

Abstract
Investigation of a series of heterobicyclic compounds with essential pharmacophoric features of the metabotropic glutamate receptor 5 (mGluR5) antagonists MPEP and MTEP provided novel structural templates with sub-micromolar affinities at the mGluR5.
Keywords: Metabotropic glutamate receptor 5, non-competitive antagonist, Suzuki-Miyaura coupling
The metabotropic glutamate receptor 5 (mGluR5) belongs to family C of the G-protein coupled receptors (GPCR) and it mediates the actions of the excitatory neurotransmitter, L-glutamate.1-3 Recently, preclinical investigation into the role of mGluR5 in anxiety, depression, pain, mental retardation and drug dependence has suggested that the mGluR5 may be a novel target for therapeutic intervention.4 In particular, studies using either an mGluR5 antagonist or mGluR5 knockout mice showed reduced locomotor stimulant effects induced by cocaine.5 Moreover, mGluR5 appears to be involved in the rewarding effects of morphine, nicotine and ethanol.6 Thus development of selective mGluR5 antagonists may provide a novel strategy toward the discovery of medications for various CNS disorders.
The alkyne based non-competitive mGluR5 antagonists MPEP (1) and MTEP (2) have been used to investigate the role of mGluR5 in CNS disorders.7 A series of compounds with different structural templates incorporating tetrazoles, phenyl ureas, thiopyrimidines and arylmethoxy pyridines have been reported recently as mGluR5 antagonist.8
To further explore the SAR at mGluR5 and to seek compounds with novel structural templates, we have designed a series of compounds based on the SAR developed from our diaryl- and heterobiaryl amides.9 Herein, we describe the discovery of novel heterobicyclic leads and preliminary SAR studies on these compounds.
The allosteric ligand binding site of MPEP-like antagonists at mGluR5 has been predicted to be in the transmembrane region of the receptor.10 The binding site consists of two hydrophobic regions, wherein the aromatic rings (a and b) of the compounds interact, with a linker in between, to position these groups in proper orientation. The pyridyl ‘N’ is essential for activity, and there is a limited tolerance of substitutions on both the aromatic rings. Bearing this in mind, we introduced the pyridyl ring in a bicyclic ring system (heterobicyclic compounds, Figure 2), whereby the important pharmacophoric groups were kept intact. Thus a series of quinoline, benzothiazole and pyridothiazole analogues were synthesized, as depicted in Schemes 1-4, and evaluated for binding at mGluR5.
Figure 2.

Design of heterobicyclic mGluR5 antagonsits.
Scheme 1.
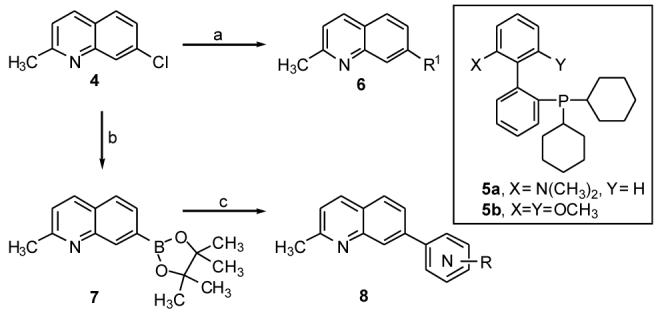
Synthesis of compounds 6-8. Reagents and conditions: (a) Aryl boronic acid, Pd(OAc)2, 5a-b, K3PO4, toluene, ethanol, 100°C, overnight, 55-95%. (b) Bis(pinacolato)diboron, KOAc, 5b, Pd(OAc)2, 100°C, 60 h, 95%; (c) Ar-Br, PdCl2(dppf), 2M Na2CO3, dioxane, 100°C, 40-88%; Ar-Cl, Pd(OAc)2, 5b, K3PO4, dioxane, H2O, 100°C, overnight, 43-86%.
Scheme 4.

Synthesis of compounds 22-24. Reagents and conditions: (a) Conc. HNO3, Conc. H2SO4; (b) (i) Benzoyl chloride, toluene, dioxane, reflux, 24 h, 35%, (ii) 10% Pd/C, methanol, H2, 60 psi, rt, ∼98%; (c) NaOAc, gl. HOAc, 90%; (d) NaH, aryl/alkyl halides, DMF, ∼40%; (e) (i)Benzaldehyde, 4A molecular sieves, methanol, (ii) NaBH3CN, rt, 1h. (iii) NaOAc, gl. HOAc, overall 86%.
The 7-substituted quinolines were obtained as shown in Scheme 1. The 7-chloro-2-methyl quinoline 4 was treated with various substituted aryl boronic acids under Suzuki-Miyuara conditions using the biphenyl phosphine ligands 5a-b to give a set of substituted quinolines 6.11 In further exploration of the SAR, boronic ester 7 was synthesized using the biphenyl phosphine catalyst and coupled with either heteroaryl bromides or chlorides.
Similarly, benzothiazole compounds (10-12) were synthesized (Scheme 2) by Suzuki coupling on compound 9. Additionally the boronic ester (11) was synthesized under standard Miyura borylation conditions and then subjected to Suzuki coupling with various heteroaryl bromides or chlorides.12
Scheme 2.
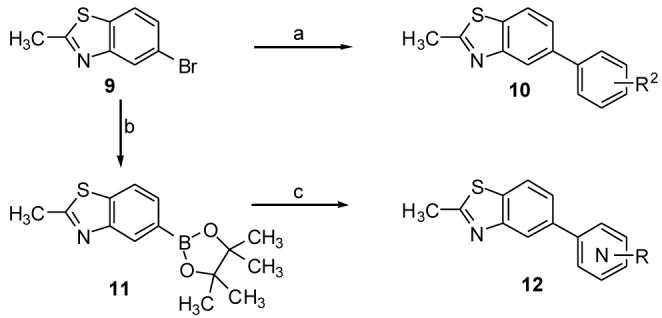
Synthesis of compounds 10-12. Reagents and conditions: (a) Aryl boronic acid, Pd(PPh3)4, 2M aq Na2CO3, toluene, ethanol, reflux 5 h, 65-70%; (b) Bis(pinacolato)diboron, KOAc, PdCl2(dppf), DMF, 105°C, 3 h, 95%; (c) Ar-Br, PdCl2(dppf), 2M Na2CO3, dioxane, 100°C, 65%; Ar-Cl, Pd(OAc)2, 5b, K3PO4, dioxane, H2O, 100°C, overnight, 40%.
In addition, the pyridothiazole and imidazopyridine compounds were synthesized as shown in Scheme 3 and 4. wherein, 6-methyl 2-amino pyridine 13 was brominated with excess N-bromo succinimide (NBS). The benzamide (15) was then prepared with relative ease, which underwent cyclization in the presence of Lawesson's reagent to obtain bromopyridothiazole 16.13 The compound 16 was then treated with various reagents to give a small set of substituted pyridothiazoles (17).
Scheme 3.

Synthesis of compounds 15-17. Reagents and conditions: (a) NBS, DMF, rt, 1h, 95%; (b) Benzoyl chloride, pyridine, reflux, 80%; (c) Lawesson's reagent, HMPA, 140°C, 1 h, ∼95%; (d) (i) Anhydrous NaOAc, 10% Pd/C, methanol, 60 psi, 45 h or (ii) Sodium methoxide, Cu(I)Br, DMF, 120°C, 30 min or (iii) Aryl boronic acid, Pd(PPh3)4, 2M aq Na2CO3, toluene, ethanol, reflux 5 h, 50-90%.
The imidazopyridines were obtained by nitrating the 6-methyl-2-amino pyridine 13 to obtain a mixture of regioisomeric nitropyridines (18-20), which were purified with extreme care by steam distillation.14 The 3-nitro compound 19 was treated with benzoyl chloride to obtain the benzamide, which was cyclized into imidazopyridine 22, after reduction of the nitro group.15 The substituted imidazopyridines were synthesized by treating compound 22 with various alkyl or arylalkyl bromides in the presence of NaH. Compound 24 was obtained by treating the benzamide 21 with benzaldehyde under reductive amination conditions, followed by cyclization.16
The compounds were evaluated for biological activity in a rat brain membrane preparation using [3H]MPEP as the radioligand to measure binding affinity at mGluR5. Data for all novel compounds described herein were obtained through the NIMH PDSP program using methods cited17 in Tables 1-3 and compared with reference compounds 1 and a diarylamide analogue 3a.9a
Table 1.
Structures and mGluR5 binding affinities of substituted quinolines.






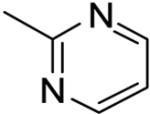
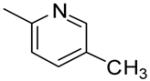

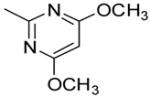


Table 3.
Representative structures and mGluR5 binding affinities of pyridothiazole or imidazopyridine compounds.
 | ||||
|---|---|---|---|---|
| Comp. | R3 | X | Y | mGluR5 Binding Affinity Ki ±S.E.M. (μM) |
| 16 | Br | S | N | 1%a |
| 17a | H | S | N | 16%a |
| 17b | OCH3 | S | N | <1%a |
| 17c | Ph | S | N | 33.48 ± 2.24b |
| 17d | 4'F-Ph | S | N | 3%a |
| 17e | 3'Cl-Ph | S | N | 1%a |
| 17f | 3'CN-Ph | S | N | 13%a |
| 17g | 2-Thioph- | S | N | >10 |
| 22 | H | N | NH | >10 |
| 23a | H | N | NCH2Ph | >10 |
| 23b | H | N | NCH2-c-C3H5 | <1%a |
| 23c | H | N | NCH3 | <1%a |
| 24 | H | NCH2Ph | N | >10 |
Percent inhibition at 10 μM
IC50 (μM) using methods described in ref. 9a.
Substitution of a phenyl ring at the 7 position of quinoline, (compound 6a) showed 50% radioligand displacement at a concentration of 10μM at mGluR5; a substantial decrease in affinity as compared to MPEP. Moreover, substitution with the bioisosteric thiophene (6b) completely eliminated activity. Substitution on the 7-phenyl ring however improved activity for some analogues, e.g. 6c with a 3-CN group showed a Ki= 110 nM in the mGluR5 binding assay. A similar improvement in affinity was observed in the diarylamide series, wherein the 3-CN compound (3a) showed improved activity over the unsubstituted lead compound.9a In a related series of compounds, substitution with pyridine and pyrimidine rings as a part of aryl ring ‘b’ improved the mGluR5 affinity.9b Thus a small set of substituted pyridines/pyrimidines (8a-8h) at the 7-position of the quinoline ring were prepared. Nevertheless, none of these compounds showed any improvement in mGluR5 activity, thereby illustrating a limited tolerance of substitution with inconsistent SAR across different templates.
Substitution on the appended phenyl ring (compounds 10b-e) provided benzothiazoles with moderate affinity (Table 2). The 3-CN compound 10e showing the highest affinity for mGluR5 in this set. Substitution with either pyridyl or pyrimidinyl rings (12a-c) did not improve mGluR5 binding affinity, as observed previously in the quinoline series.
Table 2.
Representative structures and mGluR5 binding affinities of substituted benzothiazoles.
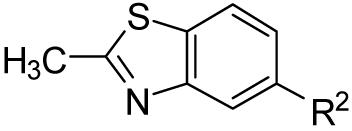 | ||
|---|---|---|
| Compound | R2 | mGluR5 Binding Affinity Ki ±S.E.M. (μM) |
| 10a | 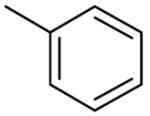 |
NAa |
| 10b | 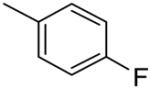 |
NAa |
| 10c |  |
43.39 ± 3.29b |
| 10d |  |
43.05 ± 2.47b |
| 10e |  |
2.10 ± 0.58 |
| 12a |  |
17 %c |
| 12b |  |
29 %c |
| 12c |  |
39 %c |
Not active at 100 μM
IC50 (μM) using methods described in ref. 9a
Percentage inhibition at 10 μM.
Further investigation into other related heterocyclic ring systems that could be modified was undertaken. In this pursuit, compounds with a pyridothiazole or imidazopyridine scaffold were synthesized. However, none of this series (16-24) gave improved mGluR5 activity. In the pyridothiazole series, substitution with additional aromatic rings (17c-g) to potentially improve the activity by gaining additional hydrophobic interactions did not yield any tractable SAR. Similarly exploration of binding sites in the linker region (23a-c and 24) on the imidazopyridine template showed no improvement in activity.
Thus, modifications to seek novel structural templates yielded some substituted quinolines and benzothiazoles with moderate mGluR5 binding affinities. However there was a limited tolerance of substitution with little discernable SAR in these series of compounds. Similarly, no SAR could be obtained from a large set of compounds recently reported by Zhao et al. as positive allosteric modulators at mGluR5.18 The discovery of only a few active compounds from exhaustive efforts in structural modifications depict the inherent difficulty in optimizing the activity at this receptor. In the present study, as observed previously substitution with a cyano group at the 3' position of the appended phenyl ring provided some improvements in mGluR5 activity. A superimposition of these templates on the MPEP 1 and compound 3a analogues shows good overlap of the pharmacophoric features (see Figure 3). Although there are few minor differences in the overlap, it is difficult to account for the dramatic changes in activity across different templates.
Figure 3.

Superimposition of MPEP 1, compounds 3, 6a (green), 10a (blue) and 17a (red).
In summary, we identified two structural templates with moderate mGluR5 antagonist activity. Most structural modification did not yield compounds with any improvement in mGluR5 affinity. In the functional assay measuring the hydrolysis of phosphoinositide at mGluR5 in CHO cells, compound 6c showed antagonist activity (IC50=0.26±0.05 μM)19 hence this template may provide a new lead for further SAR investigation.
Supplementary Material
Figure 1.

Non-competitive antagonists of mGluR5: MPEP 1 and MTEP 2.
Acknowledgements
SSK was supported by a NIH-visiting fellowship. We thank Dr. Mu-Fa Zou for critically reading this manuscript. We acknowledge Ms. Barbara Nightingale for technical assistance, Dr. Bryan Roth, Director of the National Institute on Mental Health-Psychoactive Drug Screening Program (PDSP) for his support and Ms. Estela Lopez for help with the PDSP database. This work is supported by the NIDA-IRP, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Conn PJ, Pin JP. Annu. Rev. Pharmacol. Toxicol. 1997;37:205. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 2.Spooren WPJM, Gasparini F, Salt TE, Kuhn R. TIPS. 2001;22(7):331. doi: 10.1016/s0165-6147(00)01694-1. [DOI] [PubMed] [Google Scholar]
- 3.Swanson CJ, Bures M, Johnson MP, Linden AM, Monn JA, Schoepp DD. Nature Rev. Drug Discov. 2005;4:131. doi: 10.1038/nrd1630. [DOI] [PubMed] [Google Scholar]
- 4.(a) Brodkin J, Busse C, Sukoff SJ, Varney MA. Pharmacol. Biochem. Behav. 2002;23:207. doi: 10.1016/s0091-3057(02)00828-6. [DOI] [PubMed] [Google Scholar]; (b) Tatarczynska E, Klodzinska A, Chojnacka-Wojcik E, Palucha A, Gasparini F, Kuhn R, Pilc A. Br. J. Pharmacol. 2001;132:1423. doi: 10.1038/sj.bjp.0703923. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dolan S, Kelly JG, Monteiro AM, Nolan AM. Pain. 2003;106:501. doi: 10.1016/j.pain.2003.09.017. [DOI] [PubMed] [Google Scholar]; (d) Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Neuropharmacology. 2005;49:1053. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]; (e) Li X, Need AB, Baez M, Witkin JM. J. Pharmacol. Exp. Ther. 2006;319:254. doi: 10.1124/jpet.106.103143. [DOI] [PubMed] [Google Scholar]; (f) Bear MF. Genes Brain Behav. 2005;4:393. doi: 10.1111/j.1601-183X.2005.00135.x. [DOI] [PubMed] [Google Scholar]
- 5.Chiamulera C, Eping-Jordon MP, Zocchi A, Marcon C, Cottiny C, Tacconi S, Corsi M, Orzi F, Conquet F. Nature Neurosci. 2001;4:873. doi: 10.1038/nn0901-873. [DOI] [PubMed] [Google Scholar]
- 6.(a) Popik P, Wrobel M. Neuropharmacology. 2002;43:1210. doi: 10.1016/s0028-3908(02)00309-x. [DOI] [PubMed] [Google Scholar]; (b) Paterson NE, Semenova S, Gasparini F, Markou A. Psychopharmacology(Berl.) 2003;167:257. doi: 10.1007/s00213-003-1432-z. [DOI] [PubMed] [Google Scholar]; (c) Backstrom P, Bachteler D, Koch S, Hyytia P, Spanagel R. Neuropsychopharmacology. 2004;29:921. doi: 10.1038/sj.npp.1300381. [DOI] [PubMed] [Google Scholar]
- 7.(a) Gasparini F, Floersheim P, Flor PJ, Heinrich M, Inderbitzin W, Ott D, Pagano A, Stierlin C, Stoehr N, Vranesic I, Kuhn R. Farmaco. 2001;56:95. doi: 10.1016/s0014-827x(01)01008-4. [DOI] [PubMed] [Google Scholar]; (b) Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I, Rao S, Washburn M, Varney MA. J Med Chem. 2003;46:204. doi: 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- 8.(a) Roppe J, Smith ND, Huang D, Tehrani L, Wang B, Anderson J, Brodkin J, Chung J, Jiang X, King C, Munoz B, Varney MA, Prasit P, Cosford ND. J. Med. Chem. 2004;47:4645. doi: 10.1021/jm049828c. [DOI] [PubMed] [Google Scholar]; (b) Wallberg A, Nilsson K, Osterlund K, Peterson A, Elg S, Raboisson P, Baur U, Hammerland LG, Mattsson JP. Bioorg. Med. Chem. Lett. 2006;16:1142. doi: 10.1016/j.bmcl.2005.11.092. [DOI] [PubMed] [Google Scholar]; (c) Hammerland LG, Johansson M, Malmstrom J, Mattsson JP, Minidis ABE, Nilsson K, Peterson A, Wensbo D, Wallberg A, Osterlund K. Bioorg. Med. Chem. Lett. 2006;16:2467. doi: 10.1016/j.bmcl.2006.01.100. [DOI] [PubMed] [Google Scholar]; (d) Buttelmann B, Peters JU, Ceccarelli S, Kolczewski S, Vieira E, Prinssen EP, Spooren W, Schuler F, Huwyler J, Porter RHP, Jaeschke G. Bioorg. Med. Chem. Lett. 2006;16:1892. doi: 10.1016/j.bmcl.2005.12.088. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kulkarni SS, Nightangle B, Dersch CM, Rothman RB, Newman AH. Bioorg. Med. Chem. Lett. 2006;16:3371. doi: 10.1016/j.bmcl.2006.04.032. [DOI] [PubMed] [Google Scholar]; (b) Kulkarni SS, Newman AH. Bioorg. Med. Chem. Lett. 2007 doi: 10.1016/j.bmcl.2006.12.083. manuscript in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, Floersheim P, Prezeau L, Carroll F, Pin JP, Cambria A, Vranesic I, Flor PJ, Gasparini F, Kuhn R. J. Biol. Chem. 2000;275:33750. doi: 10.1074/jbc.M006230200. [DOI] [PubMed] [Google Scholar]; (b) Malherbe P, Kratochwil N, Zenner MT, Piussi J, Diener C, Kratzeisen C, Fischer C, Porter RH. Mol. Pharmacol. 2003;64:823. doi: 10.1124/mol.64.4.823. [DOI] [PubMed] [Google Scholar]
- 11.(a) Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]; (b) Wolfe JP, Singer RA, Yang BH, Buchwald SL. J. Am. Chem. Soc. 1999;121:9550. [Google Scholar]
- 12.Ishiyama T, Murata M, Miyuara N. J. Org. Chem. 1995;60:7508. [Google Scholar]
- 13.Hutchinson I, Chua MS, Browne HL, Trapani V, Bradshaw TD, Westwell AD, Stevens MFG. J. Med. Chem. 2001;44:1446. doi: 10.1021/jm001104n. [DOI] [PubMed] [Google Scholar]
- 14.Parker ED, Shive W. J. Am. Chem. Soc. 1947;69:63. [Google Scholar]
- 15.Molina P, Alajarin M, Vidal A. Tetrahedron. 1989;45:4263. [Google Scholar]
- 16.All compounds were purified by flash chromatography and characterized by spectroscopic and microanalytical techniques. The final products were crystallized as the HBr salts for biological evaluation. The spectral data supported the assigned structures, for example, 2-methyl-7-(pyrimidin-2-yl)quinoline, 8c, mp 276-278°C; 1HNMR (400 MHz, CDCl3) δ 2.78 (s, 3H), 7.22-7.24 (t, J = 4.8 Hz, 1H), 7.31-7.33 (d, J = 8.8 Hz, 1H), 7.87-7.89 (d, J = 8.4 Hz, 1H), 8.07-8.09 (d, J = 8.4 Hz, 1H), 8.53-8.56 (dd, J = 2, 8.4 Hz, 1H), 8.86-8.87 (d, J = 4.8 Hz, 2H), 9.18 (s, 1H); 13CNMR (101 MHz, CDCl3) δ 25.66, 119.60, 123.09, 125.12, 127.96, 128.11, 129.33, 136.08, 138.87, 148.24, 157.60, 159.79, 164.70; IR (Neat, cm−1) 3401.1, 1606.4, 1566.4, 1554.6, 1420.2, 1406.9; GC-MS (EI) m/z 221 (M+); Anal. (C14H11N3.HBr.H2O) C, H, N. The experimental and spectral data for all other compounds are reported in the supporting information.
- 17.Binding affinities were determined by the National Institute on Mental Health-Psychoactive Drug Screening Program as described in http://pdsp.med.unc.edu/indexR.html.
- 18.Zhao Z, Wisnoski DD, O'Brien JA, Lemaire W, Williams DL, Jacobson MA, Wittman M, Ha SN, Schaffhauser H, Sur C, Pettibone DJ, Duggan ME, Conn PJ, Hartman GD, Lindsley CW. Bioorg. Med. Chem. Lett. 2006;17:1386. doi: 10.1016/j.bmcl.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 19.Functional assay data was provided by the National Institute on Mental Health-Psychoactive Drug Screening Program (PDSP) using methods described in Shi Q, Savage JE, Hufeisen SJ, Rauser L, Grajkowska E, Ernsberger P, Wroblewski JT, Nadeau JH, Roth BL. J. Pharmacol. Exp. Ther. 2003;305:131. doi: 10.1124/jpet.102.047092.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.