

Abstract
Mammalian 3β-hydroxysteroid dehydrogenase/isomerase (3β-HSD) is a member of the short chain dehydrogenase/reductase. It is a key steroidogenic enzyme that catalyzes the first step of the multienzyme pathway conversion of circulating dehydroepiandrosterone and pregnenolone to active steroid hormones. A three dimensional model of a ternary complex of human 3β-HSD type 1 (3β-HSD_1) with an NAD cofactor and androstenedione product has been developed based upon X-ray structures of the ternary complex of E. coli UDP-galactose 4-epimerase (UDPGE) with an NAD cofactor and substrate (PDB_AC: 1NAH) and the ternary complex of human type 1 17β-hydroxysteroid dehydrogenase (17β-HSD_1) with an NADP cofactor and androstenedione (PDB_AC: 1QYX). The dimeric structure of the enzyme was built from two monomer models of 3β-HSD_1 by respective 3D superposition with A and B subunits of the dimeric structure of Streptococcus suis DTDP-D-glucose 4,6-dehydratase (PDB_AC: 1KEP). The 3D model structure of 3β-HSD_1 has been successfully used for the rational design of mutagenic experiments to further elucidate the key substrate binding residues in the active site as well as the basis for dual function of the 3β-HSD_1 enzyme. The structure based mutant enzymes, Asn100Ser, Asn100Ala, Glu126Leu, His232Ala, Ser322Ala and Asn323Leu, have been constructed and functionally characterized. The mutagenic experiments have confirmed the predicted roles of the His232 and Asn323 residues in recognition of the 17-keto group of the substrate and identified Asn100 and Glu126 residues as key residues that participate for the dehydrogenase and isomerization reactions respectively.
Keywords: 3β-hydroxysteroid dehydrogenase, short-chain oxidoreductase, 3D model structure, bioinformatics, rational proteomics, structure based mutagenesis, structure-function relationship
1. Introduction
Human 3β-hydroxysteroid dehydrogenase/isomerase type 1 (3β-HSD_1) contains both dehydrogenase and isomerase activities on a single enzyme protein (Fig. 1) and is a member of the short chain dehydrogenase/reductase family [1]. Human type 1 (placenta, mammary gland, prostate) and type 2 (adrenals, ovary, testis) isoforms of 3β-HSD are encoded by two distinct genes which are expressed in a tissue-specific pattern [2]. Human 3β-HSD_1 catalyzes the conversion of dehydroepiandrosterone (DHEA) to androstenedione (mammary gland, prostate) or pregnenolone to progesterone (placenta). Human 3β-HSD_2 converts 17α-hydroxypregnenolone or pregnenolone to ultimately produce cortisol or aldosterone (adrenal), and converts DHEA to androstendione, the precursor of estradiol in the ovary or testosterone in the testis. In placenta, androstenedione is converted by aromatase (CYP19) and 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD_1) to estradiol, which participates in the cascade of events that initiates labor in humans. The conversion of pregnenolone to progesterone by placental 3β-HSD_1 helps to maintain the uterus in a quiescent state throughout human pregnancy [3,4]. In addition to placenta, mammary gland and prostate, 3β-HSD_1 is selectively expressed in breast tumors [5] and prostate tumors [6,7], where it catalyzes a key step in the conversion of circulating DHEA to estradiol or testosterone to promote tumor growth. DHEA-sulfate is the major circulating steroid in humans and sulfatase produces free DHEA in the target tissues [3].
Fig. 1.
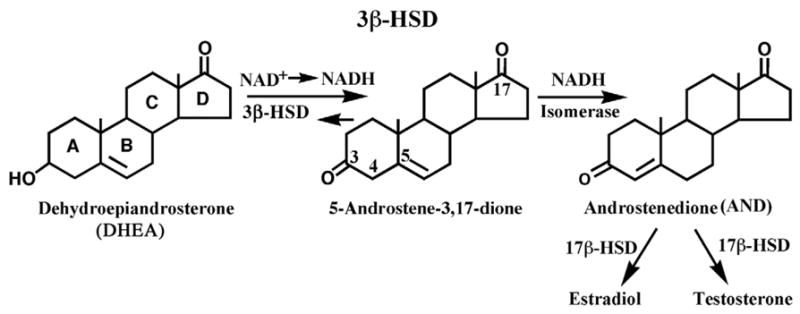
Two sequential oxidoreductase and isomerase reactions, catalyzed by a single 3β-HSD_1 enzyme using dehydroepiandrosterone as a substrate.
Knowledge of the structure/function relationships of human 3β-HSD may result in the development of specific inhibitors of 3β-HSD_1 to block the biosynthesis of estradiol from DHEA in breast tumors or of testosterone from DHEA in prostate tumors. In our recent studies of the structure and function of human 3β-HSD, we reported that it is possible to specifically inhibit purified human 3β-HSD_1 without affecting the activity of human 3β-HSD_2 [8]. We also reported that 3β-HSD_1 transfected into human breast tumor MCF-7 tet-off cells has a 12- to 17-fold greater affinity for substrate (DHEA) and inhibitor (epostane, trilostane) steroids compared to the 3β-HSD_2 that we transfected into the MCF-7 tet-off cells [9]. These observations identify human 3β-HSD_1 as a target enzyme for the inhibition of estradiol production in breast tumors and testosterone production in prostate tumors. Developing a homology model of the three-dimensional structure of human 3β-HSD_1 and testing the accuracy of the model using rational mutagenesis provides a valuable tool that can be used to design enzyme inhibitors with greater specificity for 3β-HSD_1 in hormone-sensitive tumors compared that of the existing compounds, epostane and trilostane. These inhibitors can help control the timing of labor and slow the growth of hormone-sensitive tumors without inhibiting 3β-HSD_2, so that the adrenal production of cortisol and aldosterone is not compromised.
In this article, we describe the three dimensional model of human 3β-HSD_1 complexed with an NAD cofactor and androstenedione steroidal product which was developed using a combination of bioinformatics, molecular graphics and computational biochemistry methods. Based on this three-dimensional structure, the results of our rationally designed mutagenic experiments reveal the key substrate recognition/binding residues and the key residues responsible for the 3β-HSD_1 and isomerization reactions.
2. Methods and materials
2.1 Bioinformatics/Computational Biochemistry/Graphics
A three dimensional model of a ternary complex of human 3β-HSD_1 with an NAD cofactor and the androstenedione product has been developed based upon X-ray structures of two related enzymes: the ternary complex of E-coli UDP-galactose 4-epimerase (UDPGE) with an NAD cofactor and substrate (PDB_AC: 1NAH) [10] and the ternary complex of human 17β-hydroxysteroid dehydrogenase (17β-HSD_1) with NADP and androstenedione (PDB_AC: 1QYX) [11]. The dimer structure of 3β-HSD_1 with a stereochemically optimal interface was built from two monomer models by respective 3D superposition with A and B subunits of the dimeric structure of Streptococcus suis DTDP-D-glucose 4,6-dehydratase (PDB_AC: 1KEP) [12].
The coordinates and the corresponding amino acid sequences of the homologous templates were taken from the Protein Data Bank [13] and the combined SWISS-PROT/TrEMBL database [14] respectively. The programs CLUSTALW (version 1.81) [15] and PHD [16] were used for pairwise sequence alignment and secondary structure prediction respectivly. The graphics program CHAIN (version 7.2) [17] was used for 3D modeling/docking of the 3β-HSD_1+NAD+androstenedione ternary complex. The ‘lsqkab’ [18] and ‘molrep’ [19] programs of the crystallographic package CCP4 (version 5.0.2) [20] were used for 3D superposition of protein structures. Molecular mechanics energy optimization of the model with CHARMM [22] structure was performed by the XPLOR_3.1 [21] program parameterization. In addition to the graphics program CHAIN, the programs LIGPLOT (version 4.0) [23] and HBPLUS (version 3.06) [24] were used to study protein-substrate interactions and generate schematic diagrams. The graphics program SETOR (version 4.14.21k) [25] was used to prepare three-dimensional stereo illustrations of the enzyme ternary complex with cofactor and substrate.
2.2. Mutagenesis/Expression/Kinetic study
2.2.1 Site-directed mutagenesis
Using the Advantage cDNA PCR kit (BD Biosciences Clontech, Palo Alto, CA) and pGEM-3βHSD_1 as template [26], double-stranded PCR-based mutagenesis was performed with the primers in Table 1 to create the cDNA encoding the N100S, N100A, E126L, H232A, S322A and N323L and mutant enzymes. The presence of the mutated codon and integrity of the entire mutant 3β-HSD cDNA were verified by automated dideoxynucleotide DNA sequencing using the Big Dye Terminator Cycle Sequencing Ready Reaction kit (PE Applied Biosystems, Foster City, CA). Chou-Fasman and Garnier-Osguthorpe-Robson analysis of each mutant enzyme was used to choose amino acid substitutions that produced no apparent changes in the secondary structure of the protein (Protylze program, Scientific and Educational Software, State Line, PA)
Table 1.
Oligonucleotide primers used for site-directed mutagenesis
| Mutation | Direction | Nucleotide sequence of primer1 |
|---|---|---|
| N100A | Forward | 5′-TGAATGTCGCTGTGAAAGGTACCCAGC-3′ |
| Reverse | 5′-CTTTCACAGCGACATTCATGATAGACTCT-3′ | |
| N100S | Forward | 5′-GAATGTCAGTGTGAAAGGTACCCAGC-3′ |
| Reverse | 5′-CTTTCACACTGACATTCATGATAGACTC-3′ | |
| H232A | Forward | 5′-TGGGCCGCCATTCTGGCCTTGAGG-3′ |
| Reverse | 5′-CAGAATGGCGGCCCAGGCCACATTGC-3′ | |
| S322A | Forward | 5′-CACATTGGCAAATAGCGTATTCAC-3′ |
| Reverse | 5′-CGCTATTTGCCAATGTGACTATGTG-3′ | |
| N323L | Forward | 5′-CATTGTCACTTAGCGTATTCACCTTCTC-3′ |
| Reverse | 5′-TACGCTAAGTGACAATGTGACTATGTGG-3′ | |
| E126L | Forward | 5′-AGCATACTGGTAGCCGGGCCCAACTC-3′ |
| Reverse | 5′-GGCTACCAGTATGCTACTGGTGTAG-3′ |
The mutated codons are underlined.
2.2.2. Expression and purification of the mutant and wild-type enzymes
The mutant 3β-HSD_1 cDNA was introduced into baculovirus as previously described [26]. Recombinant baculovirus was added to 1.5 × 109 Sf9 cells (1L) at a multiplicity of infection of 10 for expression of each mutant enzyme. The expressed mutant and wild-type enzymes were separated by SDS-polyacrylamide (12%) gel electrophoresis, probed with our anti-3β-HSD_1 polyclonal antibody and detected using the ECL western blotting system with antirabbit, peroxidase-linked secondary antibody (Amersham Pharmacia Biotech, Piscataway, NJ). Each expressed enzyme was purified from the 100,000 g pellet of the Sf9 cells (2 L) by our published method [1, 27] using Igepal CO 720 (Rhodia, Inc., Cranbury, NJ) instead of the discontinued Emulgen 913 detergent (Kao Corp, Tokyo). Each expressed, purified mutant and wild-type enzyme produced a single major protein band (42.0 kDa) on SDS-polyacrylamide (12%) gel electrophoresis that co-migrated with the human wild-type 1 control enzyme. Protein concentrations were determined by the Bradford method using bovine serum albumin as the standard [28].
2.2.3. Kinetic studies
Michaelis-Menten kinetic constants for the 3β-HSD_1 substrate were determined for the purified mutant and wild-type enzymes in incubations containing dehydroepiandrosterone (3–100 μM) plus NAD+ (0.2 mM) and purified enzyme (0.04 mg) at 27°C in 0.02 M potassium phosphate, pH 7.4. The slope of the initial linear increase in absorbance at 340 nm per min (due to NADH production) was used to determine 3β-HSD_1 activity. Kinetic constants for the isomerase substrate were determined at 27° C in incubations of 5-androstene-3,17-dione (20–100 μM), with or without NADH (0.05 mM) and purified enzyme (0.01–0.04 mg) in 0.02 M potassium phosphate buffer, pH 7.4. Isomerase activity was measured by the initial absorbance increase at 241 nm (due to androstenedione formation) as a function of time. Blank assays (zero-enzyme, zero-substrate) assured that specific isomerase activity was measured as opposed to non-enzymatic, “spontaneous“ isomerization [27]. In addition, isomerase incubations without added coenzyme (NADH) were used to measure any basal (zero-coenzyme) isomerase activity in the mutants, and this basal activity was subtracted as a blank. Changes in absorbance were measured with a Varian (Sugar Land, TX) Cary 300 recording spectrophotometer. The Michaelis-Menten constants (Km, Vmax) were calculated from Lineweaver-Burke (1/S vs. 1/V) plots and verified by Hanes-Woolf (S vs. S/V) plots. The kcat values (min−1) were calculated from the Vmax values (nmol/min/mg) and represent the maximal turnover rate (nmol product formed/min/nmol enzyme dimer).
Kinetic constants for the 3β-HSD_1 cofactor were determined for the purified mutant and wild-type enzymes in incubations containing NAD+ (10–200 μM), DHEA (100 μM) and purified enzyme (0.04 mg) in 0.02 M potassium phosphate, pH 7.4, at 27°C using the spectrophotometric assay at 340 nm. Kinetic constants for the isomerase cofactor as an allosteric activator were determined in incubations of NADH (0–50 μM), 5-androstene-3,17-dione (100 μM) and purified enzyme (0.01–0.04 mg) in 0.02 M potassium phosphate buffer, pH 7.4 at 27°C using the spectrophotometric assay at 241 nm. Zero-coenzyme blanks were used as described above for the substrate kinetics.
3. Results and discussion
3.1. Ternary complex of 3β-HSD_1 with the NAD and androstenedione product
One of the major goals of this research was to develop a useful tool for 3D structure based rational mutagenic study to characterize the functionally important residues in human 3β-HSD_1. A three dimensional model of a ternary complex of 3β-HSD_1 complexed with an NAD cofactor and androstenedione (AND) product has been developed based upon X-ray structures of two related enzymes, the monomeric ternary complex of UDPGE (E.coli) with an NAD cofactor and substrate (AC_PDB: 1NAH [10]) and the monomeric ternary complex of 17β-HSD_1 (human) with NADP and androstenedione (AND) (AC_PDB: 1QYX [11]) (Fig. 2). These enzymes share ~24% and ~16% overall sequence identity with 3β-HSD_1. Attempts to use any of the UDPGE or 17β-HSD_1 structures alone for the enzyme modeling were unsuccessful because of unfavorable stereochemical interactions that could not be avoided. The ~160 residue N-terminal sequence comprising the NAD binding site perfectly matches to that of the UDPGE. The essential part of the active site better matches that of 17β-HSD_1 which shares a steroid ligand specificity and dehydrogenase function with 3β-HSD_1 (Fig. 1).
Fig. 2.

The 3D amino acid sequence alignment of 3β-HSD_1 with corresponding constituent parts of UDPGE and 17β-HSD_1. The experimental (X-ray) and predicted (PHD method [16]) secondary structure elements are indicated along the sequence The sequences and individual numbering of 3β-HSD_1, UDPGE and 17β-HSD_1 are shown in black, blue and green respectively. The yellow color marks the region, deletion of which does not cause the loss of enzyme activity.
Other indicative symbols:
#(in magenta) - oxidoreductase catalytic residues, Ser124, Tyr154, Lys158
^ (in blue) - residues lining the NAD binding shell
x (in magenta) – residues lining the substrate binding shell
$ (in magenta) – proposed 3D structure based isomerase catalytic (Glu126) and substrate recognition/binding (His232, Asn323) residues in the active site.
Initially, the crystallographically observed structures of the UDPGE and 17β-HSD_1 complexes were superimposed based on the Cα atoms of residues from the secondary structure elements of the Rossman-fold common to the two structures. Additionally, this superposition included the Ser/Tyr/Lys catalytic triad, and the atoms of the nicotinamide ribose of the cofactor which have similar 3D positions in both crystal structures. The backbone fold of the central region corresponding to amino acid residues ~169–243 of 3β-HSD_1, an essential part of the substrate binding pocket, was significantly different in UDPGE and 17β-HSD_1, reflecting the corresponding significant difference in substrate size and specificity. For this reason, the substrate binding region of UDPGE, corresponding to residues 154–254 of 3β-HSD_1, was replaced by the substrate binding region of 17β-HSD_1 (Fig. 2, 3). The crystal structure of human 17β-HSD_1 (PDB_AC: 1QYX) used for this region contains androstenedione bound in a reverse orientation compared to estradiol [11], so that His221 forms a H-bonds to the 17-keto group of the androgen, and this allows a direct comparison with androstenedione bound in human 3β-HSD_1.
Fig. 3.
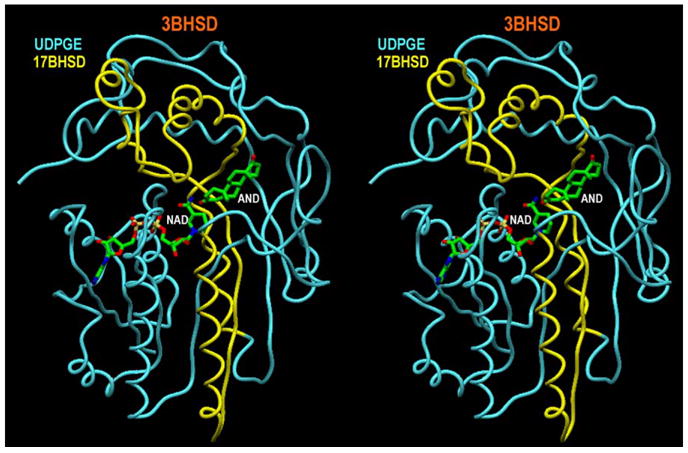
A stereoview (program SETOR [25]) of the 3β-HSD_1 fold with NAD cofactor and AND product (in green). The constituent skeleton parts of UDPGE and 17β-HSD_1 are shown in blue and yellow respectively.
The spatially overlapping backbone transition sections between the UDPGE and 17β-HSD_1 binding pockets, corresponding to residues 147–168 and 244–265 of 3β-HSD_1, have significant sequence homology, 32% identity, 18% strong similarity and 23% identity, 36% strong similarity, respectively. The transition sections permitted smooth splicing of the parent backbones and incorporated the substrate binding pocket of 17β-HSD_1 into the UDPGE structure. The 3β-HSD_1 sequence was threaded using CHAIN [17] through the spliced UDPGE/17β-HSD_1 backbone (Fig. 3). Adjustment for insertions and deletions (detected by CLUSTALW [15]) was achieved by shifting them from secondary structure elements to the nearest loops. Similarity in 2D structure motifs provides supporting evidence of 3D structural similarity. The orientations of side chains in the model and the position of androstenedione (AND) in the active site were adjusted on the basis of stereochemical requirements. At the final stage, 1000 cycles of potential energy optimization (XPLOR [21]) were applied to the model.
The final 3β-HSD_1 structure reveals no unfavorable stereochemical interactions and the stereochemistry of cofactor and substrate binding resembles that observed in the corresponding sites in the UDPGE and 17BHSD_1 structures. The reliability of the model is supported by the sequence alignment and secondary structural motif (Fig. 2). The most uncertain part of the model (residues 283 to 310) which contains a 28 residues insertion (Fig. 2) is on surface of the structure. It was constructed to satisfy stereochemical requirements of the amino acid residues and their interactions with one another and their surroundings. The model was validated by the mutagenic studies described below.
All enzyme contacts involving the NAD cofactor and the AND molecule in a 3.9 shell are shown in the LIGPLOT drawings (Fig. 4a,b) and a stereo-illustration of the ternary complex is presented in Fig. 5. These functionally important areas of the enzyme correspond to the most reliably modeled portions of our 3D model. Five of six 3β-HSD_1 residues that form side chain specific H-bonds with the NAD cofactor (Asp35, Asp61, Ser124, Tyr154, Lys158) are identical to those observed in the UDPGE structure and the sixth, Thr122, is highly similar (Fig. 2, 4a). This subset includes the catalytic triad (Ser124, Tyr154, Lys158) and Asp35 which determines cofactor (i.e., NAD) preference [29, 30]. Three additional less specific H-bonds from the enzyme backbone to the cofactor are formed by the residues Phe13, Leu14, Cys83, two of which are similar to those present in the UDPGE structure. Seven other residues that make hydrophobic contacts to NAD complete the cofactor binding shell.
Fig. 4.
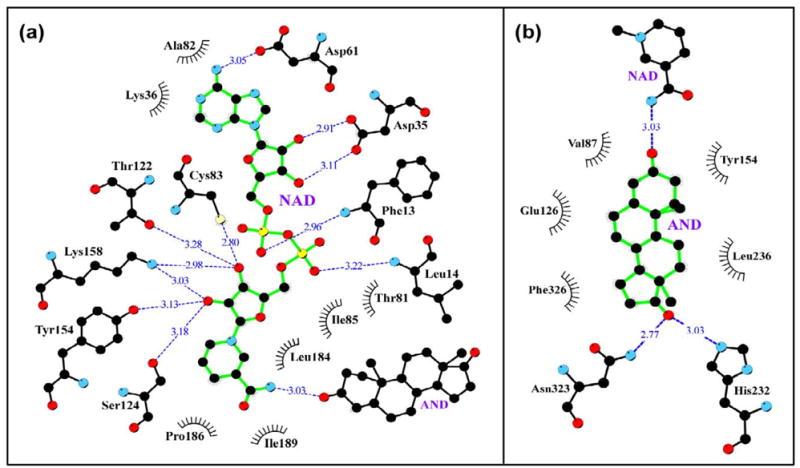
A schematics (programs LIGPLOT/HBPLUS [23,24]) of 3.9Ǻ binding shell for NAD cofactor (a) and AND product (b) in 3β-HSD_1 enzyme. The H-bonds are shown by dashed blue lines. The black eyelash shape marks the residues making hydrophobic contacts with the cofactor.
Fig. 5.
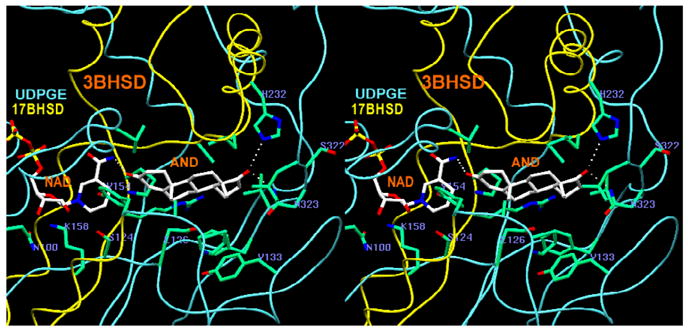
A stereoview (program SETOR [25]) of the active site area in ternary complex of 3β-HSD_1 enzyme with AND product and NAD cofactor. The constituent skeleton parts of UDPGE and 17β-HSD_1, used in modeling, are shown in blue and yellow respectively.
The binding shell of androstenedione (AND) (Fig. 4b) includes three specific H-bonds to the NAD cofactor and the side chains of His232 and Asn323, and five nonspecific mostly hydrophobic contacts. The catalytic Tyr154 forms an H-bond with the cofactor and potentially can also form an H-bond with the C3-carbonyl of the AND. The majority of the residues contacting the substrate are identical or similar to those of the 17β-HSD_1 parent structure (Fig. 2). When the seven amino acids in the substrate binding shells in the 3β-HSD_1 model and the observed structure of 17β-HSD_1 are compared, three are the same (Tyr154, His232, and Phe236) and three are similar in nature (Val87Leu, Leu236Val, and Asn323Glu). Only the Glu126Leu variation is a significant change. The residues Glu/Asp and Tyr are often found to be the catalytic residues in isomerase reactions [31–34]. The presence of the Glu126 in the active site of the 3β-HSD_1 suggested that it could be critical to isomerase activity (Fig. 1), which is absent in 17β-HSD_1 possibly due to a Leu residue in this position [11]. In the active site Glu126 is near the isomerization target C4-C5=C6 (Fig. 5). . The proton donor side chains of the His232, Asn323 and adjacent Ser322 residues may contribute to the recognition and/or H-bonding of the substrate C17-carbonyl. In our model Ser322 does not make a direct H-bond to AND but may compete with the adjacent Asn323 at the initial steps of substrate recognition.
3.2. Dimeric organization of 3β-HSD_1
It has been shown experimentally that 3β-HSD_1 is active as a dimer [1]. The construction of the dimer model was described in the ‘methods’ section. The program “molrep” [19], utilizing the Patterson function to represent all interatomic vectors for the structures was employed. In the 3β-HSD_1 dimer (Fig. 6) the interface is composed of two pairs of helices (95–114 and 155–171) related by a 2-fold symmetry axis. The interface is occupied by a symmetry related double set of side chains Arg93, Glu94, Met97, Asn98, Val101, Lys102, Gln105, Leu106, Glu109, Glu147, Trp150, His156, Lys159, Leu160, Lys163, Ala164, Leu166, Ala167. Dimerization is stabilized by a double set of salt bridges (Arg93…Glu109, Glu94…Lys102, Glu147…Lys163), H-bonds (Arg93…Gln105 or His156…Gln105 [33]) and numerous hydrophobic interactions.
Fig. 6.
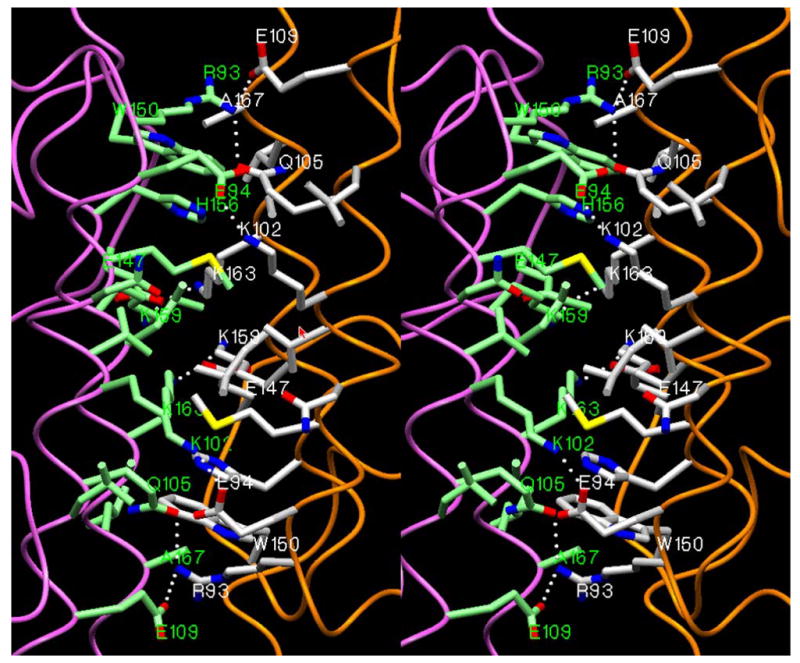
A stereoview (program SETOR [25]) of the interface area in a homodimer structure of the 3β-HSD_1. The interacting side chains from both monomers (in orange and violet) are shown in white and green colors respectively.
3.3. Structure-Function Relations and Structure Based Site Directed Mutagenesis
The 3D arrangement of oxidoreductase catalytic triad Ser124, Tyr154, Lys158 around the substrate and cofactor in the 3β-HSD_1 model is almost identical to that of other members of the short chain oxidoreductase (SCOR) family. Each of the single mutations Ser124Ala, Tyr154Phe, Lys158Gln results in the complete loss of dehydrogenase activity [8, 36, 37]. An associated reduction in isomerase activity is probably due to partial destabilization of cofactor binding, in which the catalytic triad is actively involved, and associated substrate binding (Fig. 4a,b).
Dehydrogenase and isomerase activities are completely abolished in the Asp257Leu and Asp258Leu mutants [30, 36]. In our model, these Asp residues are exposed to possible interaction with water and their replacement with hydrophobic Leu residues is thermodynamically unfavorable. We can’t exclude the possibility that they play a critical role in the protein folding process.
Destabilization of interactions at the dimer interface (Fig. 6) due to Gln105Met and His156Tyr mutations [8, 35] was demonstrated in a dramatic increase in the substrate Km and inhibitor Ki values of 3β-HSD_1 to equal those measured for 3β-HSD_2. In the dimer model, the side chain of Gln105 forms an intersubunit H-bond with the side chain of Arg93. In an alternate conformation it has the potential to form an inter-monomer H-bond with the side chain of His156. This H-bond is disrupted in 3β-HSD_1 by the replacement of Gln105 with Met and His156 with Tyr, which is the residue present in 3β-HSD_2 [35].
The conversion of the cofactor dependence from NAD to NADP caused by the double mutations Asp35Ala/Lys36Arg [30] (Fig 4a) is consistent with the established correlation between Asp and Arg residues in the cofactor binding pocket and NAD or NADP preference respectively [29]. The double mutant has no 3β-HSD or isomerase activity in the presence of NAD(H) and both activities are found with 3-fold decrease in Vmax in the presence of NADP(H). The local enhancement of a positively charged field around the O2′ adenine atom favors the binding of the NADP with its negatively charged phosphate substituent. The importance of long-range electrostatic interatomic interactions to cofactor recognition and binding has been demonstrated [38].
The retention of enzyme activity after the deletion of residues 283 to 310 (Fig. 2) is consistent with the relatively short distance of 6.7Ǻ between residues 282 and 311 in the model. This deletion mutant of 3β-HSD_1 had substrate and cofactor kinetics that were almost identical to those of the wild-type enzyme [39].
Rationally designed mutants based upon the model that successfully elucidate the mechanism of the dual activity of 3β-HSD_1 and permit the design of selective inhibitors are the best test of the model. The main focus was on identification of key residues responsible for substrate recognition and isomerase activity that are located in the regions of 3D model that are the most reliably modeled. The 3β-HSD_1 point mutants Asn100Ser, Asn100Ala, Glu126Leu, His232Ala, Ser322Ala and Asn323Leu were constructed, expressed, and purified, and the substrate and cofactor kinetic constants for each mutant were measured.
Asn100Ala and Asn100Ser mutants revealed an important functional role for the Asn100 residue (Tables 2 and 3). In the 3β-HSD_1 model structure (Fig. 5), this residue forms a direct H-bond (~3Å) with the side chain of the catalytic Lys158 stabilizing its position with respect to the catalytic triad and the cofactor, which is also H-bonded to Lys158. In accordance with the proposed mechanism of action for the SCOR enzymes [40] Tyr154 acts as a catalytic base in the enzyme transition state, while Ser124 binds to the targeted oxo group to stabilize the substrate. The Lys158 binds to the nicotinamide ribose and lowers the pKa of the phenolic hydroxyl group of the Tyr154 to promote the proton transfer. Replacement of Asn100 disrupts the network of H- bonds resulting in the loss of oxidoreductase activity and reduction of isomerase activity. Recent studies of other SCOR enzymes indicate that the Asn residue completes a catalytic tetrad critical to function [41].
Table 2.
Substrate kinetics for the 3β-HSD and isomerase activities of the purified mutant and wild-type enzymes.
| 3β-HSD1 | Isomerase2 | |||||
|---|---|---|---|---|---|---|
| Purified Enzyme | Km μM | kcat min−1 | kcat/Km min−1 μM−1 | Km μM | kcat min−1 | kcat/Km min−1 μM−1 |
| 3β-HSD_1 | 3.7 | 3.3 | 0.90 | 27.9 | 50 | 1.79 |
| H232A | N.D.3 | N.D. | N.D. | 99.4 | 2.1 | 0.02 |
| N323L | 96.5 | 4.6 | 0.05 | 68.5 | 30 | 0.44 |
| S322A | 7.8 | 0.9 | 0.05 | 32.4 | 23 | 0.71 |
| N100A | N.D. | N.D. | N.D. | 180 | 1.0 | 0.01 |
| N100S | N.D. | N.D. | N.D. | 35.1 | 1.6 | 0.05 |
| E126L | 11.9 | 1.4 | 0.12 | 64.6 | 1.6 | 0.02 |
Kinetic constants for the 3β-HSD substrate were determined in incubations containing dehydroepiandrosterone (DHEA, 2–100 μM), NAD+ (100 μM), and purified enzyme (0.03 mg) in 0.02 M potassium phosphate, pH 7.4. The values represent the means values from duplicate assays with standard deviations ≤ 7%.
Kinetic constants for the isomerase substrate 5-androstene-3,17-dione (16.7–100 μM) were determined in incubations of NADH (50 μM) and purified enzyme (0.035 mg) in 0.02 M potassium phosphate buffer, pH 7.4.
N.D., not determined because there was no detectable activity with DHEA substrate.
Table 3.
Cofactor kinetics for the 3β-HSD and isomerase activities of the purified mutant and wild-type enzymes.
| 3β-HSD1 | Isomerase NADH2 | |||||
|---|---|---|---|---|---|---|
| Purified Enzyme | Km μM | kcat min−1 | kcat/Km min−1 μM−1 | Km μM | kcat min−1 | kcat/Km min−1 μM−1 |
| 3β-HSD_1 | 34.1 | 3.5 | 0.10 | 4.6 | 45 | 9.8 |
| H232A | N.D.3 | N.D. | N.D. | 2.0 | 1.2 | 0.6 |
| N323L | 94.3 | 2.9 | 0.03 | 2.9 | 33 | 11 |
| S322A | 19.0 | 1.0 | 0.05 | 3.3 | 16 | 4.8 |
| N100A | N.D. | N.D. | N.D. | N.S.4 | N.S. | N.S. |
| N100S | N.D. | N.D. | N.D. | N.S. | N.S. | N.S. |
| E126L | 16.8 | 1.2 | 0.07 | N.S. | N.S | N.S. |
Kinetic constants for the 3β-HSD cofactor were determined in incubations containing NAD+ (13–200 μM), dehydroepiandrosterone (DHEA, 100 μM) and purified enzyme (0.03 mg) in 0.02 M potassium phosphate, pH 7.4. The values represent the means values from duplicate assays with standard deviations ≤ 6%.
Kinetic constants for the isomerase cofactor were determined in incubations of NADH (2–50 μM), 5-androstene-3,17-dione (50 μM) and purified enzyme (0.035 mg) in 0.02 M potassium phosphate buffer, pH 7.4.
N.D., not determined because there was no detectable activity with DHEA substrate.
N.S., NADH did not stimulate isomerase above the low basal activity with substrate alone (2–3% of wild-type isomerase)
The model suggests that the spatially close His232, Asn323 and Ser322 residues may play a role in substrate recognition and binding and that the Glu126 residue may be directly involved in the isomerization reaction. Mutagenic experiments have been designed to test these possibilities. In the modeled ternary complex, the proton donor side chains of the His232, Ser322 and Asn323 are positioned on a long α-helix and a short β-segment at the opening to the active site. The locations of the His232 and Asn323 residues are similar to those of His221 and Glu282 in the human 17β-HSD_1 ternary complex (AC_PDB: 1QYX), and both are within H-bonding distance from the C17 keto group of the AND substrate (Fig. 4b). The Glu126 residue is in close proximity to the δ 5-δ 4-ene double bond of the steroid, which is the site of isomerization. In 17β-HSD, which lacks isomerase activity, this position is occupied by Leu149 [11].
The His232 and Asn323 residues were found to be essential for productive substrate binding. The His232Ala mutation causes the complete loss of dehydrogenase activity and a significant reduction in isomerase activity based on the Kcat values (Tables 2 and 3). For the Asn323Leu mutant the Km values for both 3β-HSD and isomerase substrate utilization increased greatly indicating a corresponding reduction of binding affinity (Table 2). However, the Km values of His232Ala and Asn323Leu for the NADH cofactor are similar to the values measured for the wild-type enzyme (Table 3). The changes in kinetic constants suggest that neither mutant binds the substrate productively and support the model based prediction that His232 and Asn323 play a critical role in substrate binding (Fig. 4b). The reduction of both activities for the Ser322Ala mutant may indicate its direct involvement in substrate recognition or an indirect effect on the critically important neighboring Asn323 (Tables 2 and 3).
Kinetic studies of the Glu126Leu mutant show a dramatic reduction of the isomerase activity, suggesting direct participation of Glu126 in the isomerase reaction (Table 2). In the modeled structure, the side chain of the Glu126 is close to the δ 5-δ 4-ene double bond of the steroid and may serve as a proton acceptor in catalysis. The retention of a trace of isomerase activity in the mutant may suggest an additional, less effective mechanism of proton transfer via water. It is not clear, whether Glu126 is the only residue involved in catalysis, as in δ 3-δ 2-enoyl-CoA isomerase [34] or if it requires a proton donor H-bonded to C3 carbonyl, as the Tyr residue in δ 5-3-ketosteroid isomerase [26]. In our model, only the catalytic Tyr154 for the 3β-HSD reaction is in a position to form a H-bond with the C3 carbonyl of the isomerase substrate, 5-androstene-3, 17-dione. However, a 4-fold reduction of the corresponding kcat value for the isomerase reaction in the Tyr154Phe mutant with significant residual activity does not fully support its involvement in isomerization [8].
3.4. Conclusions
A reliable three dimensional model of a ternary complex of human 3β-HSD_1 complexed with an NAD and androstenedione (the coordinates are available on request; e-mail: pletnev@hwi.buffalo.edu) has been developed in two forms (monomer and dimer) based upon three X-ray structures of the ternary complexes of E-coli UDP-galactose 4-epimerase and human 17β-hydroxysteroid dehydrogenase type 1 (for monomeric form), and the dimer of Streptococcus suis DTDP-D-glucose 4,6-dehydratase. The 3D model correlates with most of the existing functional mutagenic data and has been successfully used to design additional rational mutagenic experiments to elucidate key substrate binding residues in the active site and the basis for dual function ability of the 3β-HSD_1 enzyme. Mutant enzymes designed on the basis of the model, Asn100Ser, Asn100Ala, Glu126Leu, His232Ala, Ser322Ala and Asn323Leu, have been expressed and functionally characterized. These experiments confirmed the predicted functional roles of the His232 and Asn323 residues in substrate recognition and identified Asn100 and Glu126 as the residues involved in oxidoreductase and isomerization reactions, respectively. This 3D model of the enzyme provides a reliable structural basis for additional rationally designed mutagenic experiments to map the structurally and functionally important residues in the substrate/cofactor binding sites and dimer interface as well as for the design of a highly selective inhibitor of 3β-HSD_1 to block the growth of hormone-sensitive breast and prostate tumors.
Acknowledgments
This research was supported by NIH grants DK026546 (WLD) and CA114717 (JLT). The assistance of Melda Tugac is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thomas JL, Myers RP, Strickler RC. Human placental 3β-hydroxy-5-ene-steroid dehydrogenase and steroid 5-4-ene-isomerase: purification from mitochondria and kinetic profiles, biophysical characterization of the purified mitochondrial and microsomal enzymes. J Steroid Biochem. 1989;33:209–217. doi: 10.1016/0022-4731(89)90296-3. [DOI] [PubMed] [Google Scholar]
- 2.Rheaume E, Lachance Y, Zhao HF, Breton N, Dumont M, de Launoit Y, Trudel C, Luu-The V, Simard J, Labrie F. Structure and expression of a new complementary DNA encoding the almost exclusive 3 beta-hydroxysteroid dehydrogenase/delta 5- delta 4-isomerase in human adrenals and gonads. Mol Endocrinol. 1991;5:1147–1157. doi: 10.1210/mend-5-8-1147. [DOI] [PubMed] [Google Scholar]
- 3.Rainey WE, Carr BR, Sasano H, Suzuki T, Mason JI. Dissecting human adrenal androgen production. Trends Endocrinol Metab. 2002;13:234–239. doi: 10.1016/s1043-2760(02)00609-4. [DOI] [PubMed] [Google Scholar]
- 4.Kacsoh B. Reproductive Endocrinology. In: Kacsoh B, editor. Endocrine Physiology. McGraw-Hill Companies, Inc.; New York: 2000. pp. 566–567. [Google Scholar]
- 5.Gingras S, Moriggl R, Groner B, Simard J. Induction of 3beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase type 1 gene transcription in human breast cancer cell lines and in normal mammary epithelial cells by interleukin-4 and interleukin-13. Mol Endocrinol. 1999;13:66–81. doi: 10.1210/mend.13.1.0221. [DOI] [PubMed] [Google Scholar]
- 6.Geldof AA, Dijkstra I, Newling DW, Rao BR. Inhibition of 3beta-hydroxysteroid-dehydrogenase: an approach for prostate cancer treatment? Anticancer Res. 1995;15:1349–1354. [PubMed] [Google Scholar]
- 7.Gingras S, Simard J. Induction of 3beta-hydroxysteroid dehydrogenase/isomerase type 1 expression by interleukin-4 in human normal prostate epithelial cells, immortalized keratinocytes, colon, and cervix cancer cell lines. Endocrinology. 1999;140:4573–4584. doi: 10.1210/endo.140.10.7038. [DOI] [PubMed] [Google Scholar]
- 8.Thomas JL, Mason JI, Brandt S, Spencer BR, Norris W. Structure/function relationships responsible for the kinetic differences between human type 1 and type 2 3beta-hydroxysteroid dehydrogenase and for the catalysis of the type 1 activity. J Biol Chem. 2002;277:42795–42801. doi: 10.1074/jbc.M208537200. [DOI] [PubMed] [Google Scholar]
- 9.Thomas JL, Umland TC, Scaccia LA, Boswell EL, Kacsoh B. The higher affinity of human type 1 3β-hydroxysteroid dehydrogenase (3β-HSD1) for substrate and inhibitor steroids relative to human 3β-HSD2 is validated in MCF-7 tumor cells and related to subunit interactions. Endocrine Res. 2004;30:935–941. doi: 10.1081/erc-200044164. [DOI] [PubMed] [Google Scholar]
- 10.Thoden JB, Frey PA, Holden HM. Crystal structures of the oxidized and reduced forms of UDP-galactose 4-epimerase isolated from Escherichia coli. Biochemistry. 1996;35:2557–2566. doi: 10.1021/bi952715y. [DOI] [PubMed] [Google Scholar]
- 11.Shi R, Lin SX. Cofactor hydrogen bonding onto the protein main chain is conserved in the short chain dehydrogenase/reductase family and contributes to nicotinamide orientation. J Biol Chem. 2004;279:16778–16785. doi: 10.1074/jbc.M313156200. [DOI] [PubMed] [Google Scholar]
- 12.Allard STM, Beis K, Giraud MF, Hegeman AD, Gross JW, Wilmouth RC, Whitfield C, Graninger M, Messner P, Allen AG, Maskell DJ, Naismith JH. Toward a structural understanding of the dehydratase mechanism structure. 2001;10:81–92. doi: 10.1016/s0969-2126(01)00694-3. [DOI] [PubMed] [Google Scholar]
- 13.Bernstein FC, Koetzle TF, Williams GJB, Meyer ER, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The Protein Data Bank: a computer- based archival file for macromolecular structures. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 14.Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rost B, Sander C. Improved prediction of protein secondary structure by use of sequence profiles and neural networks. Proc Natl Acad Sci USA. 1993;90:7558–7562. doi: 10.1073/pnas.90.16.7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sack JS. CHAIN-A crystallographic modeling program. J Mol Graphics. 1988;6:224–225. doi: 10.1016/s0076-6879(97)77011-3. [DOI] [PubMed] [Google Scholar]
- 18.Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Cryst. 1976;A32:922–923. [Google Scholar]
- 19.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 20.Collaborative Computational Project Number 4. The CCP4 suite: programs for protein Crystallography. Acta Cryst D Biol Crystallogr. 1994;50(Pt5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 21.Brunger AT. X-PLOR (version 3.1) Manual. Yale University; New Haven, CT: 1992. pp. 1–382. [Google Scholar]
- 22.MacKerell AD, Jr, Brooks B, Brooks CL, Nilsson L, Roux B, Won Y, Karplus M. CHARMM: The energy function and its parameterization with an overview of the program. In: Schleyer PR, et al., editors. The Encyclopedia of Computational Chemistry. Vol. 1. John Wiley & Sons; Chichester: 1998. pp. 271–277. [Google Scholar]
- 23.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 24.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential. J Mol Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 25.Evans SV. SETOR: hardware lighted three-dimensional solid model representation of macromolecules. J Mol Graphics. 1993;11:134–138. doi: 10.1016/0263-7855(93)87009-t. [DOI] [PubMed] [Google Scholar]
- 26.Thomas JL, Evans BW, Blanco G, Mercer RW, Mason JI, Adler S, Nash WE, Isenberg KE, Strickler RC. Site-directed mutagenesis identifies amino acid residues associated with the dehydrogenase and isomerase activities of human type I (placental) 3β-hydroxysteroid dehydrogenase/isomerase. J Steroid Biochem Molec Biol. 1998;66:327–334. doi: 10.1016/s0960-0760(98)00058-2. [DOI] [PubMed] [Google Scholar]
- 27.Thomas JL, Berko EA, Faustino A, Myers RP, Strickler RC. Human placental 3β-hydroxy-5-ene-steroid dehydrogenase and steroid 5-4-ene-isomerase: purification from microsomes, substrate kinetics, and inhibition by product steroids. J Steroid Biochem. 1988;31:785–793. doi: 10.1016/0022-4731(88)90287-7. [DOI] [PubMed] [Google Scholar]
- 28.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 29.Duax WL, Pletnev V, Addlagatta A, Bruenn J, Weeks CM. Rational Proteomics I. Predicting fold and cofactor preference in the short-chain oxidoreductase (SCOR) enzyme family. Proteins: Struct Funct Genet. 2003;53:931–943. doi: 10.1002/prot.10512. [DOI] [PubMed] [Google Scholar]
- 30.Thomas JL, Duax WL, Addlagatta A, Brandt S, Fuller RR, Norris W. Structure/function relationships responsible for coenzyme specificity and the isomerase activity of human type 1 3β-hydroxysteroid dehydrogenase/isomerase. J Biol Chem. 2003;37:35483–35490. doi: 10.1074/jbc.M304752200. [DOI] [PubMed] [Google Scholar]
- 31.Kuliopulos A, Talalay P, Mildvan AS. Combined effects of two mutations of catalytic residues on the ketosteroid isomerase reaction. Biochemistry. 1990;29:10271–10280. doi: 10.1021/bi00496a017. [DOI] [PubMed] [Google Scholar]
- 32.Kuliopulos A, Mildvan AS, Shortle D, Talalay P. Kinetic and ultraviolet spectroscopic studies of active mutants of δ 5-3-ketosteroid isomerase. Biochemistry. 1989;28:149–159. doi: 10.1021/bi00427a022. [DOI] [PubMed] [Google Scholar]
- 33.Partanen ST, Novikov DK, Popov AN, Mursula AM, Hiltunen JK, Wierenga RK. The 1.3Å Crystal structure of human mitochondrial δ 3-δ 2-enoyl-CoA isomerase shows a novel mode of binding for the acyl group. J Mol Biol. 2004;342:1197–1208. doi: 10.1016/j.jmb.2004.07.039. [DOI] [PubMed] [Google Scholar]
- 34.Kim SW, Cha SS, Cho HS, Kim JS, Ha NC, Cho MJ, Joo S, Kim KK, Choi KY, Oh BH. High-resolution crystal structures of δ 5-3-ketosteroid isomerase with and without a reaction intermediate analogue. Biochemistry. 1997;36:14030–14036. doi: 10.1021/bi971546+. [DOI] [PubMed] [Google Scholar]
- 35.Thomas JL, Boswell EL, Scaccia LA, Pletnev V, Umland TC. Identification of key amino acids responsible for the substantially higher affinities of human type 1 3β-hydroxysteroid dehydrogenase/isomerase (3β-HSD1) for substrates, coenzymes and inhibitors relative to human 3β-HSD2. J Biol Chem. 2005;280:21321–21328. doi: 10.1074/jbc.M501269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas JL, Duax WL, Addlagatta A, Kacsoh B, Brandt S, Norris W. Structure/function aspects of human 3β-hydroxysteroid dehydrogenase. Mol Cell Endocrinol. 2004;215:73–82. doi: 10.1016/j.mce.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 37.Thomas JL, Duax WL, Addlagatta A, Scaccia L, Frizzell KA, Carloni SB. Serine 124 completes the Tyr, Lys and Ser triad responsible for the catalysis of human type 1 3β-hydroxysteroid dehydrogenase. J Mol Endocrinol. 2004;33:253–261. doi: 10.1677/jme.0.0330253. [DOI] [PubMed] [Google Scholar]
- 38.Pletnev VZ, Weeks CM, Duax WL. Rational proteomics II: Electrostatic nature of cofactor preference in the short chain oxidoreductase (SCOR) enzyme family. Proteins: Structure, Function, and Bioinformatics. 2004;57:294–301. doi: 10.1002/prot.20205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomas JL, Mason JI, Blanco G, Veisaga ML. The engineered, cytosolic form of human type I 3β-hydroxysteroid dehydrogenase/isomerase: purification, characterization and crystallization. J Mol Endocrinol. 2001;27:77–83. doi: 10.1677/jme.0.0270077. [DOI] [PubMed] [Google Scholar]
- 40.Jornvall H, Persson B, Krook M, Atrian S, Gonzalez-Duarte R, Jeffery J, Ghosh D. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 41.Filling C, Berndt KD, Benach J, Knapp T, Prozorovski T, Nordling E, Ladenstein R, Jornvall H, Oppermann U. Critical residues for structure and catalysis in short chain dehydrogenases/reductases. J Biol Chem. 2002;277:25677–25684. doi: 10.1074/jbc.M202160200. [DOI] [PubMed] [Google Scholar]