

SUMMARY
Pairing between U2 snRNA and the branch site of spliceosomal introns is essential for spliceosome assembly and is thought to be required for the first catalytic step of splicing. We have identified an RNA comprising the 5’ end of U2 snRNA and the 3’ exon of the ACT1-CUP1 reporter gene, resulting from a trans-splicing reaction in which a 5’ splice site-like sequence in the universally conserved branch site-binding region of U2 is used in trans as a 5’ splice site for both steps of splicing in vivo. Formation of this product occurs in functional spliceosomes assembled on reporter genes whose 5’ splice sites are predicted to bind poorly at the spliceosome catalytic centre. Multiple spatially disparate splice sites in U2 can be used, calling into question both the fate of its pairing to the branch site and the details of its role in splicing catalysis.
Keywords: U2 snRNA, branch site, trans-splicing, bulged duplex model, splicing catalysis
INTRODUCTION
The removal of introns from pre-mRNA is catalysed by the spliceosome -a large, conformationally and compositionally dynamic ribonucleoprotein complex comprising five small nuclear RNAs (snRNAs) and more than a hundred proteins (Nilsen, 1998). The catalytic phase of splicing consists of two consecutive transesterification reactions: in the first step the 2’ hydroxyl of the branch site adenosine (BS-A) attacks the phosphodiester bond at the 5’ splice site (5’SS), yielding a lariat intermediate and a free 5’ exon, which attacks the 3’SS in the second step to produce an excised lariat intron and spliced mRNA. Catalysis is preceded by an extended assembly phase: the 5’SS and BS are bound by U1 and U2 snRNPs, respectively, and the 3’SS by protein factors; the [U4/U6•U5] tri-snRNP joins the complex and a series of ATP-dependent conformational rearrangements results in the release of U1 then U4 (Burge et al., 1999). The recruitment of the CDC5L complex (NTC in S. cerevisiae) completes the formation of the catalytically competent spliceosome (Makarov et al., 2002).
The pairing established between U2 snRNA and the branch site (BS) during the early stages of spliceosome assembly (Parker et al., 1987; Wu and Manley, 1989; Zhuang and Weiner, 1989) is thought to persist unaltered until after the first catalytic step of splicing, and possibly beyond. Experiments involving arabinosyl adenosine substitution at BS support the hypothesis that BS-A is activated for first step catalysis by being bulged from the BS-U2 duplex (Query et al., 1994), and that this duplex therefore represents the catalytically relevant state of BS (Fig. 1A). Further support for this model comes from NMR studies on model duplexes, which indicate that the conserved pseudouridine modification at U2 position 35 (S. cerevisiae numbering) selectively stabilises the form of the duplex in which BS-A is bulged from, rather than intercalated into, the helix (Newby and Greenbaum, 2002).
Figure 1. Trans-splicing in S. cerevisiae can generate an RNA species comprising the 5’ end of U2 snRNA and the 3’ exon of the ACT1-CUP1 reporter.
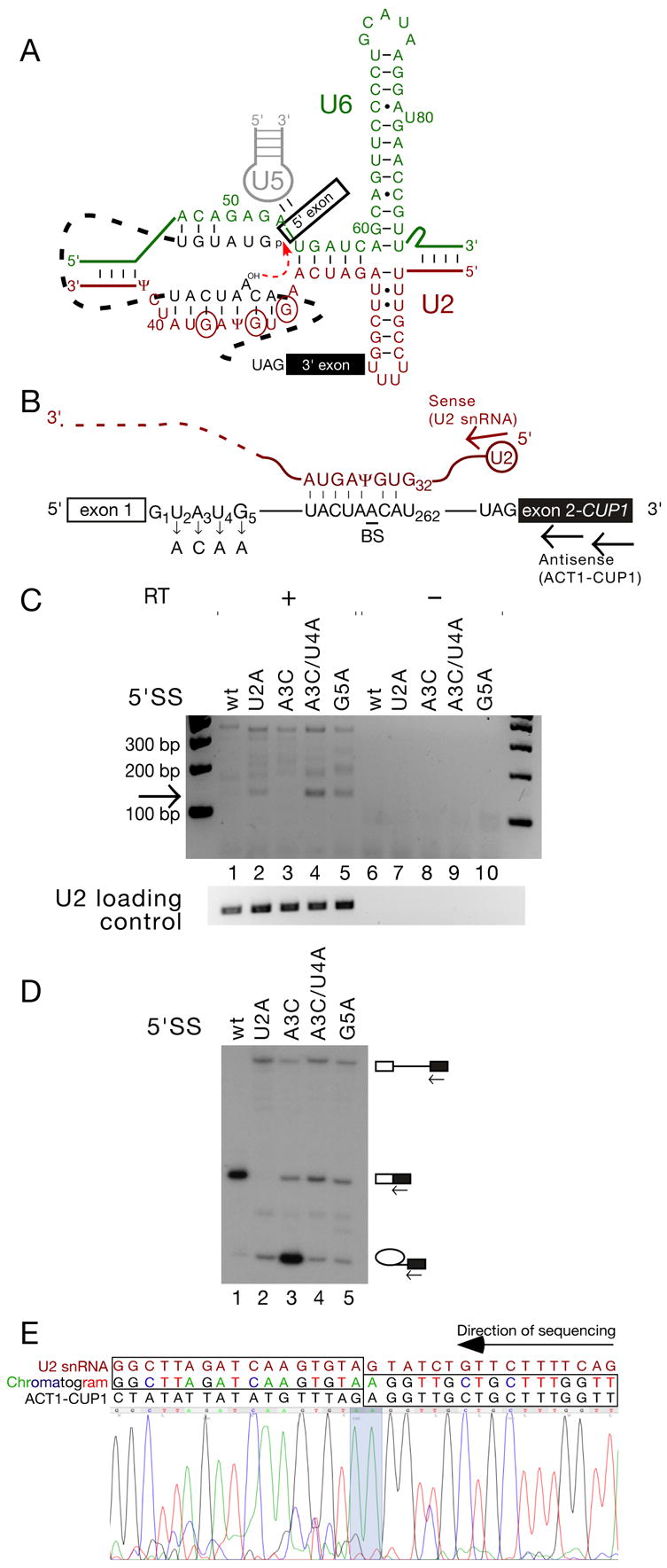
(A). Schematic of RNA-RNA interactions that contribute to the first step of splicing (modified from Konarska et al., 2006) with the initial G of each of the three 5’ splice site-like sequences in the BS-binding region of U2 snRNA indicated by circles.
(B). Schematic of ACT1-CUP1 reporter pre-mRNA and U2 snRNA, indicating the mutations used in panels C and D, and the location of the RT and PCR primers (arrows) used in panel C.
(C). RT-PCR using the primers indicated in Fig. 1B can amplify a product, of the size expected for a trans-splicing product generated using the BS-binding region of U2 as a 5’SS, from total RNA from S. cerevisiae Y04999 cells carrying reporters with 5’SS mutations as indicated.
(D). Primer extension analysis of RNA recovered from cells containing the ACT1-CUP1 reporters as indicated. Primer complimentary to the 3' exon was used to reveal levels of pre-mRNA, mRNA, and lariat intermediate. Strain Y04999 (Δdbr1) was used in order to accurately monitor the efficiency of the first step.
(E). The 130 bp RT-PCR product corresponds to trans-spliced U2-ACT1-CUP1. Reverse-complemented sequencing trace from the purified 130 bp product. U2 snRNA and ACT1-CUP1 sequence, as well as the chromatogram read, are indicated above the trace; concordance between chromatogram and gene sequence is indicated by boxes, and the splice junction is highlighted on the chromatogram trace.
U2 and U6 snRNAs appear to have intrinsic catalytic potential in combination with splicing substrates. In vitro-transcribed snRNAs, when incubated with a transcript containing a branch site sequence, induce the formation of a covalent bond proposed to be a phosphotriester between the canonical BS-A and the guanosine in the universally conserved U6 AGC triad (Valadkhan and Manley, 2003). This reaction is stimulated by pseudouridylation of U2 position 35, suggesting that bulging BS-A from a duplex may be important for the observed reactivity. This reaction may have further biological relevance, as the site of reactivity in U6 is adjacent to the location of an intron in the S. pombe U6 snRNA gene (Tani and Ohshima, 1991), thought to have arisen due to an aberrant splicing event followed by retrotransposition of the resulting product into the genome.
U6 reactivity in a spliceosomal context has been observed directly (Yu et al., 1993). In an in vitro system designed to reconstitute nematode cis- and trans-splicing using Ascaris lumbricoides extracts, Nilsen and co-workers detected branched and linear RNA products indicating the use of U6 as both a branch acceptor and a 5’ exon in a 5’SS-independent splicing reaction. The reaction appears to represent an aberrant first step followed by a quasi-normal second step, as the canonical BS and 3’SS nucleotides are used, and U6 mutants that abolish the ability to reconstitute normal cis- and trans-splicing also render U6 trans-splicing undetectable.
Although U6 is the only snRNA that has been demonstrated to be reactive, the presence of multiple 5’SS-like sequences in the BS-binding region of U2 (Fig. 1A), and the presence of a spliceosomal intron close to this region in Rhodotorula hasegawae (Takahashi et al., 1993) prompted us to search for the products of a trans-splicing reaction involving U2 in S. cerevisiae. Here we describe the formation of an RNA product comprising the highly conserved 5’ end of U2 and the 3’ exon of the ACT1-CUP1 reporter gene in intact S. cerevisiae cells. The formation of this product requires full spliceosome assembly, and is only detectable when the reporter has a mutant 5’SS predicted to bind poorly at the spliceosome catalytic centre. The predominant 5’SS used in U2 is in the middle of the predicted BS-U2 duplex, but multiple sites can be used; these data suggest that BS-U2 pairing is unlikely to be required for the second catalytic step of splicing, and that it may be at least partially disrupted prior to the first step.
RESULTS
The BS-binding region of U2 snRNA can be used in trans as a 5’ exon for both steps of splicing
Attack of U2 snRNA by the branch site nucleophile would be expected to generate a truncated U2 molecule covalently linked via its new 5’ end to the 2’ hydroxyl of BS, detectable as a strong stop by primer extension. An initial screen for such branched derivatives of U2 snRNA in S. cerevisiae total RNA was inconclusive due to unavoidable low levels of degradation (data not shown). We therefore reasoned that any ‘free 5’ exon’ generated by such aberrant attack may be able to undergo the second step of splicing in trans to the 3’SS of a reporter gene, generating a novel linear RNA detectable by RT-PCR. We performed nested RT-PCR on DNaseI-treated total RNA from strains carrying ACT1-CUP1 reporter genes (Lesser and Guthrie, 1993) with various mutations, using antisense primers complementary to the CUP1 3’ exon and a sense primer corresponding to the 5’ end of U2 snRNA (Fig. 1B). For multiple intron 5’SS mutants (G1C, G1U, U2A, A3G, [A3C+U4A], G5A, G5C), we were able to detect a ~130 bp PCR product whose size corresponded to that expected for the ACT1-CUP1 3’ exon appended to approximately 40 nucleotides of U2 (Fig. 1C, lanes 2, 4, 5 and data not shown). The 130 bp product was not generated from equivalent amounts of total RNA from strains carrying wild-type or 5’SS A3C ACT1-CUP1 reporter, both of which show an efficient first step of canonical cis-splicing (Fig. 1C&D, lanes 1 and 3).
Direct sequencing of the putative trans-splicing product indicated that it comprised the 5’-terminal 36 nt of U2 joined to the 3’ exon of ACT1-CUP1 (Fig. 1E). The homogeneity of the sequencing trace indicates a strong splice site preference, with the wild-type ACT1-CUP1 3’SS and the A/GUAUCΨ site in U2 (the best match in this region of U2 to G/GUAUGU, the S. cerevisiae consensus) apparently used substantially more efficiently than other sites.
The trans-splicing reaction appears to be very inefficient, with steady state levels of the U2-ACT1-CUP1 product apparently lower than one molecule per cell (data not shown). We estimate that, in the case of the G5A mutant, trans-splicing represents around one per hundred thousand two-step splicing events, comparable to the error rates of other macromolecular processes such as transcription.
The generation of the U2-ACT1-CUP1 RNA by an aberrant but otherwise mechanistically normal splicing reaction should also produce a second species –a branched RNA comprising the 5’ exon and intron of ACT1-CUP1 joined via the 2’ hydroxyl of BS-A to the 3’ 1139 nt of U2, and an intermediate consisting of the ‘5’ exon’ portion of U2 with a 3’ hydroxyl; although we attempted to detect these products using RT-PCR- and Klenow-based methods, respectively, we were unable to do so. This may in part be due to the rapid degradation of internally cleaved U2 snRNAs (McPheeters et al., 1989).
Trans-splicing to U2 snRNA requires poor binding of the canonical 5’SS at the spliceosomal catalytic centre
To investigate whether trans-splicing to U2 snRNA is simply a result of inhibition of the first step of the canonical splicing reaction, we analysed the effects of non-5’SS reporter gene mutations on the efficiency of trans-splicing. Mutations at and around BS (C256G, U257A, U257C, BS-C, BS-G – Fig. 2A), which virtually abolish productive splicing of the reporter transcript (Fig. 2C) did not promote trans-splicing (Fig. 2B, lanes 1–7). When combined with a 5’SS G5A mutation, which alone showed easily detectable levels of trans-splicing, diverse mutations at and around BS inhibited the trans-splicing reaction to the extent that the product was no longer detectable (Fig. 2B, lanes 15–19). Even mutations that show only mild inhibition of the first step of canonical cis-splicing of G5A mutants, such as an extension of the pairing interaction between BS and U2 snRNA, abolished trans-splicing (Fig. 2B&C, lanes 15–16). 3’SS mutants exhibited behaviour identical to that of BS mutants, failing to promote trans-splicing in an otherwise wild-type reporter, and reducing it to undetectable levels in the context of a G1C mutation (data not shown). BS and 3’SS mutations exacerbate the inhibition of canonical mRNA splicing caused by the G5A mutation (Fig. 2C, lanes 15–19 and data not shown), so their failure to further stimulate the aberrant trans-splicing reaction indicates that the requirements for this reaction extend beyond simple first step inhibition.
Figure 2. Trans-splicing does not occur in reporter genes with mutations outside the 5’SS.
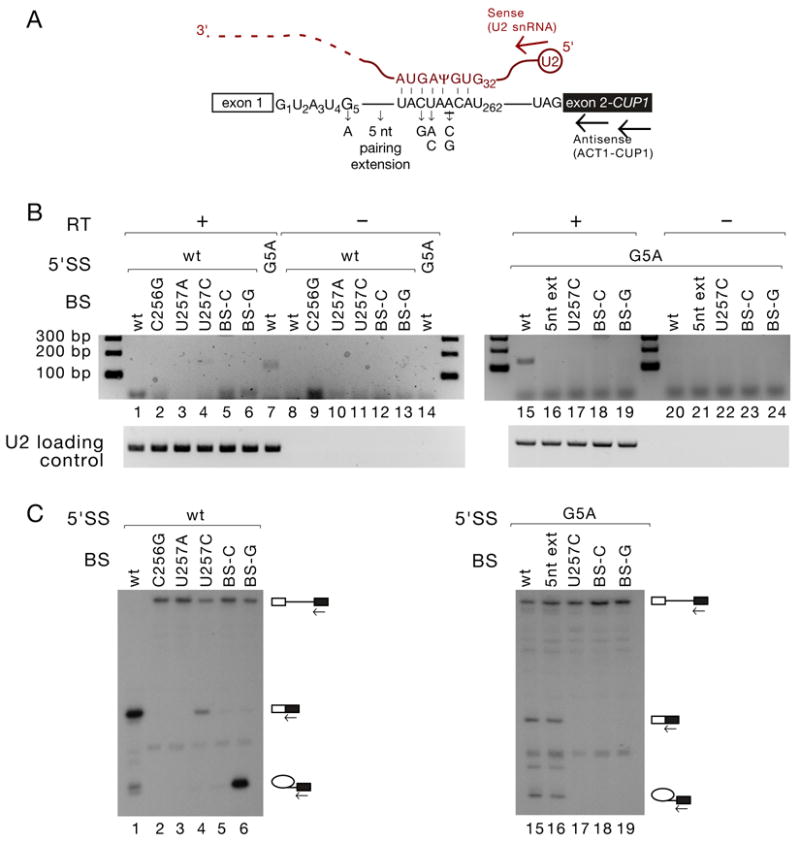
(A). Schematic of ACT1-CUP1 reporter pre-mRNA and U2 snRNA, indicating the mutations used in panels C and D, and the schematic location of RT and PCR primers (arrows) used in panel B.
(B). Mutations at and around the branch site do not stimulate trans-splicing in an otherwise wild-type context, and inhibit it in the context of an accompanying 5’SS mutation. RT-PCR analysis of RNA recovered from Y04999 cells containing the ACT1-CUP1 reporters as indicated, as in Fig. 1C.
(C). Primer extension analysis of RNA recovered from Y04999 cells containing the ACT1-CUP1 reporters as indicated, as in Fig. 1D.
Even among 5’SS mutants, the degree of first step inhibition is not a good predictor of trans-splicing efficiency. For example, splicing is much more strongly inhibited by the 5’SS U2A mutation than by [A3C+U4A] (Fig. 1D, lanes 2 and 4), yet the extent of trans-splicing to U2 snRNA is greater in the [A3C+U4A] mutant (Fig. 1C, lanes 2 and 4). [A3C+U4A] has been shown to affect splicing predominantly by altering 5’SS binding at the catalytic centre of the spliceosome (Konarska et al., 2006). Formation of the observed U2-ACT1-CUP1 RNA therefore appears to result from the aberrant attack by BS on U2 in the presence of a 5’SS that is poorly bound within the spliceosome catalytic centre, rather than simply from an inefficient first step of splicing.
The functional U2 snRNA component of the spliceosome is the substrate for trans-splicing
To verify that the U2 snRNA portion of the trans-spliced product was derived from the U2 component of a functional spliceosome (rather than, for example, a second molecule of U2 displacing the reporter gene 5’SS) we took advantage of previously characterised compensatory mutations in U2 and U6 snRNAs. Mutation of U6 positions 56 and 57 from AU to UA is lethal except in the context of the compensatory, and likewise lethal in isolation, AU to UA mutations at positions 27 and 28 in U2, predicted to restore pairing in helix Ib (Madhani and Guthrie, 1992, and Fig. 3A). S. cerevisiae U2/U6 double knockout strains were constructed in which plasmid-borne copies of both wild-type and mutant U2, and either wild-type or mutant U6 were present. In these strains, the identity of the U2 snRNA component of functional spliceosomes is expected to depend on the identity of the U6 snRNA gene carried, i.e. spliceosomes in the strain with wild-type U6 should contain wild-type U2, and those in the strain with mutant U6 should contain mutant U2 (Fig. 3B&C). In both cases, the pool of ‘free U2’, as well as early spliceosome assembly intermediates, should contain both wild-type and mutant U2. The ‘wild-type spliceosome’ and ‘mutant spliceosome’ strains were transfected with G5A ACT1-CUP1 reporter, and the U2-ACT1-CUP1 trans-spliced product from each strain amplified by RT-PCR. Sequencing indicated that the U2 snRNA portion of the trans-spliced product differed as predicted. The strain carrying a wild-type U6 snRNA gene generated a trans-spliced product in which U2 sequence was wild-type (Fig. 3D), and that carrying the mutant U6 snRNA gene generated product with mutant U2 sequence (Fig. 3E), indicating that the trans-splicing reaction involves nucleophilic attack of the U2 snRNA component of a fully assembled, catalytically competent spliceosome, and therefore of the BS-binding region of U2 to which BS was bound.
Figure 3. U2 snRNA in functional spliceosomes is the substrate for trans-splicing.
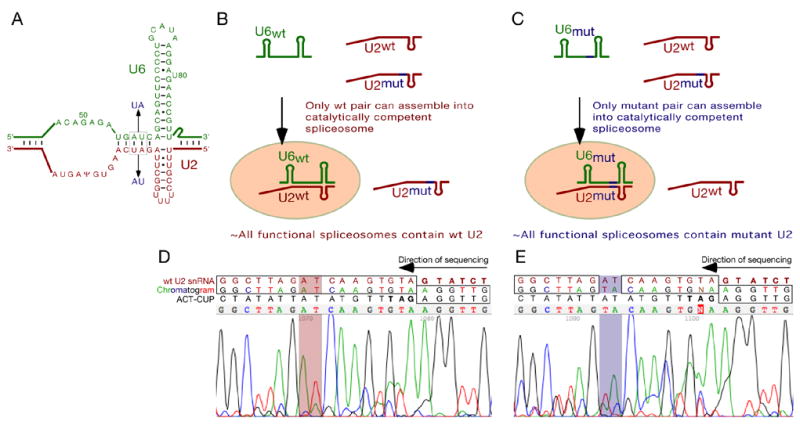
(A). Compensatory U2 and U6 snRNA mutations used in panels C and D.
(B, C). Predicted behaviour of wild-type and mutant U2 snRNAs in strains carrying a wild-type (B) or mutant (C) U6 snRNA gene.
(D, E). The trans-spliced product is generated by attack of U2 snRNA in functional spliceosomes. Reverse-complemented sequencing traces from purified trans-splicing product from yCQ62 cells carrying G5A ACT1-CUP1 reporter, both wild-type and mutant U2 snRNA genes, and a wild-type (D) or mutant (E) U6 snRNA gene. Concordance between chromatogram and wild-type gene sequence is indicated by boxes, and the mutated/wild-type helix Ib U2 snRNA nucleotides highlighted.
Splicing can occur to all three predicted 5’SS in U2 snRNA
Although direct sequencing of the purified trans-splicing product had indicated the preferred use of CAAGUGΨA/GUAUCΨ as a 5’SS, and of the wild -type ACT1-CUP1 3’SS, we reasoned that the use of less-favoured sites may be apparent if individual DNAs were analysed. We therefore cloned the product into the pDrive sequencing vector (Qiagen), and sequenced multiple clones. As expected, the majority of these clones (9/13) had used the preferred 5’SS in U2; however, one had used the GΨ/AG site one nt upstream, two the GU/GU site a further two nt upstream of this, and one the AA/GU a further two nt upstream (splice site usage in U2 snRNA is shown in Fig. 4E). All clones had used the wild-type ACT1-CUP1 3’SS. Due to our inability to detect reaction intermediates, we do not know whether the levels of trans-spliced products reflecting the use of distinct 5’ splice sites within U2 snRNA indicate differences in the efficiencies of the first step, the second step, or both, for each potential 5’SS. The 5’ splice sites observed to be used span half a helical turn of RNA: the use of such spatially disparate 5’ splice sites is inconsistent with nucleophilic attack by the canonical BS-A within an intact, unaltered and undisrupted BS-U2 duplex. Given that, even in the context of BS mutations that dramatically reduce splicing efficiency (BS-C, BS-U), S. cerevisiae introns have invariably been observed to branch from the pyrimidine nucleotide rather than the adjacent adenosine (Vijayraghavan et al., 1986 and DS, CQ and MK, unpublished data), at least some transient unwinding of the BS-U2 duplex seems more likely than aberrant attack by a non-canonical BS nucleotide.
Figure 4. U2 snRNA sequence is a suboptimal 5’SS in the context of a reporter gene.
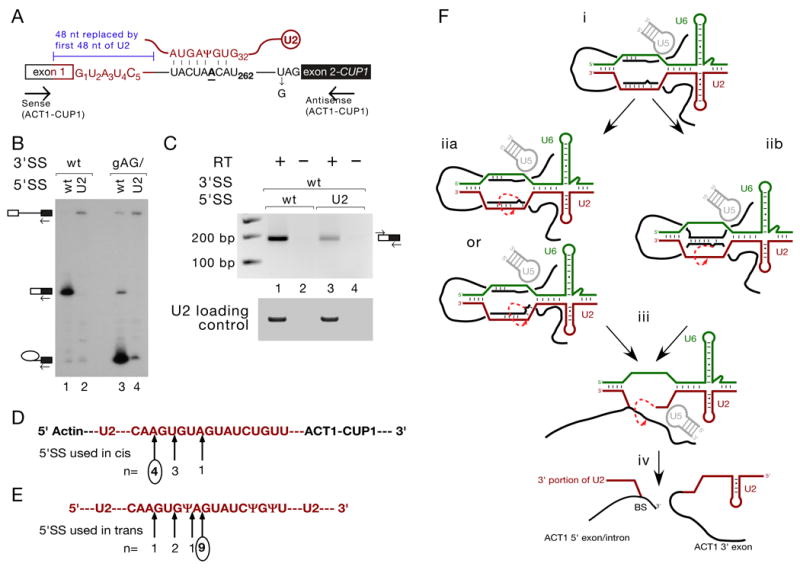
(A). Schematic of ACT1-CUP1 reporter pre-mRNA and U2 snRNA, indicating the U2 sequence substitution into ACT1-CUP1 and 3’SS gAG/ mutation used in panels B and C, and the RT-PCR primers used in panel C.
(B). Primer extension analysis of RNA recovered from Y04999 cells containing the ACT1-CUP1 reporters as indicated, as in Fig. 1D.
(C). Spliced ACT1-U2-ACT1-CUP1 mRNA can be detected by RT-PCR (lane 3). Wild-type mRNA is shown for comparison (lane 1).
(D, E). Schematic indicating 5’SS usage in U2 sequence in cis in the context of ACT1-U2-ACT1-CUP1 (D) and in trans in the context of U2 snRNA (E). n refers to the number of plasmids sequenced containing trans-spliced product that had used the 5’SS indicated by the arrow above, and circles indicate the predominant 5’SS detected in directly sequenced PCR products.
(F). Proposed mechanisms of trans-splicing: (i) RNA-RNA interactions in the fully assembled spliceosome, with BS paired to U2. (iia) BS and U2 unpair and re-pair potentially inaccurately, with BS nucleophilically attacking U2 from within the resulting helix, or (iib) BS and U2 unpair, with new pairing established between BS and the 5’SS; BS, bulged from this new helix, attacks U2. (iii) 5’SS-U6 and BS-U2 pairing both having been disrupted, the ‘U2 5’ exon’ generated in step ii attacks the reporter gene 3’SS, generating (iv) a branched product comprising the 3’ end of U2 appended to the 5’ exon and intron of the reporter gene, and a linear product comprising the 5’ end of U2 appended to the reporter 3’ exon.
The BS-binding region of U2, in the context of the ACT1-CUP1 reporter gene, is inefficiently used in cis as a 5’SS and shows a 5’SS preference distinct from that observed in trans-splicing
Given the tandem arrangement of three 5’SS-like sequences in the BS-binding region of U2 snRNA, and their use as 5’ splice sites in the context of reporter gene mutations such as [A3C+U4A] and G5A that do not dramatically reduce the overall efficiency of cis-splicing, we investigated the efficiency with which U2 sequence could be used as a 5’SS in the context of the ACT1-CUP1 reporter. 48 nt of reporter sequence around the 5’SS was replaced with sequence corresponding to the first 48 nt of S. cerevisiae U2 snRNA, such that the /GUAUCU 5’SS from U2 replaced the /GUAUGU 5’SS in the reporter (Fig. 4A). Primer extension analysis of total RNA from strains expressing these ACT1-U2-ACT1-CUP1 constructs revealed that this U2-substituted 5’SS was used inefficiently (Fig. 4B). A small amount of spliced product could be detected by RT-PCR (Fig. 4C); this RT-PCR product was cloned into pDrive (Qiagen). Sequencing of eight clones revealed that 5’SS usage in U2 sequence in the context of the reporter gene (Fig. 4D) was different from that in the context of U2 (Fig. 4E). 5’SS usage in the U2-substituted reporter gene correlated relatively well with predicted U1 snRNA binding affinity (Seraphin and Rosbash, 1989), with the upstream-most 5’SS (AA/GUGUA) used in 4/8 clones and the downstream-most 5’SS (UA/GUAUCU) in only 1/8 (cf. 9/13 in the context of trans-splicing to U2). This suggests that, while this region of U2 snRNA sequence can be recognised by U1 snRNA and participate to a low level in a canonical mRNA splicing reaction when placed in the context of an otherwise wild-type reporter gene, it is a suboptimal substrate for splicing. This rationalises the observation that U2 cannot compete effectively with a wild-type 5’SS. In addition, the different 5’SS preference observed between the cis- and trans-splicing reactions indicates that pairing between U1 and U2 snRNAs does not determine 5’SS usage in U2 snRNA in trans-splicing; the most likely candidate for such a determinant of 5’SS preference in trans-splicing is spatial proximity to the BS nucleophile. The use of multiple 5’ splice sites in the trans-splicing reaction is therefore indicative of a high degree of flexibility in the spliceosomal catalytic centre.
DISCUSSION
The nature of the trans-splicing reaction
The data presented here demonstrate the formation of a trans-spliced RNA product comprising the 5’ end of U2 and the 3’ exon of an ACT1-CUP1 reporter with a 5’SS predicted to bind poorly at the spliceosome catalytic centre. The possibility of this product being generated by an RT artefact is ruled out by its strict dependence on 5’SS mutation. In strains expressing two distinct U2 snRNAs, the identity of the U2 portion of the trans-spliced product was observed to be dependent on the identity of the U6 expressed by the strain, with the potential for helix I formation between U2 and U6 an essential requirement for trans-splicing. Interactions between U2 and U6 are established during the late stages of spliceosome assembly, so the requirement for such interactions indicates that U2 snRNA within a functional spliceosome is nucleophilically attacked, presumably by the branch site it recruited.
We were able to detect trans-splicing using reporter genes with wild-type 3’ splice sites, but not with 3’SS mutants. In addition, all trans-splicing events observed involved the same 3’SS as is used in canonical cis-splicing of the reporter gene. These observations support the hypothesis that the trans-splicing reaction is essentially identical to cis-splicing, but with an aberrant first catalytic step in which U2, rather than the 5’SS, is attacked; the exon generated by this aberrant first step proceeds through the second step in a quasi-normal fashion. As we were unable to detect reaction intermediates, the efficiency of this second step could not be investigated. Our inability to detect a branched intermediate also renders uncertain the identity of the first-step nucleophile, although the inhibition of trans-splicing by mutations at and around BS supports a 2’-5’ branching reaction. A hydrolytic first step analogous to that observed in group II introns (Podar et al., 1998) cannot be ruled out by our data, but we feel that the existence of such a first step is unlikely and would not substantially affect our interpretation.
Flexibility in the spliceosome catalytic centre, and the fate of the BS-U2 duplex for the first catalytic step of pre-mRNA splicing
Splicing is a multi-step reaction that necessarily involves conformational rearrangements as well as complex assembly and disassembly phases; global conformational flexibility is clearly required for such a reaction. In addition to this necessary flexibility, however, splicing is characterised by the need for nucleotide precision; this is especially true in S. cerevisiae, an organism with strong splice site consensuses in which functionally important alternative splicing has not been demonstrated. Therefore, while both global flexibility throughout the assembly-reaction-disassembly pathway, and local flexibility at the catalytic centre between the two chemical steps are inevitable, tight substrate binding between rearrangements is expected. As noted by Nilsen and co-workers (Yu et al., 1993) snRNA reactivity in spliceosomes is indicative of catalytic centre flexibility. The reactivity of an snRNA region intimately involved in substrate binding during assembly, and possibly also catalysis, extends our view of this flexibility to include pairing interactions important for splicing fidelity. The nucleophilic attack of four sites in U2 is inconsistent with reactivity within a BS-U2 duplex that has remained intact from early spliceosome assembly until the catalytic phase, even in the unlikely scenario of a hydrolytic first step. To rationalise the reactivity of multiple sites in U2, it is therefore necessary to invoke at least transient disruption of the interaction between BS and U2. Following this unpairing, three possibilities exist between which we cannot currently distinguish: the branch site, the presumed nucleophile for the first step of the trans-splicing reaction, could attack U2 while in a single-stranded state; this seems unlikely, as such circumvention of the need for nucleophile activation would be highly surprising in a reaction as important and precise as splicing. A second possibility is that BS may simply re-pair somewhat inaccurately with U2, and attack from within the resulting re-formed duplex (Fig. 4F, iia). The third possibility is that BS may pair to another RNA strand and attack U2 from within this duplex (Fig. 4F, iib). Given the similarity between the sequences of the yeast consensus 5’SS and the BS-binding region of U2, the 5’SS represents an obvious candidate for such a partner. While a complete strand exchange reaction, in which U2 and the 5’SS are able to swap positions and roles after assembly, does not appear consistent with the wide range of 5’SS mutations that facilitate trans-splicing, partial strand exchange is not ruled out. We are currently investigating the possibility of partial displacement of U2 from the BS-U2 duplex by the 5’SS prior to the first catalytic step of canonical pre-mRNA splicing.
The fate of the BS-U2 duplex for the second catalytic step of pre-mRNA splicing
The persistence after the first step of splicing of the entire U2-BS helix, established during the early stages of spliceosome assembly, is not inconsistent with any previously published data. Our data, however, clearly suggest that the persistence of this helix is at least not required for the second step of splicing. As indicated in figures 1A and 4E, the predominant 5’SS used in U2 lies in the middle of this helix, which must therefore be at least partially disrupted in order for the ‘U2 5’ exon’ to be free to attack the 3’SS during the second step. Given that the structural consequences of the absence of a helix normally present in the second step conformation of the spliceosome would likely cause a severe, if not terminal, second step block, and that the second step of the trans-splicing reaction appears to be analogous to the second step of canonical cis-splicing, we hypothesise that the U2-BS helix does not normally persist during the second step, and that the removal of the lariat intermediate, generated in the first step, from the catalytic centre prior to the second step may be linked to the unpairing of its BS portion from U2 snRNA as well as the disruption of the 5’SS-U6 snRNA interaction (Konarska et al., 2006). Although these conclusions are based on an infrequent trans-splicing reaction, the capacity of the spliceosome to support such a reaction has important implications for physiologically relevant pre-mRNA splicing.
MATERIALS AND METHODS
Strains and reporter plasmids
S. cerevisiae strains used in this study were Y04999 [MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0; YKL149c::kanMX4] (derived from BY4741), 46ΔCUP [MATa, ade2 cup1Δ::ura3 his3 leu2 lys2 trp1 ura3, GAL+] (Lesser and Guthrie, 1993) and yCQ62 [MATa ade2 cup1Δ::ura3 his3 leu2 lys2 trp1 ura3 snr6Δ::loxP snr20Δ::loxP, pU6+U2 (SNR6 SNR20 URA3 CEN ARS)]. ACT1-CUP1 reporter plasmids (Lesser and Guthrie, 1993) were as described (Query and Konarska, 2004) or prepared by overlapping PCR and in vivo gap repair cloning.
RT-PCR
Yeast total RNA was treated with DNaseI (Sigma) according to the manufacturer’s instructions. Reverse transcription was carried out using primer YAC94 5’-CGTCGCTGTTACACCC-3’. Reverse-transcribed DNA was amplified by PCR using primers YAC94 and yU2-22 5’-ACGAATCTCTTTGCCTTTTGGC , diluted 1:1000 and amplified using primers yU2-22 and YAC6 5'-GGCACTCATGACCTTC-3'. Products were separated in 2% agarose gels. RT-PCRs for loading controls were performed with yU2-138-154 5’-AAAGTCTCTTCCCGTCC-3’ and yU2-22 as antisense and sense primers, respectively. PCR products were purified for sequencing and cloning using Qiaquick gel extraction kits (Qiagen).
Primer extension
Primer extensions were carried out as described (Siatecka et al., 1999), using primer YAC6, complementary to exon 2 of ACT1-CUP1. Extension products were separated in 7% polyacrylamide/8 M urea gels and visualized by autoradiography.
Acknowledgments
We thank Joe Osmundson for critical reading of the manuscript. This work was supported by NIH grant GM49044 to M.M.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Burge CB, Tuschl TH, Sharp PA. Splicing of precursors to mRNAs by the spliceosomes. In: Gesteland RF, Cech TR, Atkins JF, editors. The RNA World. 2. New York: Cold Spring Harbor Laboratory Press; 1999. pp. 525–560. [Google Scholar]
- Konarska MM, Vilardell J, Query CC. Repositioning of the reaction intermediate within the catalytic center of the spliceosome. Mol Cell. 2006;21:543–553. doi: 10.1016/j.molcel.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Lesser CF, Guthrie C. Mutational analysis of pre-mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics. 1993;133:851–863. doi: 10.1093/genetics/133.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani HD, Guthrie C. A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell. 1992;71:803–817. doi: 10.1016/0092-8674(92)90556-r. [DOI] [PubMed] [Google Scholar]
- Makarov EM, Makarova OV, Urlaub H, Gentzel M, Will CL, Wilm M, Lührmann R. Small nuclear ribonucleoprotein remodeling during catalytic activation of the spliceosome. Science. 2002;298:2205–2208. doi: 10.1126/science.1077783. [DOI] [PubMed] [Google Scholar]
- McPheeters DS, Fabrizio P, Abelson J. In vitro reconstitution of functional yeast U2 snRNPs. Genes Dev. 1989;3:2124–2136. doi: 10.1101/gad.3.12b.2124. [DOI] [PubMed] [Google Scholar]
- Newby MI, Greenbaum NL. Sculpting of the spliceosomal branch site recognition motif by a conserved pseudouridine. Nat Struct Biol. 2002;9:958–965. doi: 10.1038/nsb873. [DOI] [PubMed] [Google Scholar]
- Nilsen TW. RNA-RNA interactions in nuclear pre-mRNA splicing. In: Simons RW, Grunberg-Manago M, editors. RNA Structure and Function. New York: Cold Spring Harbor Laboratory Press; 1998. pp. 279–307. [Google Scholar]
- Parker R, Siliciano PG, Guthrie C. Recognition of the TACTAAC box during mRNA splicing in yeast involves base pairing to the U2-like snRNA. Cell. 1987;49:229–239. doi: 10.1016/0092-8674(87)90564-2. [DOI] [PubMed] [Google Scholar]
- Podar M, Chu VT, Pyle AM, Perlman PS. Group II intron splicing in vivo by first-step hydrolysis. Nature. 1998;391:915–918. doi: 10.1038/36142. [DOI] [PubMed] [Google Scholar]
- Query CC, Konarska MM. Suppression of multiple substrate mutations by spliceosomal prp8 alleles suggests functional correlations with ribosomal ambiguity mutants. Mol Cell. 2004;14:343–354. doi: 10.1016/s1097-2765(04)00217-5. [DOI] [PubMed] [Google Scholar]
- Query CC, Moore MJ, Sharp PA. Branch nucleophile selection in pre-mRNA splicing: evidence for the bulged duplex model. Genes Dev. 1994;8:587–597. doi: 10.1101/gad.8.5.587. [DOI] [PubMed] [Google Scholar]
- Seraphin B, Rosbash M. Mutational analysis of the interactions between U1 small nuclear RNA and pre-mRNA of yeast. Gene. 1989;82:145–151. doi: 10.1016/0378-1119(89)90039-5. [DOI] [PubMed] [Google Scholar]
- Siatecka M, Reyes JL, Konarska MM. Functional interactions of Prp8 with both splice sites at the spliceosomal catalytic center. Genes Dev. 1999;13:1983–1993. doi: 10.1101/gad.13.15.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Urushiyama S, Tani T, Ohshima Y. An mRNA-type intron is present in the Rhodotorula hasegawae U2 small nuclear RNA gene. Mol Cell Biol. 1993;13:5613–5619. doi: 10.1128/mcb.13.9.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani T, Ohshima Y. mRNA-type introns in U6 small nuclear RNA genes: implications for the catalysis in pre-mRNA splicing. Genes Dev. 1991;5:1022–1031. doi: 10.1101/gad.5.6.1022. [DOI] [PubMed] [Google Scholar]
- Valadkhan S, Manley JL. Characterization of the catalytic activity of U2 and U6 snRNAs. RNA. 2003;9:892–904. doi: 10.1261/rna.5440303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayraghavan U, Parker R, Tamm J, Iimura Y, Rossi J, Abelson J, Guthrie C. Mutations in conserved intron sequences affect multiple steps in the yeast splicing pathway, particularly assembly of the spliceosome. EMBO J. 1986;5:1683–1695. doi: 10.1002/j.1460-2075.1986.tb04412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Manley JL. Mammalian pre-mRNA branch site selection by U2 snRNP involves base pairing. Genes Dev. 1989;3:1553–1561. doi: 10.1101/gad.3.10.1553. [DOI] [PubMed] [Google Scholar]
- Yu YT, Maroney PA, Nilsen TW. Functional reconstitution of U6 snRNA in nematode cis- and trans-splicing: U6 can serve as both a branch acceptor and a 5' exon. Cell. 1993;75:1049–1059. doi: 10.1016/0092-8674(93)90315-h. [DOI] [PubMed] [Google Scholar]
- Zhuang Y, Weiner AM. A compensatory base change in human U2 snRNA can suppress a branch site mutation. Genes Dev. 1989;3:1545–1552. doi: 10.1101/gad.3.10.1545. [DOI] [PubMed] [Google Scholar]