

Abstract
The Ca2+/calmodulin-dependent protein phosphatase 2B or calcineurin (CN) participates in several Ca2+-dependent signal transduction cascades and, thus, contributes to the short and long term regulation of neuronal excitability. By using a specific antibody to CN, we demonstrate its absence from hippocampal interneurons and illustrate a physiological consequence of such CN deficiency. Consistent with the lack of CN in interneurons as detected by immunocytochemistry, the CN inhibitors FK-506 or okadaic acid significantly prolonged N-methyl-d-aspartate channel openings recorded in the cell-attached mode in hippocampal principal cells but not those recorded in interneurons. Interneurons were also devoid of Ca2+/calmodulin-dependent protein kinase IIα, yet many of their nuclei contained the cyclic AMP-responsive element binding protein. On the basis of the CN and Ca2+/calmodulin-dependent protein kinase IIα deficiency of interneurons, entirely different biochemical mechanisms are expected to govern Ca2+-dependent neuronal plasticity in interneurons versus principal cells.
Intracellular signal-transduction cascades play a pivotal role in the short and long term control of neuronal function. Protein phosphorylation and dephosphorylation through activation of protein kinases and phosphatases have been long recognized as a major component of these signaling cascades (1). Many of the kinases and phosphatases are activated through Ca2+-dependent pathways that link Ca2+ signaling cascades to regulation of protein phosphorylation (2–4). In particular, the serine/threonine phosphatase 2B or calcineurin (CN) activated by Ca2+/calmodulin (CaM) is an important link between these two types of neuronal signaling pathways (5, 6). Many of the specific and localized effects of CN are ensured by its binding to the A kinase anchoring protein, which also fastens two protein kinases: protein kinase A and protein kinase C (7–9). CN dephosphorylates a large number of target phosphoproteins with important consequences to neuronal function (6).
In many neurons, CN regulates the activity of various ion channels and thus controls neuronal excitability through altering the gain of synaptic transmission. CN, activated by Ca2+ entering through N-methyl-d-aspartate (NMDA) channels, shortens their open time (10), which leads to a reduction of synaptic currents (11) and can affect the function of nearby γ-aminobutyric acid (GABA) receptors (12, 13). Its role in neuronal plasticity is further underscored by the dephosphorylation of the inhibitory element of phosphatase PP-1 thought to be critically involved in hippocampal long term depression (LTD) (14), the capacity to depotentiate already long term potentiated synaptic responses (15, 16), and its possible involvement in long term potentiation (LTP) (17, 18). The long term regulation of neuronal function through gene expression is also effectively controlled by CN via activation of protein phosphatase 1 (PP1) and the subsequent dephosphorylation of the nuclear cAMP-responsive element binding protein (CREB) (19).
From their fundamental involvement in neuronal development, plasticity, and normal functioning (6), most Ca2+-dependent signaling cascades are assumed to be present in all neurons. Yet, Ca2+-dependent synaptic plasticity is not uniform in all cell types. Most interneurons show no LTP or LTD (20–22), two forms of synaptic plasticity critically dependent on the activation of Ca2+ signaling cascades (14, 23, 24). Plasticity at interneuronal synapses appears to be confined to the principal cells’ excitatory terminals that form synapses with the interneurons (25). The reason for the absence of such major forms of synaptic plasticity is unknown, but some studies point to missing Ca2+-dependent enzymes from interneurons. For example, Ca2+/CaM-dependent protein kinase II (CaMKII) is conspicuously absent from neocortical interneurons (26, 27), but the functional consequences of this finding are not well understood. Because CaMKII appears to colocalize with CN (28), we decided to investigate the possibility that GABAergic interneurons may be specifically deficient in CN. Our studies show the absence of a major Ca2+ signaling pathway consisting of Ca2+, CaM, CaMKII, and CN from interneurons and may explain the inability of synapses found on hippocampal GABAergic cells to undergo specific Ca2+-dependent forms of neuronal plasticity mediated on the postsynaptic side.
MATERIALS AND METHODS
Colchicine Injections.
Colchicine solution (1 mg/100 μl of 0.9% NaCl) was injected into the lateral ventricles in three 2- to 3-month-old Wistar rats (Charles River breeding Laboratories) under Equitesin anesthesia. A total of 10 μl was delivered through a glass capillary at the following coordinates (mm from bregma): AP, −0.9; DV, 3.5; L, 1.4. The animals were killed 2 days after the injection.
Immunohistochemistry.
Immunostaining was carried out as described elsewhere (29). Briefly, nine rats were perfused with paraformaldehyde or acrolein fixative under deep Equitesin anesthesia. Sections were cut, washed, frozen/thawed, and incubated in primary antiserum against CN (mouse anti-CN, diluted 1:2,000, Transduction Laboratories, Lexington, KY; rabbit anti-CN, only for the fluorescent double-labeling, diluted 1:2,000, ICOS, Bothell, WA), GABA (monoclonal mouse anti-GABA, diluted 1:50), CREB (diluted 1:5,000, Upstate Biotechnology), and CaMKII (diluted 1:200, Life Technologies). The specificity of primary antisera has been demonstrated by the laboratories of origin [anti-GABA (30), mouse anti-CN (31), anti-CREB (32), and anti-CaMKII (33)]. Fluorescein isothiocyanate (FITC)-conjugated anti-mouse and rhodamine-conjugated anti-rabbit (Dako, diluted 1:50) antisera or biotinylated anti-mouse (diluted 1:200, Vector Laboratories) and 1-nm gold-conjugated anti-mouse secondary antibodies (diluted 1:25, Amersham) were used. Sections containing fluorescent markers were examined with an Axioscope fluorescent microscope (Zeiss). Sections for regular immunohistochemistry were incubated with avidin-biotinylated-horseradish peroxidase complex (Elite ABC, Vector Laboratories, diluted 1:400) and visualized by using Ni2+-intensified 3,3′-diaminobenzidine tetrahydrochloride. For the preembedding immunogold staining, Intense M (Amersham) silver intensification solution was used. Sections were dehydrated and embedded in Durcupan ACM (Fluka) for subsequent electron microscopy.
Electrophysiology.
Procedures for the preparation of 350-μm-thick slices from young (20–28 days old) male Wistar rats have been described in detail (34). Fire-polished borosilicate (KG-33, 1.5 mm o.d.; Garner Glass, Claremont, CA) glass pipettes (6–10 MΩ) were filled with the recording/extracellular solution containing 110 mM Na2SO4, 5 mM Cs2SO4, 1.8 mM CaCl2, 10 mM Hepes, 10 mM glucose, 1 mM pyruvic acid, and 0.001 mM tetrodotoxin (pH 7.2–3; final osmolarity was 290–310 milliosmoles). Cell-attached recordings were obtained at room temperature from hippocampal neurons by using infrared differential interference contrast video microscopy (Zeiss Axioscope) for visualization of the target cells (35). To prevent the pronounced desensitization in cell-attached patches (10, 36, 37), the agonist (l-aspartate) was used at low concentrations (200–500 nM) with saturating glycine concentrations (10 μM). FK-506 or okadaic acid were dissolved in dimethyl sulfoxide (final concentration, <0.1%) and diluted to bath concentrations of 50–100 nM and 5–10 μM, respectively. Recordings were performed with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA), digitized at 88 kHz (Neurocorder, NeuroData, New York), and stored on videotape. Channel data were filtered at 1 kHz (8-pole Bessel), sampled at 10 kHz, and analyzed off-line. Details of the 50% threshold-crossing event detection (PAT, Single Channel Analysis Program, courtesy of J. Dempster, Department of Physiology and Pharmacology, University of Strathclyde, U.K.) and of the analyses of channel kinetics are given elsewhere (37).
RESULTS
Hippocampal Interneurons Lack CN Immunoreactivity.
CN-immunoreactive neurons were found in the CA1–CA3 area of the hippocampus, as well as in the dentate gyrus, as reported (38). The cell bodies were located in the pyramidal cell layer of the CA1–CA3 area, in the granule cell layer, in the hilus of the dentate gyrus, and occasionally in the CA1 stratum radiatum. Where present, the immunoreaction end product filled the perinuclear cytoplasm and the proximal and distal dendrites but was absent from the nuclei (Fig. 1). Scattered immunonegative somata were found in all layers of the hippocampus including the principal cell layers (Fig. 1A). In strata radiatum, oriens, and lacunosum-moleculare, the vast majority of cell bodies were devoid of immunolabeling. Glial cells were also negative for CN in all layers. In the dentate gyrus, granule cells were invariably immunopositive, whereas large somata in stratum granulosum, moleculare, and the hilus were negative for CN. Immunostaining for GABA and CN was examined in adjacent 60-μm-thick sections by using the mirror technique (39). In two animals, a total of 432 GABA-positive neurons were examined in the CA1, 118 in the CA3 region, and 504 in the dentate gyrus. All of these neurons were found to be negative for CN in the adjacent sections (Fig. 1 C and D). Because GABAergic interneurons with distant projections are known to contain low levels of GABA in their somata (29), double immunostaining for GABA and CN was also performed in three colchicine-treated animals by using fluorescein isothiocyanate- and rhodamine-conjugated secondary antisera. In a sample of 344 GABA-positive neurons taken from all layers and subfields of the hippocampus, none were double-stained for CN (data not shown).
Figure 1.
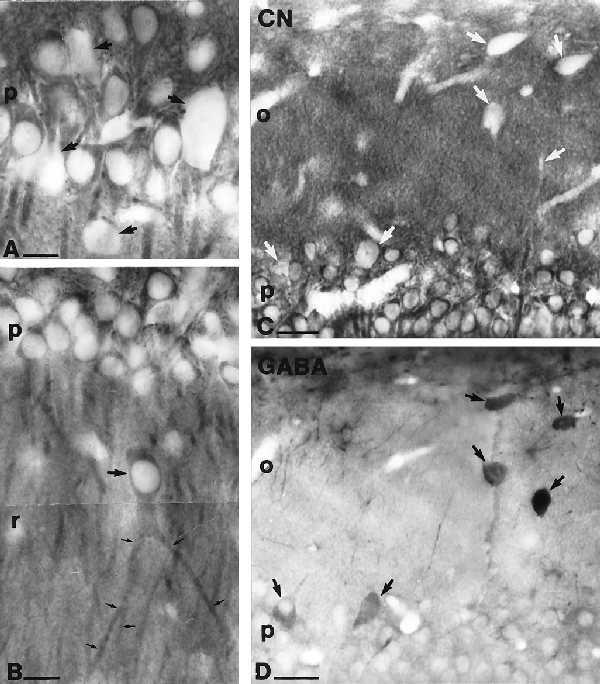
CN immunoreactivity is present in principal cells but not in GABAergic interneurons of the hippocampus. (A) High-magnification light micrograph of the stratum pyramidale from the CA1 region. The presence of the reaction product in the perinuclear cytoplasm, proximal and distal dendrites, and its absence from the nuclei of pyramidal cells is clearly visible. Both the nuclei and the cytoplasm of large neurons (showing the characteristics of interneurons) are devoid of immunoreactivity (arrows). (B) In addition to conventional pyramidal cells, a large CA1 stratum radiatum cell is also immunoreactive for CN (arrow). Note the thick bifurcated apical dendrite (small arrows) characteristic of this type of an ectopic pyramidal cell in the CA1 region. (C and D) Matching surfaces of adjacent sections, one stained for CN (C) and the other stained for GABA (D). Identification of cell bodies cut in half shows the lack of CN immunoreacivity in GABA-positive interneurons (arrows) in CA1 stratum pyramidale and oriens. Stratum oriens, o; stratum pyramidale, p; stratum radiatum, r. [Bars = 25 μm (A and B) and 50 μm (C and D).]
Subcellular Localization of CN in Hippocampal Neurons.
A fraction of cytosolic CN is anchored by the A kinase anchoring protein (7–9) in the vicinity of synapses or certain cytoplasmic organelles. Therefore, we examined the subcellular localization of CN in hippocampal sections at the electron microscopic level by using the regular immunoperoxidase (DAB) and preembedding immunogold techniques. The DAB precipitate or immunogold labeling was present in the perinuclear cytoplasm and dendrites of principal cells (Fig. 2A). Dendritic spines were also labeled throughout the dendritic layers of the hippocampus (Fig. 2 B–D). Axon initial segments were immunopositive in several cases (Fig. 2E), but glial processes were always negative (Fig. 2A). In sections stained by the preembedding immunogold method, gold particles were found in cellular structures where CN could dephosphorylate several intracellular target proteins (40–42). In dendritic spines, gold particles were often associated with small organelles identified as endoplasmic reticulum cisternae or spine apparatus (Fig. 2 C and D); i.e., structures thought to serve as intracellular Ca2+ stores (43). The presence of CN in the close proximity of these organelles may indicate its close relationship with inositol trisphosphate receptors (44). No specific association or enrichment of immunoreactivity was observed near synaptic active zones (Fig. 2B–D). Interestingly, there was no evidence for CN in axon terminals, inconsistent with the reported involvement of CN in glutamate release (45) or other types of exocytosis (46).
Figure 2.
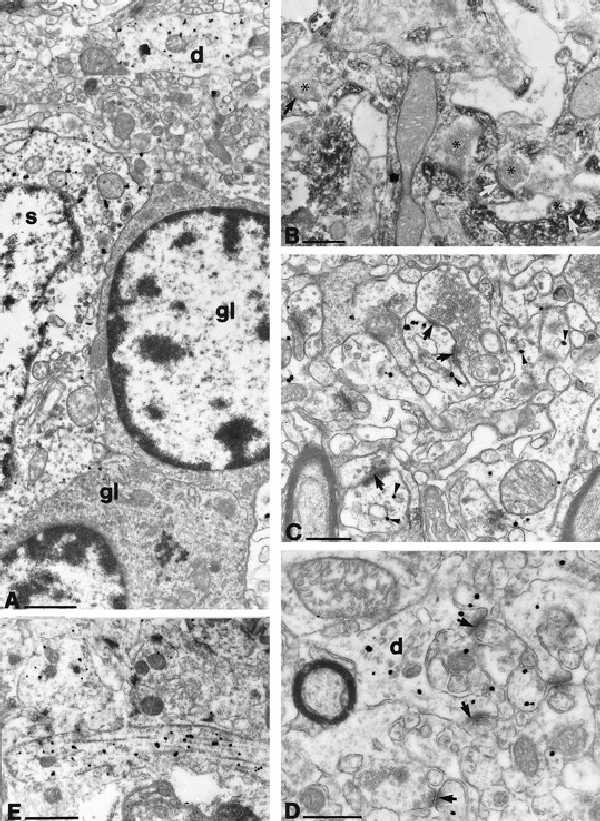
Subcellular localization of CN immunoreactivity using the preembedding immunogold method with silver intensification (A, C–E) or with the conventional immunoperoxidase technique with DAB as a chromogen (B). (A) CN immunoreactivity (black dots of irregular size) is present in the perinuclear cytoplasm of a pyramidal cell (s) and in dendrites (d) but not in glial cells (gl). (B) CN immunoreactivity, as indicated by the diffusible DAB reaction product, is present in dendrites and dendritic spines but is absent from axon terminals. Arrows indicate asymmetric synaptic contacts formed by CN-negative boutons (asterisks) on CN-positive dendritic spines. (C and D) Silver-intensified colloidal gold particles show the subcellular localization of CN in dendrites. Particles were often seen attached to small membrane-limited structures (arrowheads) that resembled saccules of smooth endoplasmic reticulum or spine apparatus fragments. Note the asymmetric synapses terminating on labeled pyramidal cell dendritic spines (arrow). (E) An axon initial segment is shown to be CN immunoreactive. [Bars = 1 μm (A, D, and E) and 0.5 μm (B and C).]
Absence of CaMKII and Presence of CREB in GABAergic Neurons.
The absence of CaMKII from GABAergic neurons in the neocortex has been reported (26, 27). To determine whether hippocampal GABAergic interneurons also lack this protein kinase, immunostaining for GABA and CaMKIIα was performed on adjacent sections from three animals. Similarly to the CN distribution, CaMKIIα-immunoreactive neurons were found in the principal cells but not in GABAergic interneurons (n = 50 GABA-immunoreactive cells tested; Fig. 3 A–D).
Figure 3.
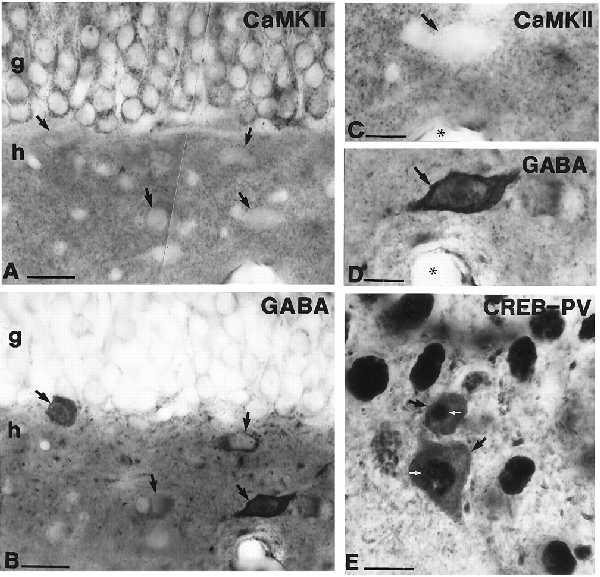
(A and B) Light micrographs of adjacent sections (mirror technique) showing the absence of CaMKIIα (A and C, at high magnification) in GABAergic (B and D, at high magnification) interneurons (arrows) in the hilar region. Capillaries (asterisks) are used as landmarks (C and D). Granule cells are positive for CaMKIIα. (E) Black DAB–Ni2+ reaction product indicates the presence of CREB immunoreactivity in the nuclei of pyramidal cells and in parvalbumin (PV)-immunoreactive GABAergic interneurons (black arrows) in the CA3 region of the hippocampus. White arrows mark the CREB-positive nuclei of PV-immunoreactive neurons. g, Granule cell layer; h, hilus. [Bars = 20 μm (A and B), 10 μm (C and D), and 25 μm (E).]
Because two major Ca2+-signaling enzymes involved in neuronal plasticity appeared to be missing from interneurons, we decided to investigate the presence of the nuclear transcription factor CREB, known to regulate gene expression and neuronal plasticity in a Ca2+/CN-dependent manner (47–50). The presence of CREB in interneurons was studied by double immunostaining using interneuron markers (parvalbumin, calretinin, or GABA) developed with DAB (brown) and CREB developed with DAB–Ni2+ (black). Double-labeled somata were seen in all layers of the hippocampus (Fig. 3E), consistent with the idea that most if not all interneuron types express CREB in their nuclei.
CN Regulates the Duration of NMDA Channel Openings in Hippocampal Principal Cells but Not in Interneurons.
A negative finding in immunocytochemical localization studies does not necessarily indicate the total absence of the protein from the neurons; therefore, we decided to examine a physiological correlate of our anatomical findings. We chose to investigate the CN-dependent regulation of NMDA channel activity, known to occur in acutely dissociated adult (10) and cultured embryonic hippocampal neurons (11). Because CN provides a negative Ca2+-dependent feedback on NMDA channel openings (10), inhibition of CN activity, if CN is present, should normally lead to a prolongation of openings. The postulated absence of CN from interneurons should render NMDA channel openings in these neurons insensitive to CN inhibition. To address this question, we examined the effect of the specific and potent CN inhibitor FK-506 on NMDA channel openings in cell-attached patches of various hippocampal neurons identified in slices by using infrared-differential interference contrast video microscopy (35). The patches were held at the cells’ resting membrane potential (−60 ± 12 mV), where unitary NMDA channel currents with similar amplitudes (3.5–4.5 pA) could be recorded. In several cases, particularly on the somata or the proximal dendrites of interneurons, more than one channel was present in the patch. This allowed us to investigate the mean open times of NMDA channels but precluded the detailed analysis of other kinetic properties.
To ensure that NMDA channels in the slice preparation behaved similarly to those previously recorded in acutely dissociated cells, we first assessed the effect of FK-506 in six granule cells. Within 6–8 min of its bath application, the membrane-permeant CN inhibitor FK-506 (50–100 nM) significantly increased the granule cells’ mean NMDA channel open time (paired t test, P < 0.01; Table 1) comparable to its effect in acutely dissociated cells (10).
Table 1.
Cell-attached recordings of NMDA channel openings in 21 hippocampal neurons identified by infrared differential interference contrast video microscopy
| Cell type | Control
|
CN inhibition
|
Ratio
|
|---|---|---|---|
| τmean, ms | τmean, ms | D/C, % | |
| GC* (n = 6) | 1.78 ± 0.51 | 3.09 ± 1.17† | 161 ± 14 |
| PC* (n = 6) | 1.37 ± 0.18 | 2.15 ± 0.29† | 157 ± 13 |
| IN* (n = 6) | 1.33 ± 0.27 | 1.31 ± 0.26 | 98 ± 3 |
| IN‡ (n = 3) | 0.98 ± 0.05 | 1.05 ± 0.08 | 107 ± 5 |
GC, dentate granule cells; PC, CA1 pyramidal cells; IN, CA1 interneurons at the border of strata radiatum and lacunosum moleculare. The drug/control (D/C) ratio represents the potentiation of the mean open time (τmean) induced by inhibition of CN. Data are the mean ± SEM.
*Cells exposed to FK-506 (50–100 μM).
Significant increment in the NMDA channel mean open times (paired t test, P < 0.01).
Cells exposed to okadaic acid (5–10 μM).
Our anatomical findings identified CN in CA1 pyramidal cells, but as of yet there is no direct evidence for a CN-dependent regulation of NMDA channel openings in these neurons. Therefore, we investigated whether CN regulates the function of NMDA channels in CA1 pyramidal cells in a manner analogous to that seen in dentate gyrus granule cells. As expected from the presence of CN in the pyramidal cells, perfusion of FK-506 (50–100 nM) had a similar effect on the NMDA channel openings in these neurons as in dentate gyrus granule cells. In six pyramidal cells, we observed a significant increase in the mean open times of NMDA channels after the drug applications (paired t test, P < 0.01; Fig. 4, also see Table 1.).
Figure 4.
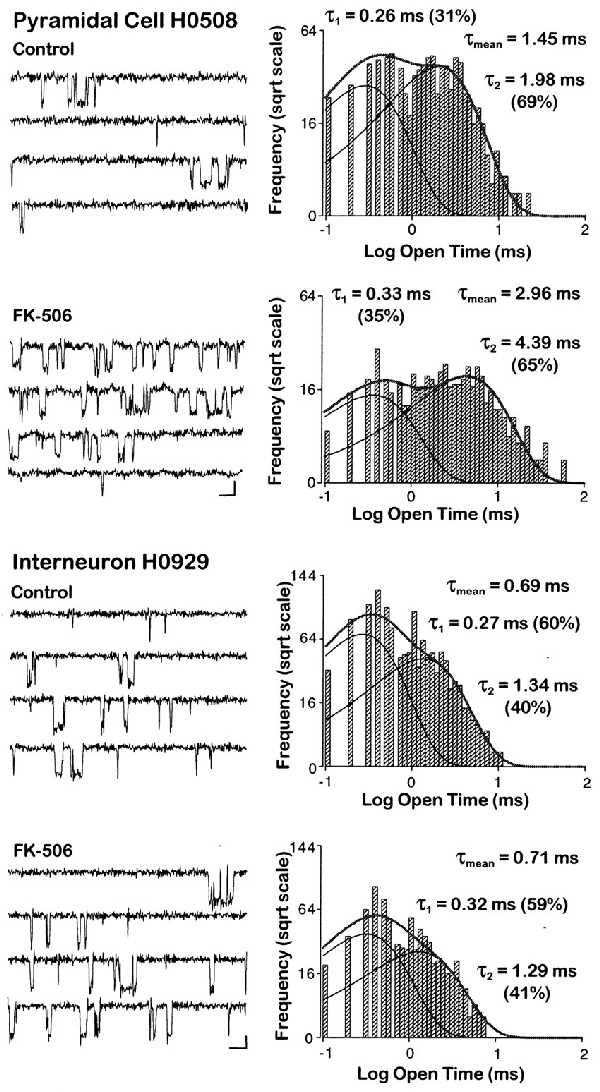
FK-506, a potent and specific CN inhibitor, prolongs NMDA channel openings in a CA1 pyramidal cell but not in an interneuron. Representative traces (Left), with openings depicted as downward deflections, and open time histograms (Right) calculated from the same patches before and after application of FK-506 (50–100 nM). Scale bars denote 10 ms and 2 pA. Within 6–8 min after bath application of the drug, the mean open time increased from 1.45 ms to 2.96 ms in this pyramidal cell. In contrast to hippocampal principal cells (also see Table 1), the mean NMDA channel open time in the interneuron did not change during the same experimental conditions. The logarithmic binned open time distributions plotted on a square root ordinate were best fitted with the sum of two exponentials and yielded mean open times of 0.69 ms under control conditions and 0.71 ms in FK-506. The individual open time components (τ1 and τ2) are indicated on the graphs with their respective amplitude components in parentheses.
If our anatomical findings indeed reflect the true absence of CN from interneurons, then the duration of NMDA channel openings in GABA-containing hippocampal neurons, in contrast to principal cells, should be independent of CN-mediated regulation. To test this hypothesis, we examined the openings of NMDA channels in CA1 interneurons in the presence of the specific CN inhibitor FK-506 and of high concentrations (>5 μM) of okadaic acid known to inhibit CN (10). Recordings were obtained from nine cells located in the stratum oriens or at the border of the strata radiatum and lacunosum-moleculare. In sharp contrast to our findings in principal cells, but consistent with our immunocytochemical results, perfusion of the CN inhibitors FK-506 (50–100 nM) or okadaic acid (5–10 μM) failed to change significantly the duration of NMDA channel openings (paired t test, P = 0.498 and P = 0.369, respectively; Table 1). The potentiation ratio of mean NMDA channel open times by CN inhibitors in interneurons (Table 1) was significantly different [post-ANOVA F(2, 18) = 13.486; Duncan’s multiple range test, P < 0.05] from those observed in principal cells.
DISCUSSION
The induction of synaptic plasticity in hippocampal principal cells primarily depends on activation of specific Ca2+-dependent signaling pathways. For example, induction of LTP in CA1 pyramidal neurons requires substantial rises in intracellular Ca2+ leading to activation of the multifunctional CaM-dependent serine/threonine protein kinase CaMKII (4, 51–55), whereas relatively smaller Ca2+ rises may trigger LTD (14) by activating CN (15, 56), an event that can also depotentiate already long term potentiated synaptic transmission (16). Although such Ca2+-dependent forms of synaptic plasticity are well established in principal cells, excitatory synapses on interneurons show little if any of such plasticity. This apparent lack of Ca2+-dependent synaptic plasticity in interneurons is even more puzzling in light of two established findings. (i) A significant NMDA-receptor-mediated component is readily found in the excitatory synaptic responses of hippocampal interneurons (21, 57, 58), and (ii) interneurons have predominantly Ca2+-permeable combinations of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (59, 60).
Our study was prompted by this conspicuous absence of a Ca2+-dependent form of synaptic plasticity from hippocampal interneurons. The LTP observed at synapses on interneurons is due to a passive propagation of plasticity through alterations in the function of the principal cells’ axon collaterals (refs. 20 and 22 but see also ref. 21). Similarly, a presynaptic decrease in glutamate release from the principal cells’ excitatory terminals has been proposed for the LTD observed in interneurons (25).
A simple explanation for the lack of any Ca2+-dependent synaptic plasticity observed so far in interneurons would be the absence of Ca2+-activated signaling pathways required for such plasticity (14, 23, 24). One clue comes from previous immunocytochemical studies in the rat and monkey neocortex showing the conspicuous absence of CaMKII immunoreactivity from GABAergic neurons (26, 27), but these studies have not identified any physiological correlates of the anatomical findings. Our present report extends the anatomical findings on CaMKII to hippocampal interneurons and, in addition, shows the GABAergic cells’ deficiency of yet another key Ca2+-dependent regulator of neuronal plasticity: the Ca2+/CaM-dependent protein phosphatase CN. We have also provided electrophysiological evidence for the absence of CN from interneurons: NMDA channel openings in these cells were not prolonged by inhibition of CN.
Lack of Regulation of NMDA Channels by CN in Interneurons.
Our anatomical findings demonstrate the absence of immunocytochemically detectable levels of CN in interneurons. Consistent with these anatomical data, the specific CN inhibitor FK-506 and high concentrations of okadaic acid, which also inhibit CN, had no significant effect on the NMDA channels of interneurons, whereas such treatments significantly prolong channel openings in principal cells (10). This finding should not be taken as evidence for a total lack of regulation of interneuronal NMDA channels by protein kinases and phosphatases. Other protein phosphatases of GABAergic neurons, such as protein phosphatases 1 or 2A (61) or tyrosine phosphatases (62, 63), may replace CN in the regulation of NMDA channels. The large diversity of hippocampal interneurons (64) raises the possibility of different NMDA channel subunit combinations being present in distinct types of GABAergic cells as single-cell reverse transcription-coupled PCR studies have shown in GABAergic neurons of the medial septum (65). We could not adequately address this issue quantitatively because most of our recordings had several channels precluding a detailed kinetic analysis of the openings. The presence of multiple channels in the patches probably results from the dense innervation of interneuron somata by excitatory terminals, unlike in pyramidal cells (66).
Physiological Significance of the Missing Ca2+ Signaling Pathway from Interneurons.
Clearly, one of the most immediate consequences of a missing major Ca2+-dependent signaling pathway in interneurons is a lack of Ca2+-dependent synaptic plasticity. We cannot exclude the involvement of other Ca2+-dependent second messenger systems in interneurons, but if two common forms of postsynaptic Ca2+-dependent synaptic plasticity (LTP and LTD) are indeed absent from GABAergic cells, changes in synaptic efficacy in these cells might be mediated predominantly by presynaptic effects. However, our immunohistochemical findings demonstrate the presence of the intranuclear regulatory element CREB in many GABAergic cells. Therefore, some aspects of lasting neuronal plasticity mediated by this intranuclear gene regulatory element should be operative in interneurons. It is interesting to note that the only GABAergic neuron known to show significant postsynaptic Ca2+-dependent plasticity is the Purkinje cell (67), and it is probably not a coincidence that both CaMKII and CN are present in this neuron type (68–70). A second important consequence of lacking two major cytoplasmic CaM-dependent enzymes is related to their affinity for CaM. The cellular concentrations of CaMKII or CN can be sufficiently large to constitute a major CaM buffer (2). CaM is present in interneurons (71), but there would be no binding to the missing CaM-dependent enzymes. This could free sufficient CaM to preferentially activate other CaM-dependent cellular processes, including the regulation of ion channels through protein–protein interactions (72–74). Thirdly, it remains to be determined how the dephosphorylation of key cytoskeletal proteins is accomplished in GABAergic cells. Microtubule-associated protein 2 (40–42), tubulin (40), and tau (75, 76) are all targets of CN, as are various phosphoprotein members of other signaling cascades (77, 78). Because many of these proteins are found in GABAergic neurons (79, 80), their dephosphorylation must depend on other phosphatases, but this will result in a Ca2+-sensitivity of the dephosphorylation process entirely different than that found in principal cells.
Activation of CN by excessive Ca2+ entry during anoxia/ischemia is thought to give rise to a cascade of events ultimately leading to neuronal death. Accordingly, antagonism of CN effectively protects against Ca2+-dependent neuronal damage (81). Therefore, the absence of CN from GABAergic cells may confer a diminished vulnerability during anoxia/ischemia. Indeed, interneurons (82) are relatively resistant to Ca2+-dependent cell damage during anoxia/ischemia. The few that die, do so by acute edemic cell death (for review, see ref. 83) and not like pyramidal cells that are killed by delayed Ca2+-dependent mechanisms. Some future neuroprotective approaches might be based on CN inhibition (81), but such strategies may not be globally effective. According to our findings, GABAergic cells would not be saved by such interventions.
In summary, our studies demonstrate the absence of a major Ca2+-signaling pathway consisting of the CaM-sensitive enzymes CaMKII and CN from GABAergic interneurons. This missing pathway may explain many special properties of interneurons including their conspicuous inability to undergo Ca2+-dependent synaptic plasticity.
Acknowledgments
We are grateful to Drs. K. G. Baimbridge, J. Rogers, I. Virtanen, and ICOS Corporation for their kind gifts of antisera against parvalbumin, calretinin, GABA, and CN. We thank E. Borók, M. Kim, B. Oyama, and A. Zöldi for excellent technical assistance. The work was supported by the Horward Hughes Medical Institute, the Human Frontier Science Program Organization, OTKA (Hungarian Science Foundation T 16942, D 25526) Hungary, ETT-073/1996, and National Institutes of Health Grant NS-27528.
ABBREVIATIONS
- CN
calcineurin
- CaM
calmodulin
- CaMKII
CaM-dependent protein kinase II
- CREB
cAMP-responsive element binding protein
- NMDA
N-methyl-d-aspartate
- DAB
3,3′-diaminobenzidine hydrochloride
- GABA
γ-aminobutyric acid
- LTP
long term potentiation
- LTD
long term depression
References
- 1.Levitan I B. Annu Rev Physiol. 1994;56:193–212. doi: 10.1146/annurev.ph.56.030194.001205. [DOI] [PubMed] [Google Scholar]
- 2.Braun A P, Schulman H. Annu Rev Physiol. 1995;57:417–445. doi: 10.1146/annurev.ph.57.030195.002221. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh A, Greenberg M E. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 4.Soderling T R. Adv Second Messenger Phosphoprotein Res. 1995;30:175–189. doi: 10.1016/s1040-7952(05)80007-2. [DOI] [PubMed] [Google Scholar]
- 5.Nairn A C, Shenolikar S. Curr Opin Neurobiol. 1992;2:296–301. doi: 10.1016/0959-4388(92)90118-5. [DOI] [PubMed] [Google Scholar]
- 6.Yakel J L. Trends Pharmacol Sci. 1997;18:124–134. doi: 10.1016/s0165-6147(97)01046-8. [DOI] [PubMed] [Google Scholar]
- 7.Coghlan V M, Perrino B A, Howard M, Langeberg L K, Hicks J B, Gallatin W M, Scott J D. Science. 1995;267:108–111. doi: 10.1126/science.7528941. [DOI] [PubMed] [Google Scholar]
- 8.Faux M C, Scott J D. Trends Biochem Sci. 1996;21:312–315. [PubMed] [Google Scholar]
- 9.Klauck T M, Faux M C, Labudda K, Langeberg L K, Jaken S, Scott J D. Science. 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- 10.Lieberman D N, Mody I. Nature (London) 1994;369:235–239. doi: 10.1038/369235a0. [DOI] [PubMed] [Google Scholar]
- 11.Tong G, Shepherd D, Jahr C E. Science. 1995;267:1510–1512. doi: 10.1126/science.7878472. [DOI] [PubMed] [Google Scholar]
- 12.Martina M, Mozrzymas J W, Boddeke H W G M, Cherubini E. Neurosci Lett. 1996;215:95–98. [PubMed] [Google Scholar]
- 13.Stelzer A, Shi H. Neuroscience. 1994;62:813–828. doi: 10.1016/0306-4522(94)90479-0. [DOI] [PubMed] [Google Scholar]
- 14.Cummings J A, Mulkey R M, Nicoll R A, Malenka R C. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- 15.Mulkey R M, Endo S, Shenolikar S, Malenka R C. Nature (London) 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 16.Muller D, Hefft S, Figurov A. Neuron. 1995;14:599–605. doi: 10.1016/0896-6273(95)90316-x. [DOI] [PubMed] [Google Scholar]
- 17.Wang J-H, Stelzer A. Neuroreport. 1994;5:2377–2380. doi: 10.1097/00001756-199411000-00041. [DOI] [PubMed] [Google Scholar]
- 18.Lu Y F, Tomizawa K, Moriwaki A, Hayashi Y, Tokuda M, Itano T, Hatase O, Matsui H. Brain Res. 1996;729:142–146. [PubMed] [Google Scholar]
- 19.Bito H, Deisseroth K, Tsien R W. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 20.Maccaferri G, McBain C J. Neuron. 1995;15:137–145. doi: 10.1016/0896-6273(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 21.Ouardouz M, Lacaille J C. J Neurophysiol. 1995;73:810–819. doi: 10.1152/jn.1995.73.2.810. [DOI] [PubMed] [Google Scholar]
- 22.Maccaferri G, McBain C J. J Neurosci. 1996;16:5334–5343. doi: 10.1523/JNEUROSCI.16-17-05334.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bliss T V, Collingridge G L. Nature (London) 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 24.Nicoll R A, Malenka R C. Nature (London) 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 25.McMahon L L, Kauer J A. Neuron. 1997;18:295–305. doi: 10.1016/s0896-6273(00)80269-x. [DOI] [PubMed] [Google Scholar]
- 26.Jones E G, Huntley G W, Benson D L. J Neurosci. 1994;14:611–629. doi: 10.1523/JNEUROSCI.14-02-00611.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X B, Jones E G. Proc Natl Acad Sci USA. 1996;93:7332–7336. doi: 10.1073/pnas.93.14.7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goto S, Nagahiro S, Korematsu K, Ushio Y, Fukunaga K, Miyamoto E, Hofer W. Neurosci Lett. 1993;149:189–192. doi: 10.1016/0304-3940(93)90768-g. [DOI] [PubMed] [Google Scholar]
- 29.Tóth K, Freund T F. Neuroscience. 1992;49:793–805. doi: 10.1016/0306-4522(92)90357-8. [DOI] [PubMed] [Google Scholar]
- 30.Szabat E, Soinila S, Happola O, Linnala A, Virtanen I. Neuroscience. 1992;47:409–420. doi: 10.1016/0306-4522(92)90255-z. [DOI] [PubMed] [Google Scholar]
- 31.Nichols RA, Suplick GR, Brown JM. J Biol Chem. 1994;269:23817–23823. [PubMed] [Google Scholar]
- 32.Ginty D, Kornhauser J M, Thompson M A, Banding H, Mayo K E, Takahasi J S, Greenberg M E. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 33.Scholz W K, Baitinger C, Schulman H, Kelly P T. J Neurosci. 1988;8:1039–1051. doi: 10.1523/JNEUROSCI.08-03-01039.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hájos N, Mody I. J Neurosci. 1997;17:8427–8442. doi: 10.1523/JNEUROSCI.17-21-08427.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakmann B, Stuart G J. In: Patch-Pipette Recordings from the Soma, Dendrites, and Axon of Neurons in Brain Slices. Sakmann B, Neher E, editors. New York: Plenum; 1995. pp. 199–211. [Google Scholar]
- 36.Gibb A J, Colquhoun D. J Physiol (London) 1992;456:143–179. doi: 10.1113/jphysiol.1992.sp019331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Köhr G, De Koninck Y, Mody I. J Neurosci. 1993;13:3612–3627. doi: 10.1523/JNEUROSCI.13-08-03612.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morioka M, Nagahiro S, Fukunaga K, Miyamoto E, Ushio Y. Neuroscience. 1997;78:673–684. doi: 10.1016/s0306-4522(96)00626-4. [DOI] [PubMed] [Google Scholar]
- 39.Kosaka T, Kosaka K, Tateishi K, Hamaoka Y, Yanaihara N, Wu J Y, Hama K. J Comp Neurol. 1985;239:420–430. doi: 10.1002/cne.902390408. [DOI] [PubMed] [Google Scholar]
- 40.Goto S, Yamamoto H, Fukunaga K, Iwasa T, Matsukado Y, Miyamoto E. J Neurochem. 1985;45:276–283. doi: 10.1111/j.1471-4159.1985.tb05504.x. [DOI] [PubMed] [Google Scholar]
- 41.Halpain S, Greengard P. Neuron. 1990;5:237–246. doi: 10.1016/0896-6273(90)90161-8. [DOI] [PubMed] [Google Scholar]
- 42.Quinlan E M, Halpain S. Neuron. 1996;16:357–368. doi: 10.1016/s0896-6273(00)80053-7. [DOI] [PubMed] [Google Scholar]
- 43.Simpson P B, Challiss R A J, Nahorski S R. Trends Neurosci. 1995;18:299–306. doi: 10.1016/0166-2236(95)93919-o. [DOI] [PubMed] [Google Scholar]
- 44.Cameron A M, Steiner J P, Roskams A J, Ali S M, Ronnett G V, Snyder S H. Cell. 1995;83:463–472. doi: 10.1016/0092-8674(95)90124-8. [DOI] [PubMed] [Google Scholar]
- 45.Victor R G, Thomas G D, Marban E, O’Rourke B. Proc Natl Acad Sci USA. 1995;92:6269–6273. doi: 10.1073/pnas.92.14.6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Renström E, Ding W G, Bokvist K, Rorsman P. Neuron. 1996;17:513–522. doi: 10.1016/s0896-6273(00)80183-x. [DOI] [PubMed] [Google Scholar]
- 47.Schwaninger M, Blume R, Oetjen E, Knepel W. Naunyn-Schmiedeberg’s Arch Pharmacol. 1993;348:541–545. doi: 10.1007/BF00173216. [DOI] [PubMed] [Google Scholar]
- 48.Ghosh A, Ginty D D, Bading H, Greenberg M E. J Neurobiol. 1994;25:294–303. doi: 10.1002/neu.480250309. [DOI] [PubMed] [Google Scholar]
- 49.Deisseroth K, Bito H, Tsien R W. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 50.Schulman H. Curr Opin Neurobiol. 1995;5:375–381. doi: 10.1016/0959-4388(95)80051-4. [DOI] [PubMed] [Google Scholar]
- 51.Malenka R C, Kauer J A, Perkel D J, Mauk M D, Kelly P T, Nicoll R A, Waxham M N. Nature (London) 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- 52.Malinow R, Schulman H, Tsien R W. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 53.Lledo P M, Hjelmstad G O, Mukherji S, Soderling T R, Malenka R C, Nicoll R A. Proc Natl Acad Sci USA. 1995;92:11175–11179. doi: 10.1073/pnas.92.24.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J H, Kelly P T. Neuron. 1995;15:443–452. doi: 10.1016/0896-6273(95)90048-9. [DOI] [PubMed] [Google Scholar]
- 55.Barria A, Muller D, Derkach V, Griffith L C, Soderling T R. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 56.Mulkey R M, Herron C E, Malenka R C. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- 57.Sah P, Hestrin S, Nicoll R A. J Physiol (London) 1990;430:605–616. doi: 10.1113/jphysiol.1990.sp018310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perouansky M, Yaari Y. J Physiol (London) 1993;465:223–244. doi: 10.1113/jphysiol.1993.sp019674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geiger J R, Melcher T, Koh D S, Sakmann B, Seeburg P H, Jonas P, Monyer H. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 60.Koh D S, Geiger J R, Jonas P, Sakmann B. J Physiol (London) 1995;485:383–402. doi: 10.1113/jphysiol.1995.sp020737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang L Y, Orser B A, Brautigan D L, MacDonald J F. Nature (London) 1994;369:230–232. doi: 10.1038/369230a0. [DOI] [PubMed] [Google Scholar]
- 62.Wang Y T, Salter M W. Nature (London) 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- 63.Wang Y T, Yu X M, Salter M W. Proc Natl Acad Sci USA. 1996;93:1721–1725. doi: 10.1073/pnas.93.4.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Freund T F, Buzsáki G. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 65.Plant T, Schirra C, Garaschuk O, Rossier J, Konnerth A. J Physiol (London) 1997;499:47–63. doi: 10.1113/jphysiol.1997.sp021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Somogyi P, Kisvarday Z F, Martin K A, Whitteridge D. Neuroscience. 1983;10:261–294. doi: 10.1016/0306-4522(83)90133-1. [DOI] [PubMed] [Google Scholar]
- 67.Eilers J, Plant T, Konnerth A. Cell Calcium. 1996;20:215–226. doi: 10.1016/s0143-4160(96)90108-6. [DOI] [PubMed] [Google Scholar]
- 68.Fukunaga K, Goto S, Miyamoto E. J Neurochem. 1988;51:1070–1078. doi: 10.1111/j.1471-4159.1988.tb03070.x. [DOI] [PubMed] [Google Scholar]
- 69.Goto S, Matsukado Y, Uemura S, Mihara Y, Inoue N, Ikeda J, Miyamoto E. Exp Brain Res. 1988;69:645–650. doi: 10.1007/BF00247316. [DOI] [PubMed] [Google Scholar]
- 70.Benson D L, Isackson P J, Gall C M, Jones E G. Neuroscience. 1992;46:825–849. doi: 10.1016/0306-4522(92)90188-8. [DOI] [PubMed] [Google Scholar]
- 71.Ikeshima H, Yuasa S, Matsuo K, Kawamura K, Hata J, Takano T. J Neurosci Res. 1993;36:111–119. doi: 10.1002/jnr.490360112. [DOI] [PubMed] [Google Scholar]
- 72.Saimi Y, Kung C. FEBS Lett. 1994;350:155–158. doi: 10.1016/0014-5793(94)00782-9. [DOI] [PubMed] [Google Scholar]
- 73.Ehlers M D, Zhang S, Bernhardt J P, Huganir R L. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- 74.Wyszynski M, Lin J, Rao A, Nigh E, Beggs A H, Craig A M, Sheng M. Nature (London) 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- 75.Carew T J. Neuron. 1996;16:5–8. doi: 10.1016/s0896-6273(00)80016-1. [DOI] [PubMed] [Google Scholar]
- 76.Harris K A, Oyler G A, Doolittle G M, Vincent I, Lehman R A, Kincaid R L, Billingsley M L. Ann Neurol. 1993;33:77–87. doi: 10.1002/ana.410330113. [DOI] [PubMed] [Google Scholar]
- 77.Biewenga J E, Schrama L H, Gispen W H. Acta Biochim Pol. 1996;43:327–338. [PubMed] [Google Scholar]
- 78.Halpain S, Girault J A, Greengard P. Nature (London) 1990;343:369–372. doi: 10.1038/343369a0. [DOI] [PubMed] [Google Scholar]
- 79.Soriano E, Del R J, Martinez A, Super H. J Comp Neurol. 1994;342:571–595. doi: 10.1002/cne.903420406. [DOI] [PubMed] [Google Scholar]
- 80.Magyar-Lehmann S, Suter C S, Stahel W, Schachner M. Eur J Neurosci. 1995;7:1449–1459. doi: 10.1111/j.1460-9568.1995.tb01140.x. [DOI] [PubMed] [Google Scholar]
- 81.Sharkey J, Butcher S P. Nature (London) 1994;371:336–339. doi: 10.1038/371336a0. [DOI] [PubMed] [Google Scholar]
- 82.Drake M, Friberg H, Boris-Moller F, Sakata K, Wieloch T. Acta Physiol Scand. 1996;158:155–159. doi: 10.1046/j.1365-201X.1996.535298000.x. [DOI] [PubMed] [Google Scholar]
- 83.Schmidt-Kastner R, Freund T F. Neuroscience. 1991;40:599–636. doi: 10.1016/0306-4522(91)90001-5. [DOI] [PubMed] [Google Scholar]