

Abstract
We investigated the role of ICAM-3 in DC-SIGN-mediated human immunodeficiency virus (HIV) infection of CD4+ T cells. Our results demonstrate that ICAM-3 does not appear to play a role in DC-SIGN-mediated infection of CD4+ T cells as virus is transmitted equally to ICAM-3+ or ICAM-3− Jurkat T cells. However, HIV-1 replication is enhanced in ICAM-3− cells, suggesting that ICAM-3 may limit HIV-1 replication. Similar results were obtained when SIV replication was examined in ICAM-3+ and ICAM-3− CEMx174 cells. Furthermore, while ICAM-3 has been proposed to play a co-stimulatory role in T cell activation, DC-SIGN expression on antigen presenting cells did not enhance antigen-dependent activation of T cells. Together, these data indicate that while ICAM-3 may influence HIV-1 replication, it does so independent of DC-SIGN mediated virus transmission or activation of CD4+ T cells.
Keywords: HIV-1, SIV, DC-SIGN, ICAM-3, T cell, activation
Introduction
Myeloid dendritic cells (DCs) are among the first cells to encounter human immunodeficiency virus type 1 (HIV-1) upon its transmission across the mucosal epithelium (Pope and Haase, 2003). DCs can transport virus to lymph nodes where a robust replication in CD4+ T cells is initiated (Hu et al., 2000; Spira et al., 1996). In vitro, DCs form an infection synapse with CD4+ T cells that facilitates viral transmission (Cameron et al., 1992; McDonald et al., 2003; Weissman et al., 1995). Within the synapse, DCs present HIV-1 to the CD4/coreceptor complex for infection of the T cells (Arrighi et al., 2004; Cameron et al., 1992; Pope et al., 1994). Similar to an immunological synapse between T cells and antigen presenting B cells (Grakoui et al., 1999), the synapse between DCs and T cells is formed through rapid relocalization of various cell surface molecules to the sites of cell-cell contact in the absence of classical antigen presentation (de la Fuente et al., 2005; Revy et al., 2001). The DC/T cell infection synapse not only includes the HIV-1 virion but also the viral receptor (CD4) and co-receptor (CCR5/CXCR4) as well as the adhesion molecule LFA-1 (McDonald et al., 2003). Additionally, the DC-specific C-type lectin, DC-SIGN (DC-Specific ICAM-3 Grabbing Non-integrin), has been shown to be present in the infection synapse (Arrighi et al., 2004). While it has been demonstrated that DC-SIGN plays a role in virion capture and transmission to CD4+ target cells (Geijtenbeek et al., 2000a; Pohlmann et al., 2001; Yu Kimata et al., 2002), how it enhances HIV-1 infection is not completely defined.
DC-SIGN is a type II transmembrane lectin binding protein that is expressed on DCs and macrophages (Curtis et al., 1992; Geijtenbeek et al., 2000b; Granelli-Piperno et al., 2005; Satomi et al., 2005). Current studies have demonstrated that the calcium dependent carbohydrate recognition domain of DC-SIGN binds high mannose residues in the HIV-1 envelope glycoprotein (Env SU) in order to capture virions (Biggins et al., 2004; Curtis et al., 1992; Geijtenbeek et al., 2000a; Mitchell et al., 2001; Yu Kimata et al., 2002). Captured virions are then transmitted to CD4+ T cells for an infection in trans (Biggins et al., 2004; Geijtenbeek et al., 2000a; Mitchell et al., 2001). Within the context of this in trans infection of the T cells, it has been suggested that virus captured by DC-SIGN has increased infectivity and the ability to evade host immune surveillance through various potential mechanisms. First, virions internalized by DC-SIGN may be protected from degradation (Blauvelt et al., 1997; Kwon et al., 2002; Nobile et al., 2005; Sol-Foulon et al., 2002) and possibly neutralizing antibodies (Ganesh et al., 2004). Second, it has been suggested that internalization of virions with subsequent recycling to the DC surface promotes infectivity (Kwon et al., 2002; Zhang et al., 1999). Alternately, infection of DCs may lead to de novo replication of the virus for transmission to CD4+ T cells (Burleigh et al., 2006; Nobile et al., 2005).
DC-SIGN is proposed to also interact transiently with intercellular adhesion molecule-3 (ICAM-3) on the surface of T cells during the initial formation of an immunological synapse (van Kooyk and Geijtenbeek, 2002). ICAM-3 is a member of the immunoglobulin superfamily and contains five immunoglobulin-like domains (de Fougerolles et al., 1993; de Fougerolles and Springer, 1992; Vazeux et al., 1992). It is constitutively expressed on the surface of resting T cells and can function as a co-stimulatory molecule during T cell activation (Berney et al., 1999; de Fougerolles et al., 1994; Fawcett et al., 1992; van Kooyk and Geijtenbeek, 2002). ICAM-3 also facilitates the initial contact between antigen presenting cells and T cells (Montoya et al., 2002). The cellular ligands LFA-1 and DC-SIGN are recognized via domains one and two, respectively (Holness et al.,1995; Geijtenbeek et al., 2002; Jimenez et al., 2005). Early studies have suggested that DC-SIGN binds to ICAM-3 in a similar fashion to the interaction with HIV-1 Env SU, and that this interaction may be important for T cell activation, as antibodies to DC-SIGN have been reported to inhibit T cell activation (Geijtenbeek et al., 2000b). Additionally, although it has been demonstrated that HIV-1 can infect resting T cells in vivo (Zhang et al., 1999; Zhang et al., 2004), infection occurs much more efficiently in activated T cells (Eckstein et al., 2001; Scales et al., 2001; Stevenson et al., 1990; Zack et al., 1990). Thus, DC-SIGN may stimulate HIV-1 infection of CD4+ T cells through an ICAM-3 dependent mechanism.
In the current study, we examined the significance of ICAM-3 during both HIV-1 infection and activation of CD4+ T cells in the presence of DC-SIGN. Our data indicate that while DC-SIGN enhances HIV-1 infection of CD4+ T cells in trans, virion transmission is not facilitated by the presence of ICAM-3. Furthermore, DC-SIGN did not enhance T cell activation. Interestingly, ICAM-3 appears to influence viral replication independent of DC-SIGN. Our data suggest that DC-SIGN functions as an antigen capture factor that is capable of transmitting virus to CD4+ T cells, but that it does not play a role in T cell activation. Instead, the effects of ICAM-3 on viral replication may occur independent of DC-SIGN.
Results
Selection of ICAM-3 negative Jurkat cell line
To examine the importance of DC-SIGN–ICAM-3 interactions for HIV-1 infection, an ICAM-3 negative T cell line was generated by sterile cell sorting for the ICAM-3 negative population. Jurkat cells were chosen because they have functional ICAM-3 costimulatory activity, capable of enhancing HIV-1 expression (Barat et al. 2004; Hayflick et al., 1997). The sorted cells were then expanded and examined by FACS to confirm the absence of ICAM-3 (ICAM-3−) expression as compared to wildtype Jurkat cells (ICAM-3+) (Fig. 1A). In addition, the cell surface expression of various T cell markers was assessed by flow cytometry. Expression levels of HIV receptor (CD4) and co-receptor (CXCR4) on the ICAM-3− Jurkat cells were comparable to those observed on wildtype Jurkat cells as indicated by similar mean fluorescence intensities (MFIs). Furthermore, the MFI of cells stained with anti-CD3 and anti-CD28 antibodies was the same for both cell lines (Fig. 1A).
Fig. 1. Selection and infection of ICAM-3 negative Jurkat cell line.
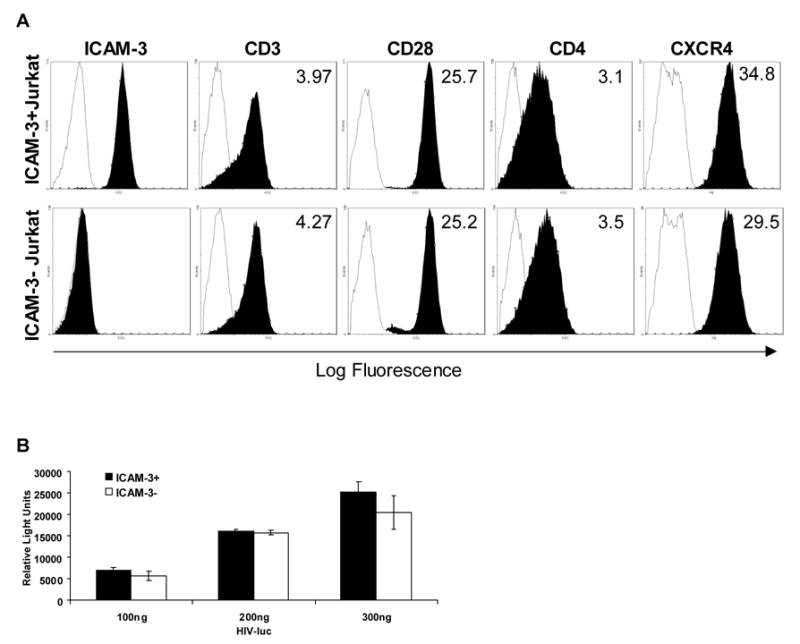
(A) Phenotype of wildtype and ICAM-3− Jurkat cells. The wildtype Jurkat cells (ICAM-3+) and mutant (ICAM-3−) cells were examined for surface expression of ICAM-3, CD3, CD28, CD4, and CXCR4 (closed curves). Isotype control antibodies are represented by open curves. The mean fluorescence intensity (MFI) of the stained cells is indicated on each histogram. (B) HIV-1 infection of wildtype and ICAM-3 negative Jurkat cells. Jurkat cells were incubated with an HIV-luciferase virus pseudotyped with a CXCR4-tropic HIV-1 envelope or left untreated for 3 hours. On the third day post-infection, cells were harvested and virus production was analyzed by measuring luciferase activity in the infected cell lysates. Mock treated cells were used for background controls. Relative luciferase activity ± standard error of the mean (SEM) is shown.
In order to confirm that the ICAM-3− Jurkat cells are still permissive for HIV-1 infection, a single cycle infection of ICAM-3− Jurkat cells was performed with an HIVΔenv-luciferase reporter virus pseudotyped with a CXCR4-tropic HIV envelope. No difference in the level of luciferase activity was observed between the wildtype and ICAM-3− Jurkat cells infected with equal amounts of virus (100–300ng p24) (Fig. 1B). When infections were allowed to proceed for seven days, there were still similar levels of luciferase activity in the ICAM-3 positive and negative Jurkat cells (data not shown). These data indicate that the ICAM-3− Jurkat cells remain permissive for HIV-1 infection and that ICAM-3 may not be required for infection of T cells.
DC-SIGN mediated transmission of HIV is independent of ICAM-3 expression
To examine whether ICAM-3 expression on target T cells is necessary for DC-SIGN–mediated infection of CD4+ T cells, we performed virion capture and transfer assays with ICAM-3+ or ICAM-3− Jurkat cells and Raji cells expressing DC-SIGN. In order to accomplish this, Raji cells stably expressing pig-tailed macaque DC-SIGN (Fig. 2A) or wildtype Raji cells were separately incubated with increasing amounts of an HIV-luciferase virus for several hours. After washing away unbound virus, the cells were co-cultured with either ICAM-3− or ICAM-3+ Jurkat cells. On the third day post-infection, a luciferase assay was performed in order to assess virus infection. For DC-SIGN–mediated capture and transfer of virus to either ICAM-3 positive or negative Jurkat cells, there was a similar 7-fold increase in luciferase activity as compared to cultures containing DC-SIGN− Raji cells, which showed low level transmission activity (Fig. 2B). This indicates that there was no difference in DC-SIGN–dependent infection of the ICAM-3 positive or negative Jurkat cells. Similar results were observed when the co-culture was allowed to proceed for seven days (data not shown). Thus, ICAM-3 does not appear to influence DC-SIGN–dependent HIV-1 infection of CD4+ T cells.
Fig. 2. DC-SIGN mediated transmission of HIV-1 to ICAM-3 positive and negative Jurkat cells.

(A) Pig-tailed macaque DC-SIGN expressing Raji cells were created via retroviral transduction. DC-SIGN expression was confirmed through FACS analysis by using an anti-DC-SIGN monoclonal antibody (filled curves) or isotype control antibody (open curves). (B) Capture and transmission of HIV from DC-SIGN+ Raji cells to ICAM-3− Jurkat cells. DC-SIGN+ Raji cells were incubated with a pseudotyped HIV-luciferase virus for 2 hours, washed and then co-cultured with target cells. On the third day post infection virus production was analyzed by measuring luciferase activity in target cells. (C) Capture and transmission of HIV-1 from DC-SIGN+ Raji cells to ICAM-3− Jurkat cells in the presence of superantigen. DC-SIGN mediated virus capture and transfer assay was performed as described above. Co-cultures were maintained in the presence or absence of staphylococcal enterotoxin D (SED). (D) Capture and transmission of HIV-1 from primary human dendritic cells to ICAM-3− Jurkat cell line. A DC-SIGN mediated virus capture and transfer assay was performed as described above except that human monocyte derived dendritic cells were substituted for DC-SIGN+ Raji cells. All of the experiments were performed in triplicate and the data are representative of at least two independent experiments. Mock treated cells were used for background direct virus infection. Relative luciferase activity ± SEM is shown.
To examine the possibility that activation of the target cells may be required in order to observe a difference in virus infection of ICAM-3− Jurkat cells as compared to the wildtype cells, we performed the DC-SIGN–mediated virus capture and transfer assay as described above in the presence of the superantigen, SED (Fig. 2C). Although there was a 2-fold increase in luciferase activity in either ICAM-3− or ICAM-3+ Jurkat cells when superantigen was added to the co-cultures as compared to those co-cultures in which superantigen was absent, the differences were not significant. More importantly, the level of luciferase activity between the ICAM-3+ and ICAM-3− cells in the presence of superantigen was similar. These results indicate that DC-SIGN–mediated HIV-1 infection occurs independently of ICAM-3 in the context of superantigen-mediated T cell activation.
To confirm the results obtained with the DC-SIGN-expressing Raji cell line, we performed a similar virus capture and transfer assay using human monocyte-derived DCs. As in the experiments with DC-SIGN-expressing Raji cells, ICAM-3+ and ICAM-3− Jurkat cells showed similar levels of infection when virus was presented by human monocyted-derived DCs, further suggesting that ICAM-3 does not play a role in DC-mediated infection of T cells (Fig. 2D). While there was an apparent 2-fold increase in transmission by DCs at the higher dose of virus, the difference is neither significant nor repeatable. Taken together, these data suggest that ICAM-3 is not important for the initial infection of CD4+ T cells, whether by direct infection or DC-SIGN–dependent transmission.
HIV and SIV replication are enhanced in the absence of ICAM-3
To determine whether ICAM-3 may have an effect on viral replication independent of DC-SIGN, we infected the ICAM-3− or ICAM-3+ Jurkat cells with replication competent HIV-1NL4-3. Interestingly, we observed about a 4-fold greater level of virus production from ICAM-3− Jurkat cells as compared to the wildtype ICAM-3+ Jurkat cells (Fig. 3). This increase is significant (p<0.05), suggesting that ICAM-3 may play a role in limiting HIV-1 replication during a spreading infection. The difference in virus production levels is not due to differences in cell viability as there is no significant difference in viable cell number between infected ICAM-3 positive and negative Jurkat cells (data not shown).
Fig. 3. HIV-1 replication is enhanced in the absence of ICAM-3.
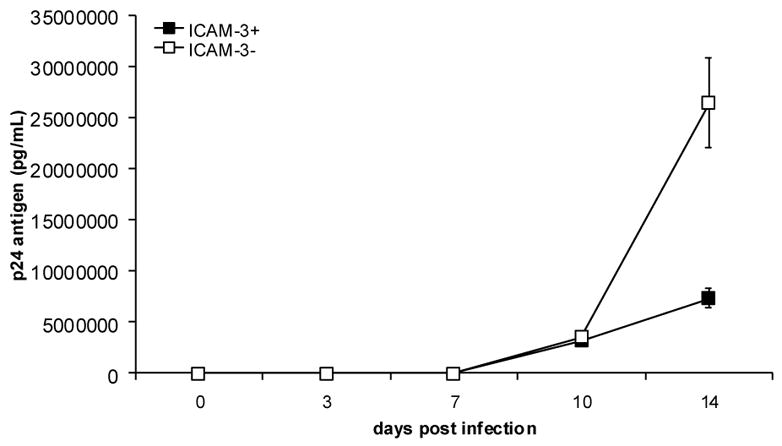
ICAM-3− or ICAM-3+ Jurkat cells were infected with HIV-1NL4-3 at a low multiplicity of infection (MOI). At various times during a two-week period post-infection supernatants were collected and virus production was measured by p24 antigen capture ELISA. Experiments were performed in triplicate, and the data are representative of three independent experiments. The average levels of p24gag ± SEM are shown.
To further investigate the effect of ICAM-3 on viral replication, we examined whether it would also limit SIV replication. For these studies, we generated an ICAM-3 negative CEMx174 cell line, since these cells are permissive for SIV replication. An ICAM-3 negative population of cells (ICAM-3−) was obtained through cell sorting (Fig. 4A). In addition, the cell surface expression of the HIV receptor (CD4) on the ICAM-3− CEMx174 cells was assessed by flow cytometry and found to be comparable to ICAM-3+ parental CEMx174 cells as demonstrated by the similar MFIs. When these cells were infected with the SIV molecular clone, SIVmne170, we observed a highly significant (p<0.001) 7-fold increase in viral production from ICAM-3− CEMX174 cells as compared to the wildtype ICAM-3+ CEMx174 cells (Fig. 4B). Additionally, after 7 days of infection, cell viability in the SIVmne170-infected ICAM-3− CEMx174 cells was decreased by approximately 10-fold more than that in the ICAM-3+ wild type cells, as a result of greater cell killing (Fig. 4C). Similar observations were made using two other molecular variants of SIVmne clones. These data further indicate that while ICAM-3 is not involved in DC-SIGN-dependent viral transmission, it may limit viral replication.
Fig. 4. Selection and infection of ICAM-3 negative CEMx174 cells.
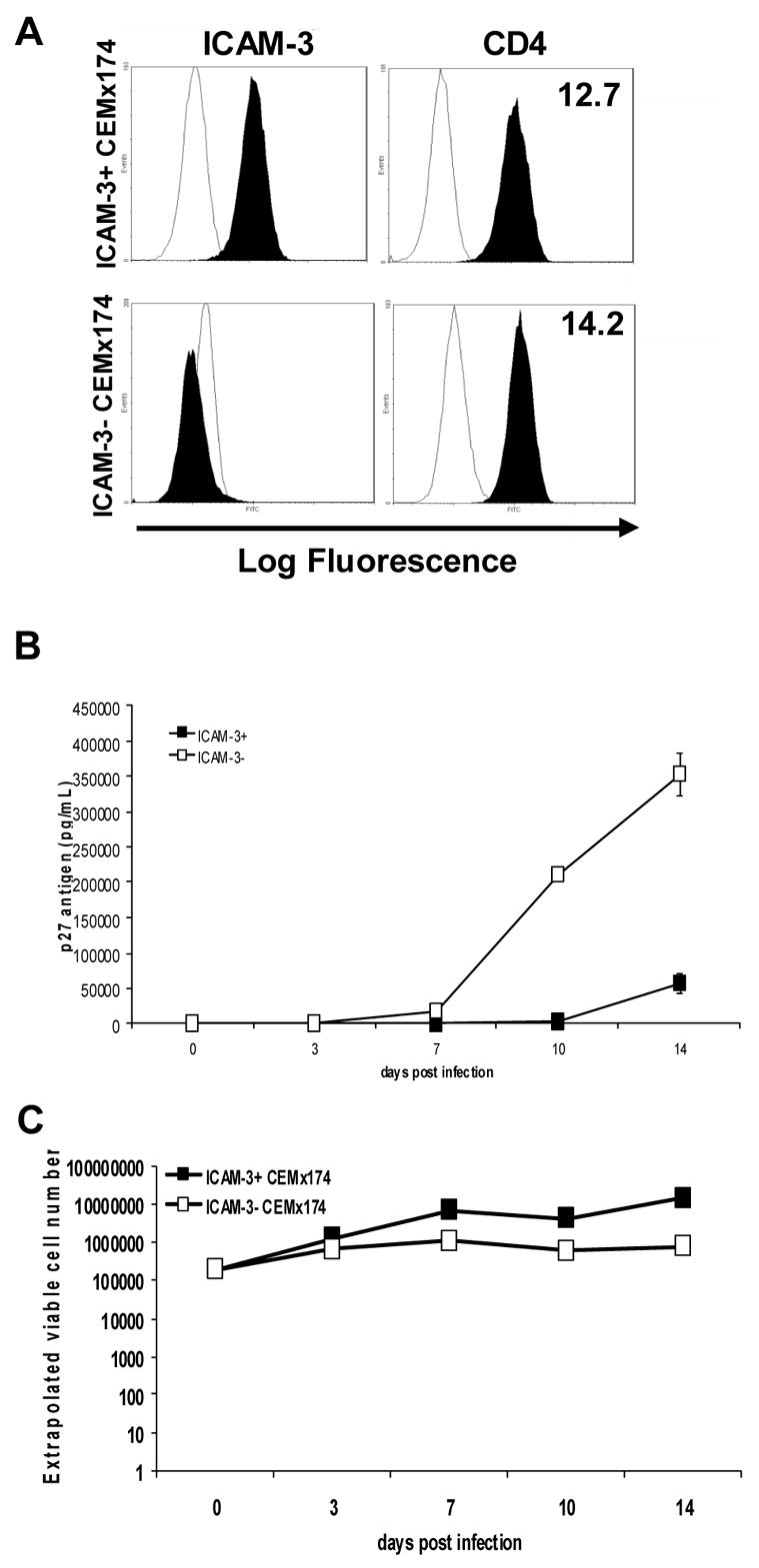
(A) Phenotype of wildtype and ICAM-3− CEMx174 cells. The wildtype CEMx174 cells (ICAM-3+) and mutant (ICAM-3−) cells were examined for surface expression of ICAM-3 and CD4 (closed curves). Isotype control antibodies are represented by open curves. The mean fluorescence intensity (MFI) of the stained cells is indicated on each histogram. (B) SIV infection of wildtype and ICAM-3− CEMx174 cells. (C) Extrapolated viable cell number of wild type and ICAM-3− CEMx174 cells. CEMx174 cells were infected with SIVmne170 at a low MOI. At various times during a two-week time period post-infection supernatants were collected and virus production was measured using an SIV p27 antigen capture ELISA. The average levels of p27gag ± SEM are shown.
DC-SIGN does not modulate T cell activation
Because it has previously been suggested that DC-SIGN may initiate contact between APCs and T cells, we also examined whether T cell activation might be influenced by the presence or absence of DC-SIGN in Jurkat:Raji co-cultures. Using the DC-SIGN-expressing Raji cell line or wildtype Raji cells, we evaluated the influence of DC-SIGN on T cell activation in the context of superantigen presentation. We found that treatment of Jurkat cells with anti-CD3 and anti-CD28 antibodies resulted in a 35-fold induction of IL-2 over unstimulated Jurkat cells (Fig. 5A CD3/CD28) and upregulation of the early activation marker CD69 from 26% to 90% of Jurkat cells (Fig. 5B CD3/CD28). Similarly, when Jurkat cells were incubated with either DC-SIGN+ or DC-SIGN− Raji cells in the presence of SED, IL-2 was induced 10-fold (Fig. 5A DC-SIGN−/+ + SED) and CD69 was upregulated to 78% of cells (Fig. 5B DC-SIGN−/DC-SIGN+ + SED), albeit to a lesser extent than the anti-CD3/anti-CD28 antibody activation control. When we compared the amount of activation marker upregulation between cultures with DC-SIGN+ Raji cells to DC-SIGN− Raji cells, we found no significant difference in IL-2 concentration (Fig. 5A DC-SIGN− + SED vs. DC-SIGN+ + SED). Likewise, a comparison of CD69 expression between DC-SIGN–expressing and wildtype Raji cell cocultures revealed no significant difference (Fig. 5B DC-SIGN− + SED vs. DC-SIGN+ + SED).
Fig. 5. DC-SIGN does not modulate T cell activation.

(A) IL-2 concentration in supernatants after activation. (B) CD69 surface expression on Jurkat cells. Jurkat cells were either cultured alone and unstimulated, co-cultured with either DC-SIGN+ or DC-SIGN− Raji cells with or without Staphylococcal enterotoxin D (SED), or cultured alone and treated with anti-CD3 and anti-CD28 antibodies. The average IL-2 levels ±SEM are shown. (C) Activation of Jurkat cells in the presence of different amounts of DC-SIGN+ Raji cells. Jurkat cells were co-cultured with either DC-SIGN+ or DC-SIGN− Raji cells and SED at the indicated ratios. After 24 hours, culture supernatants were examined for IL-2 concentration by ELISA. The average IL-2 levels ±SEM are shown.
To further investigate DC-SIGN-mediated activation of T cells we tested whether DC-SIGN+ Raji cells would be better able to activate Jurkat cells than DC-SIGN− Raji cells at higher Jurkat to Raji cell ratios. Jurkat cells were mixed with the appropriate number of either DC-SIGN+ or wildtype Raji cells at ratios of 20:1, 10:1, 2:1 or 1:1 Jurkat to Raji cells. No significant difference was observed in CD69 upregulation (data not shown) or in IL-2 concentration (Fig. 5C), regardless of the T cell:Raji ratio. Taken together, these data suggest that DC-SIGN may not play a role in altering antigen-dependent T cell activation.
Soluble DC-SIGN does not co-stimulate T cells
Alternative splicing of DC-SIGN transcripts is predicted to result in the production of a soluble form of DC-SIGN (Mummidi et al., 2001). Thus, to further characterize effects of DC-SIGN on T cell activation, we tested the ability of a recombinant soluble DC-SIGN (sDC-SIGN) molecule to co-stimulate T cells treated with anti-CD3 antibodies. sDC-SIGN was generated with a drosophila expression system and purified from culture supernatants via affinity purification (Fig. 6A). We performed immunoprecipitation experiments with our sDC-SIGN construct to confirm its ability to bind ICAM-3, but were unable to detect this interaction (data not shown). Given the high dissociation constant for interactions between recombinant DC-SIGN and ICAM-3 (KD=4.6 μM) (Satomi et al., 2005), these results were not entirely unexpected. Therefore, we assessed sDC-SIGN functionality by examining its ability to inhibit virion capture by cell-associated DC-SIGN. We found that sDC-SIGN competitively inhibited binding of HIV-1 virions to DC-SIGN-expressing cells by 75% (Fig. 6B), similar to inhibition by EGTA and mannan, other known inhibitors of DC-SIGN virion capture activity.
Fig. 6. Soluble DC-SIGN does not co-stimulate T cells.
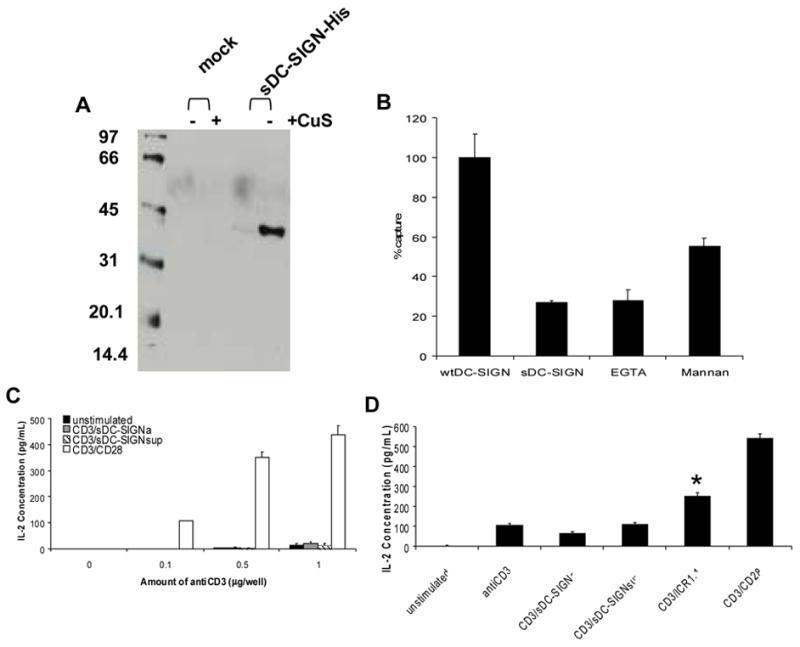
(A) Expression of sDC-SIGN. Conditioned supernatants from S2 cells stably expressing sDC-SIGN were collected and subjected to SDS-12% polyacrlyamide gel electrophoresis (PAGE). A western blot was performed and the His-tagged protein was detected by enhanced chemiluminescence. Cells were either left untreated (−) or induced to express DC-SIGN with copper sulfate (+). (B) sDC-SIGN inhibition of virion capture. DC-SIGN expressing 293T cells were incubated with indicated inhibitors for 20 minutes. The cells were then incubated with SIVmne170 for 2 hours. Cells were washed, lysed, and the amount of virus remaining bound to the cell surface was determined by antigen capture ELISA. Mock treated cells were used for background controls. Virus captured in the presence of each inhibitor is shown relative to untreated cells after correcting for background binding ± SEM. (C) Co-stimulation Assay. Jurkat cells were added to wells coated with indicated amounts of anti-CD3 antibody alone (■), anti-CD3 antibody + 1 μg sDC-SIGN (▒), coated with anti-CD3 antibody only and 1 μg sDC-SIGN added to culture medium (▧ ) or coated with anti-CD3 + 1 μg anti-CD28 antibodies (□). After 24 hours culture supernatants were examined for IL-2 concentration by ELISA. (D) ICAM-3 Activation. Jurkat cells were added to wells left untreated or coated with 1 μg/well each of anti-CD3 antibody, anti-CD3 antibody and sDC-SIGN, anti-CD3 antibody only with sDC-SIGN added to culture supernatant, anti-CD3 and anti-ICAM-3 (ICR1.1) antibodies, or anti-CD3 and anti-CD28 antibodies. After 24 hours, IL-2 concentration was measured in supernatants by ELISA. The average IL-2 levels ±SEM are shown. Student’s t-test was performed: p< .001.
To determine if purified sDC-SIGN could stimulate T cells, we incubated Jurkat cells with combinations of anti-CD3, anti-CD28 antibodies and sDC-SIGN. The results indicate that anti-CD3 antibody alone is able to induce IL-2 production, albeit at a lower level (13 pg/mL of IL-2 with 1 μg of anti-CD3) than that seen with the anti-CD3/anti-CD28 antibody (436 pg/mL) treatment (Fig. 6C). When cells were stimulated with the anti-CD3 antibody in the presence of sDC-SIGN, either adhered to the plate or added to the culture supernatant, no significant increase in IL-2 concentration was measured even with levels of sDC-SIGN reaching 6 μg per well (Fig. 6C and data not shown). To demonstrate that ICAM-3 signaling is functional in the Jurkat cell line, an activating ICAM-3 antibody (ICR1.1) was used in conjunction with the anti-CD3 antibody to stimulate Jurkat cells. While anti-ICAM-3/anti-CD3 antibody treatment is not as effective (250 pg/mL) as anti-CD3/anti-CD28 antibody co-stimulation (540 pg/mL), anti-ICAM-3/anti-CD3 antibody treatment was able to increase IL-2 concentration over that of anti-CD3 antibody alone (107 pg/mL) and, more importantly, over that seen with anti-CD3 antibody/sDC-SIGN treatment (62 pg/mL adhered sDC-SIGN, 110 pg/mL supernatant sDC-SIGN) (Fig. 6D). Statistical analysis was performed and the increase in IL-2 production by the anti-CD3/anti-ICAM-3 treatment was found to be highly significant (p-value <.001) over that of anti-CD3/sDC-SIGN co-stimulation. These results demonstrate that while ICAM-3 signaling is intact in the Jurkat cells, sDC-SIGN was unable to use this signaling pathway to enhance IL-2 production. Together, these data suggest that DC-SIGN is unable to function as a co-stimulatory molecule in either the context of the immunological synapse formed by Raji cell presentation of superantigen or when potent signaling through the TCR is present.
To further demonstrate that the interaction between DC-SIGN and ICAM-3 does not play a role in T cell activation, we incubated ICAM-3+/− Jurkat/DC-SIGN+/− Raji cell co-cultures with SED and measured IL-2 concentration. As shown in Fig. 7, there was no difference in the IL-2 production from co-cultures regardless of the status of ICAM-3 or DC-SIGN. These results further confirm that the binding of DC-SIGN to ICAM-3 does not affect IL-2 production from Jurkat cells.
Fig. 7. IL-2 production is sustained despite loss of ICAM-3-DC-SIGN interaction.

Wildtype or ICAM-3− Jurkat cells were cocultured with either wildtype or DC-SIGN+ Raji cells in the presence or absence of Staphylococcal enterotoxin D (SED). Alternately, Jurkat cells cultured alone were treated with anti-CD3 and anti-CD28 antibodies or left untreated. After 24 hours, IL-2 supernatant levels were analyzed by ELISA. The average IL-2 levels ±SEM are shown.
Discussion
DC-SIGN is a pathogen capture factor expressed on the surfaces of immature dendritic cells and macrophages that facilitates HIV-1 and SIV transmission to CD4+ T cells (Wu and KewalRamani, 2006). However, the mechanism by which DC-SIGN enhances infection is not well understood, and its involvement in DC-mediated enhancement of infection remains controversial. Initial studies showed that DC-SIGN is able to both bind HIV-1 and facilitate transmission of virions from dendritic cells to target cells in vitro. However, recent studies indicate that DC-SIGN’s primary function in this process may not be virion capture but transmission of virions to CD4+ T cells and formation of an infection synapse (Arrighi et al., 2004; Geijtenbeek et al., 2000a; Mitchell et al., 2001). Furthermore, other studies have suggested that DC-mediated enhancement of HIV-1 and SIV infection may occur by mechanisms independent of DC-SIGN (Boggiano et al., 2007; Gummuluru et al., 2003; Wu et al., 2002b). Because DC-SIGN interacts with ICAM-3, an additional proposed function is that DC-SIGN increases HIV-1 infection by promoting interactions with CD4+ T cells and activation through the cellular ligand, ICAM-3 (Geijtenbeek et al., 2000b). Here, we evaluated the importance of DC-SIGN–ICAM-3 interactions for HIV-1 infection and T cell activation. The data indicate that DC-SIGN acts as a capture factor that enhances infection in trans, but not as a costimulatory molecule that modulates the activation status of CD4+ T cells or increases infection via contact with ICAM-3. Interestingly, ICAM-3 appears to modulate HIV-1 and SIV replication independent of DC-SIGN.
We demonstrate that ICAM-3 does not play a functional role in the transmission of HIV-1 from DC-SIGN expressing cells to CD4+ T cells using ICAM-3 positive and negative Jurkat T cells as targets. Similar results were obtained whether HIV-1 was transmitted by either DC-SIGN+ Raji cells or primary human DCs or if viral infection was performed during antigen-dependent activation. These data are consistent with those of Wu et al. who demonstrated that DC-SIGN binding to ICAM-3 does not play a role in DC-SIGN–mediated virus transmission by using blocking antibodies and soluble ICAM-3, as well as engineered expression of ICAM-3 on GHOST/R5 target cells (Wu et al., 2002a). Furthermore, because we did not observe differences in virus transmission between ICAM-3 positive and negative Jurkat cells, the data suggest that if DC-SIGN is important for the formation of an infection synapse, it may be via a mechanism other than binding to ICAM-3.
In earlier studies, it had been demonstrated that DC-SIGN could induce T cell activation and proliferation in DC-T cell mixed lymphocyte reactions (Geijtenbeek et al., 2000b). Therefore, we evaluated the effects of DC-SIGN on Jurkat T cell activation in the context of superantigen presentation by Raji cells with or without DC-SIGN. The presence of DC-SIGN did not increase the induction of IL-2 or CD69 on target Jurkat T cells, suggesting that DC-SIGN was unable to actuate any signaling event that lead to any modulation in T cell activation. This was probably not likely due to an inability of DC-SIGN to bind ICAM-3, as we and others have shown that DC-SIGN is capable of binding to ICAM-3 (Biggins et al., 2004; Geijtenbeek et al., 2000b; Geijtenbeek et al., 2002). Furthermore, we show that co-stimulation through ICAM-3 with an activating anti-ICAM-3 antibody can induce IL-2 expression in Jurkat T cells, indicating that the ICAM-3 co-stimulatory pathway is functional in the Jurkat clone used in these experiments. Consistent with the cell-associated DC-SIGN experiments, we also found that a soluble DC-SIGN molecule could not costimulate Jurkat cells. Together, these data suggest that DC-SIGN is unable to initiate T cell activation through this interaction. These data are in agreement with recent studies that used anti-DC-SIGN blocking antibodies to show that DC-SIGN is not involved in the induction of T cell proliferation (Granelli-Piperno et al., 2005; Real et al., 2004). Additionally, domain I of ICAM-3 has been shown to be the activation domain, but biochemical studies suggest that DC-SIGN may interact with high mannose carbohydrate residues linked to domain II (Jimenez et al., 2005), which is not involved in transducing activation signals in T cells (Bossy et al., 1995; Hernandez-Caselles et al., 1993). Thus, DC-SIGN–ICAM-3 interactions may not result in activation events that enhance HIV-1 infection of T cells. Furthermore, that we did not observe greater infection of ICAM-3+ Jurkat cells compared to ICAM-3− cells when HIV-1 is transmitted by DC-SIGN in either the presence or absence of superantigen is further confirmation that DC-SIGN–mediated co-stimulation via ICAM-3 does not play a role in transmission of HIV-1.
A potential caveat to these experiments is that they were performed with Jurkat T cells as targets for infection. Jurkat T cells were chosen because it has been shown that they can transduce costimulatory activation signals via ICAM-3 and because HIV-1 gene expression is enhanced in Jurkat cells after costimulation via CD3 and ICAM-3 with anti-CD3 and anti-ICAM-3 monoclonal antibodies (Barat et al., 2004; Hayflick et al., 1997; Montoya et al., 2002). Nevertheless, further studies with primary resting CD4+ T cells could provide additional insight. Secondly, we primarily used macaque DC-SIGN for our studies, which may differ from human DC-SIGN in binding to human ICAM-3. However, this is not likely as we and others have previously shown that human ICAM-3 binds to both macaque DC-SIGN and human DC-SIGN, and that the interaction is likely mediated by recognition of carbohydrate residues in ICAM-3 (Baribaud et al., 2001; Yu Kimata et al., 2002; Biggins et al., 2004).
One intriguing observation of this study is that both HIV-1 and SIV show greater replication kinetics in CD4+ lymphocyte cell lines that do not express ICAM-3. The data suggest that ICAM-3 may negatively regulate T cell activities involved in viral replication. How this may occur is unclear. Furthermore, the signaling mechanisms of ICAM-3 remain poorly characterized. There is suggestion that ICAM-3 expression may play a negative regulatory role in T cell activation responses (Green et al., 1996). Given the importance of activation events for HIV-1 infection and replication, this type of activity may also diminish HIV-1 and SIV replication in CD4+ T cells. Furthermore, because luciferase expression was similar in single-round infections of the ICAM-3+ and ICAM-3− Jurkat cells using the Env pseudotyped HIV-luc vector, events in the viral life cycle leading to viral gene expression probably do not account for the differences in viral replication in the presence and absence of ICAM-3. Further confirmation of our results by siRNA knock down of ICAM-3 expression in primary CD4+ T cells may provide additional insight into the effects of ICAM-3 on viral replication.
In summary, DC-SIGN-mediated transmission of HIV-1 to CD4+ T cells occurs independent of the interaction between DC-SIGN and ICAM-3. Additionally, while ICAM-3 is known to play a co-stimulatory role in T cell activation and enhancement of HIV-1 transcription, DC-SIGN does not enhance antigen dependent or independent activation of T cells. Therefore, we conclude that DC-SIGN–ICAM-3 interactions do not lead to enhanced activation or HIV-1 infection of CD4+ T cells. Thus, the data are consistent with a model in which HIV-1 subverts the pathogen capture function of DC-SIGN for enhanced transmission to CD4+ T cells in context of an infection synapse. Finally, independent of DC-SIGN, ICAM-3 may modulate the extent of HIV-1 replication.
Materials and Methods
Cell lines and viruses
CEMx174, Jurkat, and Raji cells were cultured in RPMI 1640 medium containing 10% heat inactivated fetal bovine serum (FBS), 100 U of penicillin per mL, 100 μg of streptomycin per mL, and 2 mM glutamine (RPMI complete). DC-SIGN+ Raji cells were maintained as described below. The 293T cell line and PT67 packaging cell line were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 2 mM glutamine, 100 U of penicillin per ml, and 100 μg of streptomycin per ml (DMEM complete). Human monocyte-derived DCs were generated from peripheral blood mononuclear cells (PBMCs) as previously described (Yu Kimata et al., 2002). Briefly, CD14+ monocytes were isolated via anti-CD14 microbeads and miniMACS system according to manufacturer instructions (Miltenyi Biotech, Auburn, CA). The CD14+ monocytes were then washed with RPMI complete media and cultured with 1000U/mL each of granulocyte-macrophage colony-stimulating factor (GM-CSF) and Interleukin-4 (IL-4) (R&D Systems, Minneapolis, MN) in RPMI complete media for 7 days to generate dendritic cells. By FACS analysis greater than 95% of these cells express DC-SIGN (data not shown).
HIV-luciferase virus stocks were generated by co-transfecting 293T cells with both the plasmid expressing the CXCR4-tropic HIV envelope (pHxB2-env) and the plasmid containing the HIV-luciferase proviral genome (HIV-luc, a gift from N. Landau). The cells were washed with phosphate buffered saline (PBS) twenty-four hours later, and then cultured in fresh DMEM complete medium. The following day, supernatants were collected, passed through 0.22 μm syringe filters, and saved at −80°C until use for infection experiments. The amount of virus was determined by p24 antigen capture ELISA (Coulter-Immunotech, Miami, FL).
Replication competent HIVNL4-3 and SIVmne170 virus stocks were generated by transiently transfecting 293T cells with plasmid proviral clones, washing with PBS twenty-four hours later, and then culturing in fresh DMEM complete medium. The following day, supernatants were collected, passed through 0.22 μm syringe filters, and saved at −80°C until used for infection experiments. The amount of HIV p24gag antigen or SIV p27gag antigen was quantified using a commercial ELISA (Immunotech-Coulter, Miami, FL).
Selection of ICAM-3− cells
Approximately 30×106 Jurkat or CEMx174 cells were stained with 1 μg FITC-conjugated anti-ICAM-3 antibody (R&D Systems, Minneapolis, MN) for 30 minutes at 4°C in a FACS staining buffer (PBS supplemented with 10% FBS) without sodium azide. The cells were then sterile sorted using a Beckman-Coulter Ultra high speed, high pressure cell sorting machine for the cell population negative for ICAM-3 expression. The ICAM-3− cells were collected in RPMI complete media and allowed to expand. The absence of ICAM-3 expression was confirmed by staining the sorted cells in a FACS staining buffer containing 0.1% sodium azide for 30 min at 4°C with the same anti-ICAM-3 antibody. After washing with PBS, the cells were resuspended in 1% paraformaldehyde and analyzed with a Beckman Coulter EPICS XL-MCL. The Jurkat cells were then subjected to a second sterile sort in order to obtain a homogeneous population of ICAM-3− cells while the CEMx174 cells underwent two subsequent sterile sorts. These cells maintained a stable ICAM-3− phenotype during 6 months of continuous culturing.
Transduction and selection of DC-SIGN expressing Raji cells
The cDNA encoding pig-tail macaque DC-SIGN was inserted into the pLNCX retroviral vector (Clontech) at the HpaI and ClaI restriction sites. We previously showed that macaque DC-SIGN is structurally and functionally similar to human DC-SIGN (Baribaud et al., 2001; Yu Kimata et al., 2002). The PT67 retroviral packaging cell line was then transiently transfected via FuGene lipid carrier (Roche, Indianapolis, IN) with the DC-SIGN/pLNCX vector. Forty-eight hours post transfection the virus was harvested and Raji cells were spin-inoculated in the presence of 0.8 μg/mL polybrene at 32°C for 1 hour. The cells were then incubated overnight at 32°C and resuspended in fresh media. After an additional 48 hours, the cells were placed under 1 mg/mL G418 selection media. Viable cells were stained with an anti-DC-SIGN monoclonal antibody (8C1; (Yu Kimata et al., 2002)) and a FITC-conjugated goat anti-mouse secondary antibody (Dako Corp, Carpinteria, CA). The cells were then examined by FACS analysis as described above for expression of DC-SIGN (Fig. 2). As compared to the parental cells, greater than 98% of the transduced cells expressed DC-SIGN. The cells were also subjected to sterile sorting, as described above, in order to isolate a population of Raji cells that expressed high levels of DC-SIGN on the cell surface.
Infection of cell lines
ICAM-3+ or ICAM-3− Jurkat cells (2×105) were incubated with varying amounts of a HIV-luciferase virus pseudotyped with a CXCR4 tropic HIV envelope or left untreated for 3 hours at 37°C in a 96-well round bottom plate. The cells were then washed vigorously 3 times with RPMI complete media. Following the last wash, the cells were resuspended in 150 μL RPMI complete media and transferred to a 24-well plate where the final volume was 2 mL RPMI complete media. On day 3 or 7 post-infection, cells were harvested and a luciferase assay (Roche, Indianapolis, IN) was performed according to manufacturer instructions with a tube luminometer (Berthold Detection Systems).
HIV-1 capture and transmission assays were performed as previously described (Yu Kimata et al., 2002). Briefly, either 1×105 DC-SIGN+ or DC-SIGN− Raji cells were incubated with varying amounts of HIV-luciferase virus pseudotyped with a CXCR4 tropic HIV-1 envelope or left untreated for 2 hours. The cells were then washed 3 times and co-cultured with either 2×105 ICAM-3+ or ICAM-3− Jurkat cells. On day 3 post-infection, cells were harvested and a luciferase assay (Roche) was performed according to manufacturer instructions. Similar experiments were performed with cells harvested 7 days post-infection. For capture and transmission assays that include Staphylococcol enterotoxin D (SED; Toxin Technology, Inc., Sarasota, FL), the cells were co-cultured in the presence of 100 ng SED. For capture and transmission assays that include human DCs, a similar protocol was followed, substituting 1×105 human DCs for 1×105 Raji cells.
Direct infection with replication competent HIV NL4-3 or SIVmne170 was performed by incubating ICAM-3− or ICAM-3+ Jurkat or CEMx174 cells (2×105) with the appropriate virus (1ng p24gag antigen HIV NL4-3 or 1ng p27gag antigen SIVmne170, respectively) or DMEM complete media in a 96 well U-bottom plate for 2-4 hours at 37°C. The cells were then washed vigorously 3 times with RPMI complete media. Following the last wash, the cells were resuspended in 150 μL RPMI complete media and transferred to a 24-well plate where the final volume was 2 mL RPMI complete media. At various time points during a two week period supernatants were collected from each well and analyzed by antigen capture ELISA (Immunotech-Coulter, Miami, FL). Cell viability was determined by trypan blue dye exclusion.
Soluble DC-SIGN
In order to create a soluble DC-SIGN (sDC-SIGN) molecule the extracellular neck and CRD domains were amplified by PCR and inserted into a drosophila expression vector, pMTBiP/V5-His, which would allow a his-tagged DC-SIGN molecule to be secreted upon induction in Drosophila S2 cells (Invitrogen, Carlsbad, CA). S2 cells stably carrying the expression vector were produced by co-transfecting the expression plasmid along with a plasmid encoding for the blasticidin selectable marker. Forty-eight hours post-transfection the cells were selected in Drosophila growth media and blasticidin (25 μg/ml). To produce the soluble secreted DC-SIGN protein, expression of the sDC-SIGN was induced with copper sulfate. Twenty-four to forty-eight hours post-induction conditioned supernatants were collected and expression of his-tagged sDC-SIGN was confirmed by Western Blot. The His-tagged protein was detected with a 1:5000 dilution of a horseradish peroxidase coupled anti-His antibody by enhanced chemiluminescence. sDC-SIGN protein was purified away from the conditioned supernatant through affinity purification with a nickel chelating column and varying concentrations of imidazol according to manufacturer instructions (Invitrogen). In order to remove the imidazol, the sDC-SIGN protein was dialyzed against a native purification buffer (250 nM NaPO4, 2.5 M NaCl) that does not contain imidazole. Expression of the sDC-SIGN protein was confirmed by western blot and quantified by the Micro BCA Protein Assay Kit (Pierce, Rockford, IL).
sDC-SIGN capture inhibition assay
293T cells were transiently transfected with either a pig-tailed macaque (pt) DC-SIGN expression vector or pcDNA3 vector without insert using the FuGene 6 reagent (Roche). Twenty-four hours post-transfection the cells were replated in a 96 well U-bottom plate at a concentration of 1×105 cells per well in 100μL. The following day the cells were pretreated with purified sDC-SIGN, 20 μg/mL mannan, 5mM EGTA, or DMEM complete media for 20 minutes at 37°C. The cells were then incubated with SIVmne170 (5ng p27gag antigen) or DMEM complete for 2 hours at 37°C, washed vigorously 2 times with DMEM complete media and one time with PBS. After the last wash the cells were resuspended in PBS and transferred to a fresh 96 well U-bottom plate that contains triton-x 100 for cell lysis. The resulting lysate was examined by antigen capture ELISA to determine the amount of virus bound to the cell surface.
Co-stimulation Assays
24-well plates were coated with antibodies in a volume of 0.5 mL PBS for 3 hours at 37°C. The plates were then washed twice with PBS, blocked with 1% bovine serum albumin in PBS for 30 minutes and washed a final time with PBS. Wells used for stimulation controls were coated with 1 μg each of anti-CD3 and anti-CD28 antibodies (BD Biosciences, San Diego, CA; (Zhou et al., 2001)). For the ICAM-3 co-stimulation control, wells were coated with 1 μg each of the anti-CD3 antibody and anti-ICAM-3 antibody, ICR1.1 (Joel Hayflick, ICOS; (Hayflick et al., 1997)). For experiments with soluble DC-SIGN (sDC-SIGN), wells were coated with 1 μg of the anti-CD3 antibody and sDC-SIGN was added to the culture supernatant or adhered to the plate concurrently with the anti-CD3 antibody. Jurkat (106) cells were then added to each well in a 1 mL volume of RPMI complete media and incubated for 24 hours at 37°C. The cells were then pelleted by centrifugation and supernatants were harvested for use in an IL-2 ELISA, which was performed according to manufacturer instructions (R&D Systems). The pelleted cells were stained with anti-CD20-PE and anti-CD69-PECy5 antibodies (BD Biosciences) in FACS staining buffer for 30 minutes on ice. The cells were fixed with 1% paraformaldehyde in PBS and FACS analysis was performed.
SED Superantigen stimulation
Staphylococcal enterotoxin D (SED) is a superantigen that is able to bind the Vβ8 chain found on the Jurkat E61 T cell and induce activation (Fraser et al., 1992; Kimata et al., 1994; Marrack and Kappler, 1990). Jurkat cells and either DC-SIGN− or DC-SIGN+ Raji cells were mixed at different ratios in a volume of 2mL RPMI complete media in the presence of 100 ng of Staphylococcal enterotoxin D (SED; Toxin Technology, Inc., Sarasota, FL). DC-SIGN+ Raji cells were washed once with PBS and resuspended in RPMI complete media prior to use in co-cultures. After 24 hours the cells were then pelleted by centrifugation and the supernatants were harvested for use in an IL-2 ELISA while the pelleted cells were analyzed by flow cytometry as described above.
Acknowledgments
We thank Andy Rice and Richard Sutton for helpful suggestions and Jeff Scott and Cassandra Horne for assistance with flow cytometry. This work was supported by grants from NIH (AI47725) and the San Antonio Area Foundation (J.T. K.), and in part by the Baylor-UT Houston CFAR grant (AI36211). We also thank Joel Hayflick at ICOS for the activating ICAM-3 antibody, ICR1.1. The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: Jurkat E6-1 from A. Weiss; and phxb2-env from K. Page and D. Littman.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arrighi JF, Pion M, Garcia E, Escola JM, van Kooyk Y, Geijtenbeek TB, Piguet V. DC-SIGN-mediated Infectious Synapse Formation Enhances X4 HIV-1 Transmission from Dendritic Cells to T Cells. J Exp Med. 2004;200:1279–1288. doi: 10.1084/jem.20041356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barat C, Gervais P, Tremblay MJ. Engagement of ICAM-3 Provides a Costimulatory Signal for Human Immunodeficiency Virus Type 1 Replication in both Activated and Quiescent CD4+ T Lymphocytes: Implications for Virus Pathogenesis. J Virol. 2004;78:6692–6697. doi: 10.1128/JVI.78.12.6692-6697.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baribaud F, Pohlmann S, Sparwasser T, Kimata MTY, Choi YK, Haggarty BS, Ahmad N, Macfarlan T, Edwards TG, Leslie GJ, Arnason J, Reinhart TA, Kimata JT, Littman DR, Hoxie JA, Doms RW. Functional and Antigenic Characterization of Human, Rhesus Macaque, Pigtailed Macaque, and Murine DC-SIGN. J Virol. 2001;75:10281–10289. doi: 10.1128/JVI.75.21.10281-10289.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berney SM, Schaan T, Alexander JS, Peterman G, Hoffman PA, Wolf RE, van der Heyde H, Atkinson TP. ICAM-3 (CD50) cross-linking augments signaling in CD3-activated peripheral human T lymphocytes. J Leukoc Biol. 1999;65:867–874. doi: 10.1002/jlb.65.6.867. [DOI] [PubMed] [Google Scholar]
- Biggins JE, Yu Kimata MT, Kimata JT. Domains of macaque DC-SIGN essential for capture and transfer of simian immunodeficiency virus. Virology. 2004;324:194–203. doi: 10.1016/j.virol.2004.03.026. [DOI] [PubMed] [Google Scholar]
- Blauvelt A, Asada H, Saville MW, Klaus-Kovtun V, Altman DJ, Yarchoan R, Katz SI. Productive infection of dendritic cells by HIV-1 and their ability to capture virus are mediated through separate pathways. Journal of Clinical Investigation. 1997;100:2043–2053. doi: 10.1172/JCI119737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggiano C, Manel N, Littman DR. Dendritic cell-mediated trans-enhancement of human immunodeficiency virus type 1 infectivity is independent of DC-SIGN. J Virol. 2007;81:2519–2523. doi: 10.1128/JVI.01661-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy D, Buckley CD, Holness CL, Littler AJ, Murray N, Collins I, Simmons DL. Epitope mapping and functional properties of anti-intercellular adhesion molecule-3 (CD50) monoclonal antibodies. Eur J Immunol. 1995;25:459–465. doi: 10.1002/eji.1830250223. [DOI] [PubMed] [Google Scholar]
- Burleigh L, Lozach PY, Schiffer C, Staropoli I, Pezo V, Porrot F, Canque B, Virelizier JL, Arenzana-Seisdedos F, Amara A. Infection of Dendritic Cells (DCs), Not DC-SIGN-Mediated Internalization of Human Immunodeficiency Virus, Is Required for Long-Term Transfer of Virus to T Cells. J Virol. 2006;80:2949–2957. doi: 10.1128/JVI.80.6.2949-2957.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron PU, Freudenthal PS, Barker JM, Gezelter S, Inaba K, Steinman RM. Dendritic cells exposed to human immunodeficiency virus type-1 transmit a vigorous cytopathic infection to CD4+ T cells. Science. 1992;257:383–387. doi: 10.1126/science.1352913. [DOI] [PubMed] [Google Scholar]
- Curtis BM, Scharnowski S, Watson AJ. Sequence and Expression of a Membrane-Associated C-Type Lectin that Exhibits CD4-Independent Binding of Human Immunodeficiency Virus Envelope Glycoprotein gp120. PNAS. 1992;89:8356–8360. doi: 10.1073/pnas.89.17.8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Fougerolles AR, Klickstein LB, Springer TA. Cloning and expression of intercellular adhesion molecule 3 reveals strong homology to other immunoglobulin family counter-receptors for lymphocyte function-associated antigen 1. J Exp Med. 1993;177:1187–1192. doi: 10.1084/jem.177.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Fougerolles AR, Qin X, Springer TA. Characterization of the function of intercellular adhesion molecule (ICAM)-3 and comparison with ICAM-1 and ICAM-2 in immune responses. J Exp Med. 1994;179:619–629. doi: 10.1084/jem.179.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Fougerolles AR, Springer TA. Intercellular adhesion molecule 3, a third adhesion counter-receptor for lymphocyte function-associated molecule 1 on resting lymphocytes. J Exp Med. 1992;175:185–190. doi: 10.1084/jem.175.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente H, Mittelbrunn M, Sanchez-Martin L, Vicente-Manzanares M, Lamana A, Pardi R, Cabanas C, Sanchez-Madrid F. Synaptic Clusters of MHC Class II Molecules Induced on DCs by Adhesion Molecule-mediated Initial T-Cell Scanning. Mol Biol Cell. 2005;16:3314–3322. doi: 10.1091/mbc.E05-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstein DA, Penn ML, Korin YD, Scripture-Adams DD, Zack JA, Kreisberg JF, Roederer M, Sherman MP, Chin PS, Goldsmith MA. HIV-1 Actively Replicates in Naive CD4+ T Cells Residing within Human Lymphoid Tissues. Immunity. 2001;15:671–682. doi: 10.1016/s1074-7613(01)00217-5. [DOI] [PubMed] [Google Scholar]
- Fawcett J, Holness CLL, Needham LA, Turley H, Gattert KC, Mason DY, Simmons DL. Molecular cloning of ICAM-3, a third ligand for LFA-1, constitutively expressed on resting leukocytes. Nature. 1992;360:481–484. doi: 10.1038/360481a0. [DOI] [PubMed] [Google Scholar]
- Fraser JD, Newton ME, Weiss A. CD28 and T cell antigen receptor signal transduction coordinately regulate interleukin 2 gene expression in response to superantigen stimulation. J Exp Med. 1992;175:1131–1134. doi: 10.1084/jem.175.4.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh L, Leung K, Lore K, Levin R, Panet A, Schwartz O, Koup RA, Nabel GJ. Infection of Specific Dendritic Cells by CCR5-Tropic Human Immunodeficiency Virus Type 1 Promotes Cell-Mediated Transmission of Virus Resistant to Broadly Neutralizing Antibodies. J Virol. 2004;78:11980–11987. doi: 10.1128/JVI.78.21.11980-11987.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TBH, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GCF, Middel J, Cornelissen ILMH, Nottet HSLM, KewalRamani VN, Littman DR. DC-SIGN, a Dendritic Cell-Specific HIV-1-Binding Protein that Enhances trans-Infection of T Cells. Cell. 2000a;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek TBH, Torensma R, van Vliet SJ, van Duijnhoven GCF, Adema GJ, van Kooyk Y, Figdor CG. Identification of DC-SIGN, a Novel Dendritic Cell-Specific ICAM-3 Receptor that Supports Primary Immune Responses. Cell. 2000b;100:575–585. doi: 10.1016/s0092-8674(00)80693-5. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek TBH, van Duijnhoven GCF, van Vliet SJ, Krieger E, Vriend G, Figdor CG, van Kooyk Y. Identification of Different Binding Sites in the Dendritic Cell-specific Receptor DC-SIGN for Intercellular Adhesion Molecule 3 and HIV-1. J Biol Chem. 2002;277:11314–11320. doi: 10.1074/jbc.M111532200. [DOI] [PubMed] [Google Scholar]
- Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The Immunological Synapse: A Molecular Machine Controlling T Cell Activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- Granelli-Piperno A, Pritsker A, Pack M, Shimeliovich I, Arrighi JF, Park CG, Trumpfheller C, Piguet V, Moran TM, Steinman RM. Dendritic Cell-Specific Intercellular Adhesion Molecule 3-Grabbing Nonintegrin/CD209 Is Abundant on Macrophages in the Normal Human Lymph Node and Is Not Required for Dendritic Cell Stimulation of the Mixed Leukocyte Reaction. J Immunol. 2005;175:4265–4273. doi: 10.4049/jimmunol.175.7.4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JM, Thompson CB. Homotypic Interactions Mediated through LFA-1/ICAM-3 Decrease the Proliferative Response of Activated T Cells. Cell Immunol. 1996;171:126–131. doi: 10.1006/cimm.1996.0182. [DOI] [PubMed] [Google Scholar]
- Gummuluru S, Rogel M, Stamatatos L, Emerman M. Binding of human immunodeficiency virus type 1 to immature dendritic cells can occur independently of DC-SIGN and mannose binding C-type lectin receptors via a cholesterol-dependent pathway. J Virol. 2003;77:12865–12874. doi: 10.1128/JVI.77.23.12865-12874.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick JS, Stine J, Fox R, Hoekstra D, Gallatin WM. Functional Mapping of the Cytoplasmic Region of Intercellular Adhesion Molecule-3 Reveals Important Roles for Serine Residues. J Biol Chem. 1997;272:22207–22214. doi: 10.1074/jbc.272.35.22207. [DOI] [PubMed] [Google Scholar]
- Hernandez-Caselles T, Rubio G, Campanero MR, del Pozo MA, Muro M, Sanchez-Madrid F, Aparicio P. ICAM-3, the third LFA-1 counterreceptor, is a co-stimulatory molecule for both resting and activated T lymphocytes. European Journal of Immunology. 1993;23:2799–2806. doi: 10.1002/eji.1830231112. [DOI] [PubMed] [Google Scholar]
- Holness CL, Bates PA, Littler AJ, Buckley CD, McDowall A, Bossy D, Hogg N, Simmons DL. Analysis of the Binding Site on Intercellular Adhesion Molecule 3 for the Leukocyte Integrin Lymphocyte Function-associated Antigen 1. J Biol Chem. 1995;270:877–884. doi: 10.1074/jbc.270.2.877. [DOI] [PubMed] [Google Scholar]
- Hu J, Gardner MB, Miller CJ. Simian Immunodeficiency Virus Rapidly Penetrates the Cervicovaginal Mucosa after Intravaginal Inoculation and Infects Intraepithelial Dendritic Cells. J Virol. 2000;74:6087–6095. doi: 10.1128/jvi.74.13.6087-6095.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez D, Roda-Navarro P, Springer TA, Casasnovas JM. Contribution of N-Linked Glycans to the Conformation and Function of Intercellular Adhesion Molecules (ICAMs) J Biol Chem. 2005;280:5854–5861. doi: 10.1074/jbc.M412104200. [DOI] [PubMed] [Google Scholar]
- Kimata JT, Wong FH, Ratner L. CD3-dependent lympocyte activation by human T cell leukaemia virus type I-producing T cells. Journal of General Virology. 1994;75:2433–2437. doi: 10.1099/0022-1317-75-9-2433. [DOI] [PubMed] [Google Scholar]
- Kwon DS, Gregorio G, Bitton N, Hendrickson WA, Littman DR. DC-SIGN-Mediated Internalization of HIV Is Required for Trans-Enhancement of T Cell Infection. Immunity. 2002;16:135–144. doi: 10.1016/s1074-7613(02)00259-5. [DOI] [PubMed] [Google Scholar]
- Marrack P, Kappler J. The Staphylococcal Enterotoxins and Their Relatives. Science. 1990;248:705–711. doi: 10.1126/science.2185544. [DOI] [PubMed] [Google Scholar]
- McDonald D, Wu L, Bohks SM, KewalRamani VN, Unutmaz D, Hope TJ. Recruitment of HIV and Its Receptors to Dendritic Cell-T Cell Junctions. Science. 2003;300:1295–1297. doi: 10.1126/science.1084238. [DOI] [PubMed] [Google Scholar]
- Mitchell DA, Fadden AJ, Drickamer K. A Novel Mechanism of Carbohydrate Recognition by the C-type Lectins. J Biol Chem. 2001;276:28939–28945. doi: 10.1074/jbc.M104565200. [DOI] [PubMed] [Google Scholar]
- Montoya MC, Sancho D, Bonello G, Collette Y, Langlet C, He HT, Aparicio P, Alcover A, Olive D, Sanchez-Madrid F. Role of ICAM-3 in the initial interaction of T lymphocytes and APCs. Nat Immunol. 2002;3:159–168. doi: 10.1038/ni753. [DOI] [PubMed] [Google Scholar]
- Mummidi S, Catano G, Lam L, Hoefle A, Telles V, Begum K, Jiminez F, Ahuja SS, Ahuja SK. Extensive Repertoire of Membrane-bound and Soluble Dendritic Cell-specific ICAM-3-grabbing Nonintegrin 1 (DC-SIGN1) and DC-SIGN2 Isoforms. J Biol Chem. 2001;276:33196–33212. doi: 10.1074/jbc.M009807200. [DOI] [PubMed] [Google Scholar]
- Nobile C, Petit C, Moris A, Skrabal K, Abastado JP, Mammano F, Schwartz O. Covert Human Immunodeficiency Virus Replication in Dendritic Cells and in DC-SIGN-Expressing Cells Promotes Long-Term Transmission to Lymphocytes. J Virol. 2005;79:5386–5399. doi: 10.1128/JVI.79.9.5386-5399.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlmann S, Baribaud F, Lee B, Leslie GJ, Sanchez MD, Hiebenthal-Millow K, Munch J, Kirchhoff F, Doms RW. DC-SIGN Interactions with Human Immunodeficiency Virus Type 1 and 2 and Simian Immunodeficiency Virus. J Virol. 2001;75:4664–4672. doi: 10.1128/JVI.75.10.4664-4672.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope M, Betjes MGH, Romani N, Hirmand H, Cameron PU, Hoffman L, Gezelter S, Schuler G, Steinman RM. Conjugates of dendritic cells and memory T lymphocytes from skin facilitate productive infection with HIV-1. Cell. 1994;78:389–398. doi: 10.1016/0092-8674(94)90418-9. [DOI] [PubMed] [Google Scholar]
- Pope M, Haase AT. Transmission, acute HIV-1 infection and the quest for strategies to prevent infection. Nat Med. 2003;9:847–852. doi: 10.1038/nm0703-847. [DOI] [PubMed] [Google Scholar]
- Real E, Kaiser A, Raposo G, Amara A, Nardin A, Trautmann A, Donnadieu E. Immature Dendritic Cells (DCs) Use Chemokines and Intercellular Adhesion Molecule (ICAM)-1, But Not DC-Specific ICAM-3-Grabbing Nonintegrin, to Stimulate CD4+ T Cells in the Absence of Exogenous Antigen. J Immunol. 2004;173:50–60. doi: 10.4049/jimmunol.173.1.50. [DOI] [PubMed] [Google Scholar]
- Revy P, Sospedra M, Barbour B, Trautmann A. Functional antigen-independent synapses formed between T cells and dendritic cells. Nat Immunol. 2001;2:925–931. doi: 10.1038/ni713. [DOI] [PubMed] [Google Scholar]
- Satomi M, Shimizu M, Shinya E, Watari E, Owaki A, Hidaka C, Ichikawa M, Takeshita T, Takahashi H. Transmission of Macrophage-Tropic HIV-1 by Breast-Milk Macrophages via DC-SIGN. Journal of Infectious Disease. 2005;191:174–181. doi: 10.1086/426829. [DOI] [PubMed] [Google Scholar]
- Scales D, Ni H, Shaheen F, Capodici J, Cannon G, Weissman D. Nonproliferating Bystander CD4+ T Cells Lacking Activation Markers Support HIV Replication During Immune Activation. J Immunol. 2001;166:6437–6443. doi: 10.4049/jimmunol.166.10.6437. [DOI] [PubMed] [Google Scholar]
- Sol-Foulon N, Moris A, Nobile C, Boccaccio C, Engering A, Abastado JP, Heard JM, van Kooyk Y, Schwartz O. HIV-1 Nef-Induced Upregulation of DC-SIGN in Dendritic Cells Promotes Lymphocyte Clustering and Viral Spread. Immunity. 2002;16:145–155. doi: 10.1016/s1074-7613(02)00260-1. [DOI] [PubMed] [Google Scholar]
- Spira AI, Marx PA, Patterson BK, Mahoney J, Koup RA, Wolinsky SM, Ho DD. Cellular targets of infection and route of viral dissemination after an intravaginal inoculation of simian immunodeficiency virus into rhesus macaques. J Exp Med. 1996;183:215–225. doi: 10.1084/jem.183.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA. HIV-1 replication is controlled at the level of T cell activation and proviral integration. The EMBO Journal. 1990;9:1551–1560. doi: 10.1002/j.1460-2075.1990.tb08274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kooyk Y, Geijtenbeek TBH. A novel adhesion pathway that regulates dendritic cell trafficking and T cell interactions. Immunological Reviews. 2002;186:47–56. doi: 10.1034/j.1600-065x.2002.18605.x. [DOI] [PubMed] [Google Scholar]
- Vazeux R, Hoffman PA, Tomfta JK, Dkkinson ES, Jasman RL, St John T, Gallatin WM. Cloning and characterization of a new intercellular adhesion molecule ICAM-R. Nature. 1992;360:485–488. doi: 10.1038/360485a0. [DOI] [PubMed] [Google Scholar]
- Weissman D, Li Y, Orenstein JM, Fauci AS. Both a precursor and a mature population of dendritic cells can bind HIV. However, only the mature population that expresses CD80 can pass infection to unstimulated CD4+ T cells. J Immunol. 1995;155:4111–4117. [PubMed] [Google Scholar]
- Wu L, Martin TD, Vazeux R, Unutmaz D, KewalRamani VN. Functional Evaluation of DC-SIGN Monoclonal Antibodies Reveals DC-SIGN Interactions with ICAM-3 Do Not Promote Human Immunodeficiency Virus Type 1 Transmission. J Virol. 2002a;76:5905–5914. doi: 10.1128/JVI.76.12.5905-5914.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Bashirova AA, Martin D, Villamide L, Mehlhop E, Chertov AO, Unutmaz D, Pope M, Carrington M, KewalRamani VN. Rhesus macaque dendritic cells efficiently transmit primate lentiviruses independently of DC-SIGN. PNAS. 2002b;99:1568–1573. doi: 10.1073/pnas.032654399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, KewalRamani VN. Dendritic-cell Interactions with HIV: Infection and Viral Dissemination. Nat Rev Immunol. 2006;6:859–868. doi: 10.1038/nri1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Kimata MT, Cella M, Biggins JE, Rorex C, White R, Hicks S, Wilson JM, Patel PG, Allan JS, Colonna M, Kimata JT. Capture and Transfer of Simian Immunodeficiency Virus by Macaque Dendritic Cells Is Enhanced by DC-SIGN. J Virol. 2002;76:11827–11836. doi: 10.1128/JVI.76.23.11827-11836.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen ISY. HIV-1 entry into quiescent primary lymphocytes: Molecular analysis reveals a labile, latent viral structure. Cell. 1990;61:213–222. doi: 10.1016/0092-8674(90)90802-l. [DOI] [PubMed] [Google Scholar]
- Zhang ZQ, Schuler T, Zupancic M, Wietgrefe S, Staskus KA, Reimann KA, Reinhart TA, Rogan M, Cavert W, Miller CJ, Veazey RS, Notermans D, Little S, Danner SA, Richman DD, Havlir D, Wong J, Jordan HL, Schacker TW, Racz P, Tenner-Racz K, Letvin NL, Wolinsky S, Haase AT. Sexual Transmission and Propagation of SIV and HIV in Resting and Activated CD4+ T Cells. Science. 1999;286:1353–1357. doi: 10.1126/science.286.5443.1353. [DOI] [PubMed] [Google Scholar]
- Zhang ZQ, Wietgrefe SW, Li Q, Shore MD, Duan L, Reilly C, Lifson JD, Haase AT. Roles of substrate availability and infection of resting and activated CD4+ T cells in transmission and acute simian immunodeficiency virus infection. PNAS. 2004;101:5640–5645. doi: 10.1073/pnas.0308425101. [DOI] [PMC free article] [PubMed] [Google Scholar]