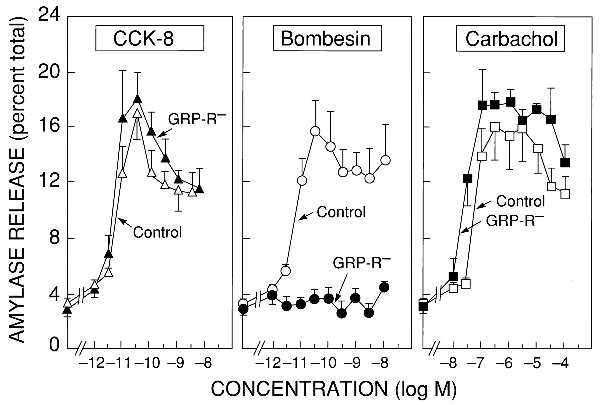
Amylase release in pancreatic acini. Abilities of CCK-8 (Left), BN (Center), and carbachol (Right) to stimulate amylase release from dispersed pancreatic acini prepared from GRP-R-targeted (−/Y) male mice (solid symbols) or control (+/Y) male littermates (open symbols). Dispersed acini were incubated with the indicated concentrations of secretagogues for 30 min at 37°C. Results are expressed as the percentage of total cellular amylase activity in the cells before the incubation that was released into the extracellular medium during the incubation and are the means ± SEM of three experiments performed in triplicate.
Phenotype of GRP-R-Deficient Mice.
No evidence for embryonic mortality was observed in GRP-R-deficient mice because offspring were born at the expected Mendelian frequencies. All offspring grew and developed normally with no gross phenotypic abnormalities. Hemizygous (−/Y) males, heterozygous (+/−) females, and homozygous (−/−) females also bred and showed no indication of reproductive deficiencies. A pathological/histological examination of all major organ systems (including brain, adrenal, pituitary, thymus, pancreas, stomach, intestine, colon, lung, liver, bladder, kidney, femur, and reproductive organs) showed no differences between GRP-R-deficient (−/Y) males (n = 2) and normal (+/Y) male littermates (n = 2) up to 10 months of age.
Loss of BN-Induced Satiety in GRP-R-Deficient Mice.
BN has many central and peripheral effects with satiety being one of the responses most widely studied (28–31). In rats, both GRP and NMB receptors have been implicated in mediating the satiety effect of peripherally administered BN (30, 31). If this response was mediated by BN binding to the GRP-R, then mice lacking a functional GRP-R should not show the BN-induced satiety observed in wild-type mice. This was in fact the case. Fig. 3 shows a comparison of glucose intake in wild-type and targeted mice after the administration of increasing amounts of BN. Even at the highest dose of BN (32 nmol/kg) given intraperitoneally, there is no evidence for a BN-induced satiety response in GRP-R-targeted mice. In contrast, a decrease in glucose intake is observed in wild-type littermates when BN as low as 3.2 nmol/kg is administered. This deficiency in mediating satiety did not appear to cause weight gain, because total body weights of GRP-R-targeted mice were not significantly different from the wild-type mice (data not shown).
Figure 3.
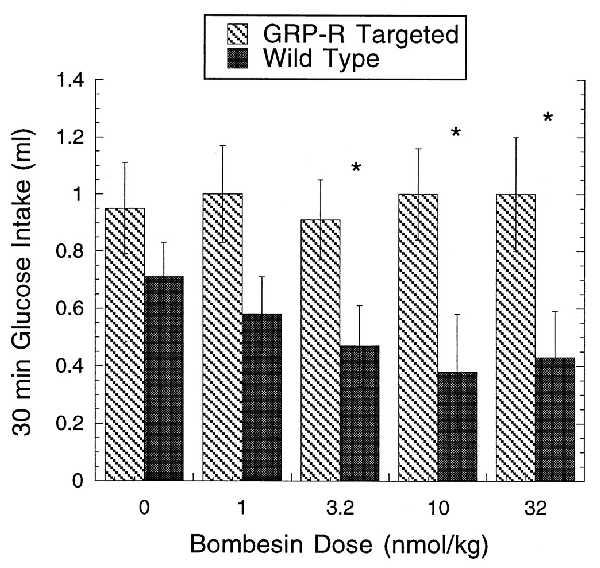
Effect of BN on glucose uptake. Thirty-minute glucose intake in GRP-R-targeted (−/Y) male mice and wild-type (+/Y) male littermates after intraperitoneal administration of BN at 0 (0.9% saline) or 1.0, 3.2, 10, and 32 nmol/kg. BN produced a significant suppression of glucose intake in wild-type mice at doses of 3.2, 10, and 32 nmol/kg but failed to suppress intake at any dose in GRP-R-targeted mice. ∗, Significantly different from 0.9% saline (P < 0.05).
DISCUSSION
We report the creation and germ-line transmission of a mouse GRP-R allele that is inactivated by targeted insertion of a neo gene in the GRP-R protein coding region. The resulting GRP-R is functionally inactive as shown by the specific loss of BN-induced amylase release from dispersed pancreatic acinar cells, whereas the carbachol- and cholecystokinin-induced amylase release remain intact in male mice hemizygous for the targeted GRP-R allele. Furthermore, no binding to GRP-R could be detected in the targeted mice, whereas binding to cholecystokinin A receptor was normal. Hemizygous males lacking a functional GRP-R allele did not show the BN-induced satiety response that was observed in their wild-type male littermates, establishing that peripherally administered BN-induced appetite suppression is mediated through the GRP-R and not any other BN receptor subtype.
Despite the fact that studies of GRP-R gene expression and functional studies using BN or BN-like peptides suggested a role for GRP-R during development, we observed no obvious gross phenotypic abnormalities in GRP-R (−/Y) males. It is possible that the loss-of-function mutation produced by gene targeting is compensated for by other intact members of the BN receptor family despite the fact that the expression patterns of the different receptors during development is not entirely overlapping; in this case, future studies of double-knockout mice homozygous for targeted alleles at the other BN receptor loci may reveal phenotypes not readily apparent when only one of the genes is disrupted. Another possible explanation for the observation that GRP-R-deficient mice show grossly normal development may be that other neuropeptide receptors, or other signaling pathways, may compensate for the function lost when GRP-R is eliminated from a developing organ system. The existence, importance, and evolutionary stability of redundancy of gene function has recently been explored in a theoretical study (32).
Mammalian BN-like peptides and their receptors have been implicated in the pathogenesis and progression of several human tumors, including a subset of human small cell lung carcinomas. If the GRP-R is critical for mediating the pathogenesis of lung malignancies, a decrease in incidence of lung malignancies may be associated with animal model systems lacking a functional GRP-R allele. This hypothesis could be tested by introducing the targeted GRP-R allele into a mouse genetic background where the susceptibility for carcinogen-induced lung malignancy is high.
Numerous pharmacological and behavioral studies have proposed that BN-like peptides may be important in mediating satiety in various species (28, 33, 34). These studies have relied mainly on pharmacological methods using BN receptor agonists and receptor-subtype-specific antagonists to block BN-induced suppression of feeding. However, because of the lack of highly selective agonists and antagonists, these type of analyses have produced equivocal results in determining the contribution of the GRP-R to the satiety actions of BN. Recent studies in humans have shown that intravenous infusions of GRP resulted in a dose-dependent reduction in caloric intake while producing sensations associated with normal satiety (35). Our data establish that the GRP-R mediates the BN-induced satiety response reported in the literature. The results are in agreement with a previous study in rats showing that a BN receptor antagonist (BW2258U89) selectively blocked the satiety response induced by BN administered peripherally (36). The present study provides unequivocal evidence that the cell surface target for the satiety response produced by peripheral administration of BN in mice is the GRP-R and not any other BN receptor subtype. Although there were no significant weight differences between the GRP-R-targeted and wild-type mice, the long-term effects of removal of a receptor involved in short-term regulation of food intake is currently unknown. It is possible that recruitment of other signals involved in meal termination play a compensatory role in animals lacking the GRP-R. In fact, preliminary data suggests that GRP-R-targeted mice have increased sensitivity to cholecystokinin, another peptide involved in regulating individual meal size. A recent report of mice that lack the BRS-3 receptor and develop obesity and metabolic defects (37) emphasizes the importance of these mammalian BN-like peptide receptors in regulating feeding and metabolism. These observations suggest that the search for small molecule agonists for the GRP-R may be a useful strategy for the development of novel therapeutic approaches to weight control disorders.
Acknowledgments
We are grateful to Dr. Victor Tibulewicz for kindly providing the pPNT vector used in these experiments and we thank Dr. Helen Hellmich for providing the mouse 129/SvJ genomic library used to identify genomic clones for constructing the GRP-R-targeting vector. Animal studies were conducted under National Institutes of Health Protocol 776–97.
ABBREVIATIONS
- BN
bombesin
- GRP
gastrin-releasing peptide
- NMB
neuromedin B
- GRP-R
GRP receptor
- NMB-R
NMB receptor
- BRS-3
BN receptor subtype 3
- ES
embryonic stem
- HSVtk
herpes simplex virus thymidine kinase
- neo
neomycin
- CCK-8
carboxyl-terminal octapeptide of cholecystokinin
References
- 1.Anastasi A, Erspamer V, Bucci M. Experientia. 1971;27:166–167. doi: 10.1007/BF02145873. [DOI] [PubMed] [Google Scholar]
- 2.McDonald T J, Jornvall H, Nilsson M, Vagne M, Ghatei M, Bloom S R, Mutt V. Biochem Biophys Res Commun. 1979;90:227–233. doi: 10.1016/0006-291x(79)91614-0. [DOI] [PubMed] [Google Scholar]
- 3.Minamino N, Kangawa K, Matuso H. Biochem Biophys Res Commun. 1983;114:541–548. doi: 10.1016/0006-291x(83)90814-8. [DOI] [PubMed] [Google Scholar]
- 4.Lebacq-Verheyden A-M, Trepel J, Sausville E A, Battey J F. In: Handbook of Experimental Pharmacology. Sporn M, Roberts A, editors. Vol. 95. Berlin: Springer; 1990. pp. 71–124. [Google Scholar]
- 5.Spindel E R. Trends Neurosci. 1986;9:130–133. [Google Scholar]
- 6.Rozengurt E, Sinnett-Smith J. Proc Natl Acad Sci USA. 1983;80:2936–2940. doi: 10.1073/pnas.80.10.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuttitta F, Carney D N, Mulshine J, Moody T W, Fedorko J, Fischler A, Minna J D. Nature (London) 1985;316:823–826. doi: 10.1038/316823a0. [DOI] [PubMed] [Google Scholar]
- 8.Spindel E R, Giladi E, Brehm P, Goodman R H, Segerson T P. Mol Endocrinol. 1990;4(12):1956–1963. doi: 10.1210/mend-4-12-1956. [DOI] [PubMed] [Google Scholar]
- 9.Battey J F, Way J M, Corjay M H, Shapira H, Kusano K, Harkins R, Wu J M, Slattery T, Mann E, Feldman R I. Proc Natl Acad Sci USA. 1991;88:395–399. doi: 10.1073/pnas.88.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wada E, Way J, Shapira H, Kusano K, Lebacq-Verheyden A-M, Coy D, Jensen R, Battey J F. Neuron. 1991;6:421–430. doi: 10.1016/0896-6273(91)90250-4. [DOI] [PubMed] [Google Scholar]
- 11.Gorbulev V, Akhundova A, Buchner H, Fahrenholz F. Eur J Biochem. 1992;208:405–410. doi: 10.1111/j.1432-1033.1992.tb17201.x. [DOI] [PubMed] [Google Scholar]
- 12.Fathi Z, Corjay M H, Shapira H, Wada E, Benya R, Jensen R, Viallet J, Sausville E A, Battey J F. J Biol Chem. 1993;268:5979–5984. [PubMed] [Google Scholar]
- 13.von Schrenck T, Wang L-H, Coy D H, Villanueva M L, Mantey S, Jensen R T. Am J Physiol. 1990;259:G468–G473. doi: 10.1152/ajpgi.1990.259.3.G468. [DOI] [PubMed] [Google Scholar]
- 14.Moran T H, Moody T W, Hostetler A M, Robinson P H, Goldrich M, McHugh P R. Peptides. 1988;9:643–649. doi: 10.1016/0196-9781(88)90177-5. [DOI] [PubMed] [Google Scholar]
- 15.von Schrenck T, Heinz-Erian P, Moran T, Mantey S A, Gardner J D, Jensen R T. Am J Physiol. 1989;256:G747–G758. doi: 10.1152/ajpgi.1989.256.4.G747. [DOI] [PubMed] [Google Scholar]
- 16.Wada E, Wray S, Key S, Battey J. Mol Cell Neurosci. 1992;3:446–460. doi: 10.1016/1044-7431(92)90056-8. [DOI] [PubMed] [Google Scholar]
- 17.Jensen R T, Coy D H, Saeed Z A, Heinz-Erian P, Mantey S, Gardner J D. Ann NY Acad Sci. 1988;547:138–149. doi: 10.1111/j.1749-6632.1988.tb23882.x. [DOI] [PubMed] [Google Scholar]
- 18.Ladenheim E E, Moore K A, Salorio C F, Mantey S A, Taylor J E, Coy D H, Jensen R T, Moran T H. Eur J Pharmacol. 1997;319:245–251. doi: 10.1016/s0014-2999(96)00854-0. [DOI] [PubMed] [Google Scholar]
- 19.Bitar K N, Zhu X-X. Gastroenterology. 1993;105:1672–1680. doi: 10.1016/0016-5085(93)91062-m. [DOI] [PubMed] [Google Scholar]
- 20.Wada E, Battey J, Wray S. Mol Cell Neurosci. 1993;4:13–24. doi: 10.1006/mcne.1993.1002. [DOI] [PubMed] [Google Scholar]
- 21.Li K, Nagalla S R, Spindel E R. J Clin Invest. 1994;94:1605–1615. doi: 10.1172/JCI117502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis L, Kuehl M, Battey J F. Basic Methods in Molecular Biology. 2nd Ed. Norwalk, CT: Appleton & Lange; 1994. pp. 397–456. [Google Scholar]
- 23.Tybulewicz V L J, Crawford C E, Jackson P K, Bronson R T, Mulligan R C. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 24.Jensen R T, Lemp G F, Gardner J D. J Biol Chem. 1982;257:5554–5559. [PubMed] [Google Scholar]
- 25.Huang S C, Yu D H, Wank S A, Gardner J D, Jensen R T. Peptides. 1990;11:1143–1150. doi: 10.1016/0196-9781(90)90144-t. [DOI] [PubMed] [Google Scholar]
- 26.Mantey S, Frucht H, Coy D H, Jensen R T. Mol Pharmacol. 1993;43:762–774. [PubMed] [Google Scholar]
- 27.Jensen R T. In: Physiology of the Gastrointestinal Tract. 3rd Ed. Johnson L R, Jacobsen E D, Christensen J, Alpers D H, Walsh J H, editors. New York: Raven; 1994. pp. 1377–1446. [Google Scholar]
- 28.Gibbs J, Fauser D J, Rowe E A, Rolls B J, Rolls E T, Maddison S P. Nature (London) 1979;282:208–210. doi: 10.1038/282208a0. [DOI] [PubMed] [Google Scholar]
- 29.Gibbs J, Smith G P. Ann NY Acad Sci. 1988;547:210–216. doi: 10.1111/j.1749-6632.1988.tb23889.x. [DOI] [PubMed] [Google Scholar]
- 30.Stratford T R, Gibbs J, Smith G P. Peptides. 1995;16(5):903–909. doi: 10.1016/0196-9781(95)00051-k. [DOI] [PubMed] [Google Scholar]
- 31.Ladenheim E E, Wirth K E, Moran T H. Pharmacol Biochem Behav. 1996;54(4):705–711. doi: 10.1016/0091-3057(96)00023-8. [DOI] [PubMed] [Google Scholar]
- 32.Nowak M A, Boerlijst M C, Cooke J, Smith J M. Nature (London) 1997;388:167–171. doi: 10.1038/40618. [DOI] [PubMed] [Google Scholar]
- 33.Cowan A, Khunawat P, Zhu X Z, Gmerek D E. Life Sci. 1985;37:135–145. doi: 10.1016/0024-3205(85)90416-3. [DOI] [PubMed] [Google Scholar]
- 34.Muurahainen N E, Kissileff H R, Pi-Sunyer F X. Am J Physiol. 1993;264:R350–R354. doi: 10.1152/ajpregu.1993.264.2.R350. [DOI] [PubMed] [Google Scholar]
- 35.Gutzwiller J-P, Drewe J, Hildebrand P, Rossi L, Lauper J Z, Beglinger C. Gastroenterology. 1994;106:1168–1173. doi: 10.1016/0016-5085(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 36.Kirkham T C, Walsh C A, Gibbs J, Smith G P, Leban J, McDermed J. Pharmacol Biochem Behav. 1994;48:809–811. doi: 10.1016/0091-3057(94)90351-4. [DOI] [PubMed] [Google Scholar]
- 37.Ohki-Hamazaki H, Watase K, Yamamoto K, Ogura H, Yamano M, Yamada K, Maeno H, Imaki J, Kikuyama S, Wada E, Wada K. Nature (London) 1997;390:165–169. doi: 10.1038/36568. [DOI] [PubMed] [Google Scholar]