

Abstract
The goal of this study was to determine whether the intracellular distribution of the proapoptotic enzyme glycogen synthase kinase-3β (GSK-3β) is dynamically regulated by conditions that activate apoptotic signaling cascades. In untreated human neuroblastoma SHSY5Y cells, GSK-3β was predominantly cytosolic, although a low level was also detected in the nucleus. The nuclear level of GSK-3β was rapidly increased after exposure of cells to serum-free media, heat shock, or staurosporine. Although each of these conditions caused changes in the serine 9 and/or tyrosine phosphorylation of GSK-3β, neither of these modifications was correlated with nuclear accumulation of GSK-3β. Heat shock and staurosporine treatments increased nuclear GSK-3β prior to activation of caspase-9 and caspase-3, and this nuclear accumulation of GSK-3β was unaltered by pretreatment with a general caspase inhibitor. The GSK-3β inhibitor lithium did not alter heat shock-induced nuclear accumulation of GSK-3β but increased the nuclear level of cyclin D1, indicating that cyclin D1 is a substrate of nuclear GSK-3β. Thus, the intracellular distribution of GSK-3β is dynamically regulated by signaling cascades, and apoptotic stimuli cause increased nuclear levels of GSK-3β, which facilitates interactions with nuclear substrates.
Glycogen synthase kinase-3β (GSK-3β)1 affects many fundamental cellular functions, such as the cell cycle, gene transcription, and cytoskeletal integrity, because of its ability to phosphorylate key proteins governing these processes (reviewed in Ref. 1). Although GSK-3β is considered a constitutively active enzyme, its activity is subject to regulation by intracellular signaling cascades. One of these regulatory mechanisms consists of site-specific phosphorylation of GSK-3β. The activity of GSK-3β is increased by phosphorylation of Tyr216, which can result from autophosphorylation (2) and from intracellular signaling mechanisms (3, 4). Conversely, phosphorylation of Ser9 inhibits GSK-3β activity. The phosphatidylinositol 3-kinase/Akt (PI3K/Akt) signaling pathway is one of the signaling systems that leads to inhibition of GSK-3β by increasing Ser9 phosphorylation. In this signaling pathway PI3K is activated by growth factors, such as insulin-like growth factor-1 (IGF-1), which leads to the phosphorylation and activation of Akt (5). Subsequently Akt can phosphorylate Ser9 of GSK-3β (6). Additionally, Ser9 of GSK-3β can be phosphorylated by protein kinase C (7), p90Rsk, p70 S6 kinase (8), and protein kinase A (9). Thus, multiple mechanisms are utilized to modify the activity of GSK-3β by phosphorylation.
In addition to phosphorylation, the actions of GSK-3β may be modulated by regulation of its subcellular distribution. GSK-3β was initially considered to be a soluble cytoplasmic kinase (10, 11). However, GSK-3β has the capacity to phosphorylate proteins within the nucleus, such as cyclin D1 (12), nuclear factor of activated T-cells (13), heat shock factor-1 (14–16), and cAMP response element-binding protein (17). This capacity implies the existence of nuclear, as well as cytosolic, GSK-3β, a localization for which there is some evidence. GSK-3β co-purified with glycogen in the rat liver nuclear pore lamina matrix (18), and nuclear GSK-3β was identified in cell cycle-arrested NIH-3T3 cells treated with serum (12), in cardiomyocytes stimulated with endothelin-1 (19), and in heat shock-treated Xenopus laevis oocytes (16). In addition, nerve growth factor withdrawal from PC12 cells and staurosporine treatment of human neuroblastoma SH-SY5Y cells increased Tyr216-phosphorylated GSK-3β in the nucleus, which may reflect changes in total GSK-3β levels, but these were not reported (20). Thus, besides the prevailing concept that GSK-3β activity can be regulated by phosphorylation, these observations suggest that GSK-3β activity may also be controlled by regulation of its intracellular localization.
In addition to its other functions, recent evidence indicates that GSK-3β is a key component of some apoptotic signaling cascades. Takashima et al. (21) reported that treatment of rat cortical neurons with β-amyloid peptide increased GSK-3β activity and induced apoptosis, both of which were inhibited by treatment with GSK-3β antisense oligonucleotides. Additionally, Pap and Cooper (22) reported that transient overexpression of GSK-3βin PC12 cells and Rat-1 fibroblasts caused cells to spontaneously undergo apoptosis, and the GSK-3β-induced apoptosis was inhibited by activation of the PI3K/Akt signaling pathway. In another cell culture model, Bijur et al. (23) reported that stable overexpression of modest levels of GSK-3β in SH-SY5Y neuroblastoma cells did not alter basal indices of apoptosis but facilitated staurosporine- and heat shock-induced apoptosis, and this facilitation was attenuated by treatment with lithium, a direct inhibitor of GSK-3β (24). Hetman et al. (25) reported that inhibition of GSK-3β by transfection of rat primary cortical neurons with GSK-3β-binding protein, a protein that inhibits GSK-3β activity, protected cells from serum withdrawal-induced apoptosis. Additionally, activation of the PI3K/Akt pathway protects cells from many apoptotic stimuli (26), an action that may be due, in part, to the inhibitory effect of Akt on GSK-3β activity (22). Further evidence has supported the proapoptotic actions of GSK-3β (26–28), and many reports have shown that lithium is protective against a variety of proapoptotic insults (reviewed in Ref. 29), an action that is suggested to result from its inhibition of GSK-3β (1).
Because GSK-3β has been identified as a contributory factor in apoptosis induced by serum withdrawal, heat shock, and staurosporine, the present study examined whether the intracellular distribution of GSK-3β was regulated by proapoptotic conditions. Using human neuroblastoma SH-SY5Y cells, we report that each of these apoptosis paradigms induces a transient nuclear accumulation of GSK-3β that appears to be independent of changes in Ser9 and Tyr216 phosphorylation of GSK-3β. Furthermore, GSK-3β nuclear accumulation occurs early in the apoptotic process, preceding activation of caspases.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatments
SH-SY5Y human neuroblastoma cells were grown as described previously (23). For serum withdrawal, adherent cells were rinsed twice with serum-free media supplemented with 2 mm L-glutamine, 100 units/ml penicillin, and 100 µg/ml streptomycin and were cultured in serum-free media for the indicated time. For heat shock treatment, 100-mm tissue culture plates containing cells in serum-containing media were sealed with Parafilm and floated in a 45 °C water bath with gentle agitation. For staurosporine treatment, cells were maintained in serum-containing media. Caspase activity was measured as described previously (23).
Purification of Nuclei
Nuclei were isolated from cells cultured in 100-mm plates by washing the adherent cells twice with phosphate-buffered saline (PBS) and adding 500 µl of lysis buffer (10 mm Tris, pH 7.5, 10 mm NaCl, 3 mm MgCl2, 0.05% Nonidet P-40, 1 mm EGTA, 1 mm sodium orthovanadate, 50 mm sodium fluoride, 100 µm phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, 10 µg/ml aprotinin, 5 µg/ml pepstatin A, and 1 nm okadaic acid). Lysed cells were collected in micro-centrifuge tubes and centrifuged at 2,700 × g for 10 min at 4 °C. The supernatant containing the cytosol was further centrifuged at 20,800 × g for 15 min at 4 °C to obtain the cytosolic fraction. The nuclei in the pellet were washed three times by gently resuspending the nuclei in 200 µl of wash buffer (10 mm PIPES, pH 6.8, 300 mm sucrose, 3 mm MgCl2, 1 mm EGTA, 25 mm NaCl, 1 mm sodium orthovanadate, 50 mm sodium fluoride, 100 µM phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, 10 µg/ml aprotinin, and 5 µg/ml pepstatin A) and centrifuging at 2,700 × g for 5 min at 4 °C. For a final wash, the nuclei were resuspended in 100 µl of wash buffer, layered over a cushion of 1 ml of sucrose buffer (1 M sucrose, 1 mm sodium orthovanadate, 50 mm sodium fluoride, 100 µm phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, 10 µg/ml aprotinin, and 5 µg/ml pepstatin A), and centrifuged at 2,700 × g for 10 min. The sucrose buffer containing non-sedimented cellular debris was discarded, and the pellet containing nuclei was washed in 100 µl of lysis buffer and centrifuged at 2,700 × g for 5 min at 4 °C to remove residual sucrose buffer. To measure relative GSK-3β immunofluorescence, nuclei were fixed with methanol-acetone (1:1) for 10 min at −20 °C, stained immunofluorescently with a mouse monoclonal anti-GSK-3β antibody (Transduction Laboratories, Lexington, KY), and stained with 10 ng/ml Hoechst 33342 (Molecular Probes, Eugene, OR) for 30 min at room temperature. The number of nuclei exhibiting GSK-3β immunofluorescence was determined using a FACSVantage flow cytometer (Becton Dickinson) by first subtracting nuclei exhibiting background fluorescence and then counting 10,000 Hoechst 33342-positive nuclei. For immunoblotting, nuclear proteins were extracted with 20 mm HEPES, pH 7.9, 300 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 0.1 mm β-glycerophosphate, 1 mm sodium orthovanadate, 50 mm sodium fluoride, 100 µm phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, 10 µg/ml aprotinin, 5 µg/ml pepstatin A, and 1 nm okadaic acid. After extraction on ice for 30 min, the samples were centrifuged at 20,800 × g for 15 min at 4 °C. Protein concentrations in the cytosolic and nuclear extracts were determined using the bicinchoninic acid method (Pierce).
Immunoblot Analysis
Cell lysates for immunoblotting were prepared as described previously (23). Proteins were resolved in SDS-polyacrylamide gels (8% for GSK-3β, Akt, and PARP, and 10% for tubulin, histone, and caspase-3 immunoblots), transferred to nitrocellulose, and incubated with mouse monoclonal anti-GSK-3β (Transduction Laboratories), anti-poly(ADP-ribose) polymerase (PARP) (Phar-Mingen, San Diego, CA), anti-phospho-(Ser9)-GSK-3β, anti-phospho- (Ser473)-Akt, anti-total Akt, anti-active caspase-3 (New England Bio-Labs, Beverly, MA), anti-histone protein (Chemicon, Temecula, CA), or anti-β-tubulin III (Sigma) antibodies. Immunoblots were developed using horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgG, followed by detection with enhanced chemiluminescence, and the protein bands were quantitated with a densitometer.
Immunofluorescence
Cells were cultured on poly-d-lysine-coated glass coverslips and subjected to treatments as indicated. The cells were washed twice with PBS and then fixed and permeabilized in ice-cold methanol-acetone (1:1) for 10 min at −20 °C. The coverslips were washed twice with PBS and incubated overnight at 4 °C with 10 µg/ml of mouse monoclonal anti-GSK-3β antibody diluted in PBS containing 2% bovine serum albumin. The coverslips were washed three times with PBS and incubated for 1 h at room temperature with 10 µg/ml fluorescein isothiocyanate-conjugated anti-mouse antibody (Chemicon) diluted in PBS containing 2% bovine serum albumin. The coverslips were washed with PBS and incubated with 21 µg/ml anti-β-tubulin III antibody for 1 h at room temperature. The coverslips were washed with PBS and incubated with 5 µg/ml Texas red-conjugated goat anti-mouse antibody (Chemicon) for 1 h at room temperature. The coverslips were washed once with PBS and twice with deionized water and incubated with 50 ng/ml 4,6-diamino-2-phenylindole stain (Molecular Probes) for 1 h at room temperature. The coverslips were washed with deionized water, mounted onto glass slides using Immu-Mount (Shandon, Pittsburgh, PA), and examined by a fluorescence microscope (Nikon) set at 400× magnification.
GSK-3β Activity Measurements
To immunoprecipitate GSK-3β, 50 µg of protein from nuclear extracts (0.5 µg/µl) was incubated with 0.75 µg of mouse monoclonal anti-GSK-3β antibody overnight at 4 °C with gentle agitation. Extracts were incubated with 30 µl of protein G-Sepharose for 1 h at 4 °C. The immobilized immune complexes were washed twice with immunoprecipitation lysis buffer and twice with kinase buffer (20 mm Tris, pH 7.5, 5 mm MgCl2, and 1 mm dithiothreitol). Kinase activity was measured by mixing immunoprecipitated GSK-3β with 25 µl of kinase buffer containing 20 mm Tris, pH 7.5, 5 mM MgCl2, 1 mM dithiothreitol, 250 µM ATP, 1.4 µCi of [γ-32P]ATP (Amersham Pharmacia Biotech), and 0.1 µg/µl recombinant tau protein (Panvera, Madison, WI). The samples were incubated at 30 °C for 15 min, and 25 µl of Laemmli sample buffer (2% SDS) was added to each sample to stop the reaction. Samples were placed in a boiling water bath for 5 min, and proteins were separated in 7.5% SDS-polyacrylamide gels. The gels were vacuum-dried, exposed to a phosphoscreen overnight, and quantitated using a PhosphorImager (Molecular Dynamics, Sunnyvale, CA). The efficiency of GSK-3β immunoprecipitation was determined by immunoblotting for GSK-3β.
RESULTS
Serum Withdrawal Induces Nuclear Accumulation of GSK-3β
Immunoblots of cytosolic and nuclear fractions prepared from untreated SH-SY5Y cells revealed that although GSK-3β was predominantly located in the cytosol, it was also present in the nuclear fraction (Fig. 1). Complete separation of the nuclear and cytosolic fractions was verified by immunoblotting each fraction for tubulin, a cytosolic protein, and histone, a nuclear protein. Fig. 1 shows that each fraction was free of contamination, as tubulin and histone were detected only in the cytosolic and nuclear fractions, respectively.
FIG. 1. GSK-3β levels in the cytoplasm and nucleus.
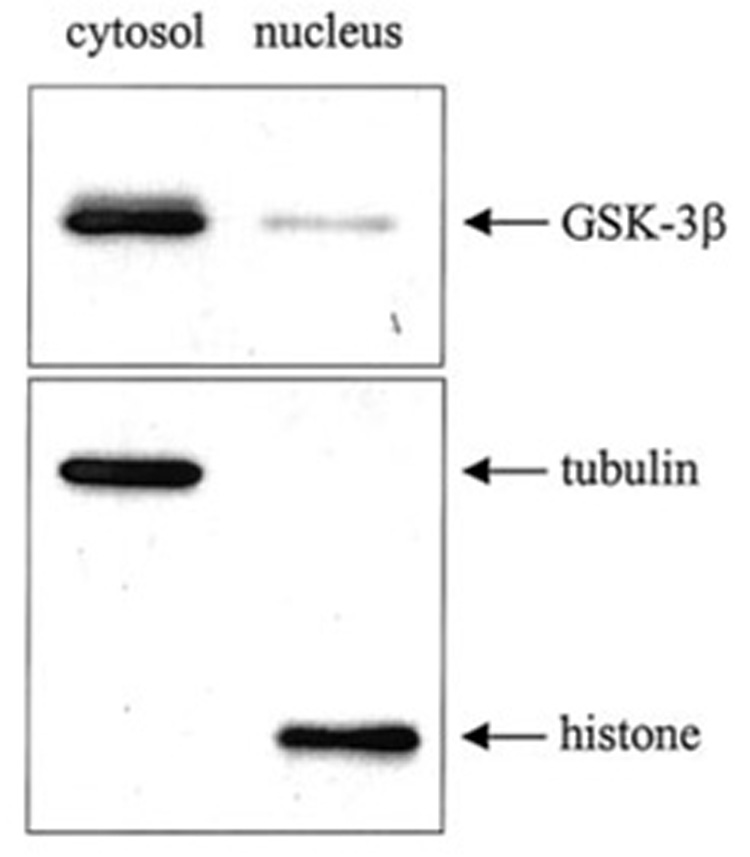
SH-SY5Y cells were fractionated into cytosolic and nuclear fractions as described under “Experimental Procedures.” Extracts (2.5 µg of protein) were immunoblotted for GSK-3β. To verify complete separation of the cytosolic and nuclear fractions, cytosolic and nuclear extracts (10 µg of protein) were immunoblotted for tubulin and histone.
To test whether the nuclear level of GSK-3β is subject to regulation by intracellular signaling cascades, SH-SY5Y cells were incubated in serum-free media, a condition known to inactivate the PI3K/Akt signaling cascade. Serum withdrawal caused a marked increase in nuclear GSK-3β, which reached a maximum 75% increase above basal levels after 60 min (Fig. 2, A and B). Serum withdrawal did not change the total level of GSK-3β in whole cell lysates. Concomitantly with the serum withdrawal-induced increase in nuclear GSK-3β, there was a rapid decrease in the activation-associated phosphorylation of Ser473 of Akt, whereas the total level of Akt was unchanged. However, whereas the nuclear GSK-3β returned toward the basal level between 60 and 240 min of incubation in serum-free media, during this time the level of phosphorylated Akt remained low, at 25% of its level in control cells. Immunoblots of tubulin and histone confirmed the complete separation of the cytosolic and nuclear fractions. Thus, these results revealed that the nuclear level of GSK-3β is dynamic and is subject to regulation by intracellular signaling activities.
FIG. 2. Serum withdrawal induces nuclear accumulation of GSK-3β.
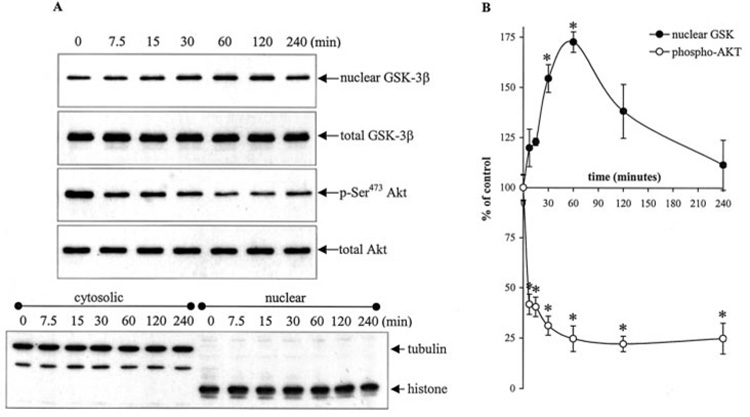
Serum-containing media was withdrawn, and cells were treated with serum-free media for 0, 7.5, 15, 30, 60, 120, and 240 min. One aliquot of cells was fractionated into cytosolic and nuclear fractions, and another aliquot of cells was used to produce total cell lysates as described under “Experimental Procedures.” A, nuclear extracts were immunoblotted for GSK-3β. Cytosolic and nuclear extracts were immunoblotted for tubulin and histone protein to verify complete separation of the cytosolic and nuclear fractions. Total cell lysates were immunoblotted for GSK-3β, p-Ser473 Akt, and total Akt. The blots shown are representative of at least three independent experiments. B, GSK-3β and p-Ser473 Akt bands were analyzed by a scanning densitometer. Values are expressed as a percent of GSK-3β nuclear levels and p-Ser473 Akt levels in untreated cells. Means ± S.E., n = three experiments; *, p < 0.05 (ANOVA) compared with untreated cells.
The correlation between the initial increase in nuclear GSK-3β and decrease in phosphorylated Akt was investigated further by applying agents that modulate the PI3K/Akt signaling pathway. Treatment of cells in serum-free media with either of two PI3K inhibitors, 20 µM LY294002 or 40 nm wortmannin, increased the nuclear level of GSK-3β and decreased phosphorylated Akt to a greater extent than that caused by serum withdrawal alone (Fig. 3, A and B). Activation of the PI3K/Akt pathway with 50 ng/ml IGF-1 after serum withdrawal increased phosphorylated Akt and lowered the nuclear GSK-3β level to that measured in untreated cells. Treatment with LY294002 or wortmannin blocked the effects of IGF-1 on both phosphorylated Akt and nuclear GSK-3β levels. These results further support the conclusion that the level of nuclear GSK-3β is subject to regulation by intracellular signaling pathways.
FIG. 3. PI3K inhibitors and IGF-1 affect serum withdrawal-induced GSK-3β nuclear accumulation.

Serum-containing media was withdrawn, and cells were incubated in serum-free media for a total time of 1 h. For combined IGF-1 and LY294002 or wortmannin treatments, cells were incubated in serum-free media for 15 min, treated with 20 µm LY294002 or 40 nm wortmannin for 15 min, and incubated with 50 ng/ml IGF-1 for 30 min. For individual treatments, cells were treated with 20 µm LY294002 or 40 nm wortmannin alone for 45 min or 50 ng/ml IGF-1 alone for 30 min. The nuclear extracts were immunoblotted for GSK-3β. Total cell lysates were immunoblotted for GSK-3β, p-Ser473 Akt, and total Akt. B, nuclear GSK-3β and p-Ser473 Akt bands were analyzed by densitometer. Means ± S.E., n = three experiments; *, p < 0.05 (ANOVA) compared with untreated cells.
Heat Shock and Staurosporine Treatments Induce Nuclear Accumulation of GSK-3β
The effects of two apoptotic stimuli, heat shock and staurosporine, were measured on the intracellular distribution of GSK-3β. Exposure of SH-SY5Y cells to heat shock (30 min at 45 °C) followed by incubation at 37 °C was shown previously to lead to apoptosis over a period of several hours (23). Therefore, the present experiments used a paradigm of incubation of cells for 30 min at 45 °C followed by 30 min at 37 °C, a time at which caspase-3 activity was only beginning to increase (23) to test whether the intracellular distribution of GSK-3β was altered. Immunofluorescent staining of GSK-3β revealed that little GSK-3β was detectable in the nucleus in control cells (Fig. 4A), but significant nuclear GSK-3β was evident after treatment with heat shock. This altered distribution of GSK-3β was assessed further by examining isolated nuclei prepared from control and heat shock-treated cells by flow cytometry. These measurements confirmed that there was a marked accumulation of GSK-3β in the nuclei after heat shock treatment (Fig. 4B), as indicated by the rightward shift and greater magnitude of the GSK-3β immunofluorescence signal in nuclei after heat shock (gray peak) compared with controls (white peak). To further confirm that heat shock treatment induces GSK-3β nuclear accumulation, cells were treated with 100 nM okadaic acid, a phosphatase inhibitor known to broadly inhibit nuclear import (30), prior to heat shock treatment. Okadaic acid alone decreased the basal level of GSK-3β in the nucleus (Fig. 4C) and inhibited the heat shock-induced GSK-3β nuclear accumulation. Treatment with 10 ng/ml leptomycin B, a direct inhibitor of the nuclear export receptor CRM1 (31), alone increased the basal level of GSK-3β in the nucleus, and treatment with leptomycin B 2.5 h prior to heat shock treatment augmented the heat shock-induced GSK-3β nuclear accumulation. Together, these results demonstrate that heat shock induces GSK-3β to enter into the nucleus, and its levels in the nucleus can be modulated by mechanisms that regulate nuclear transport.
FIG. 4. Heat shock induces GSK-3β nuclear accumulation.
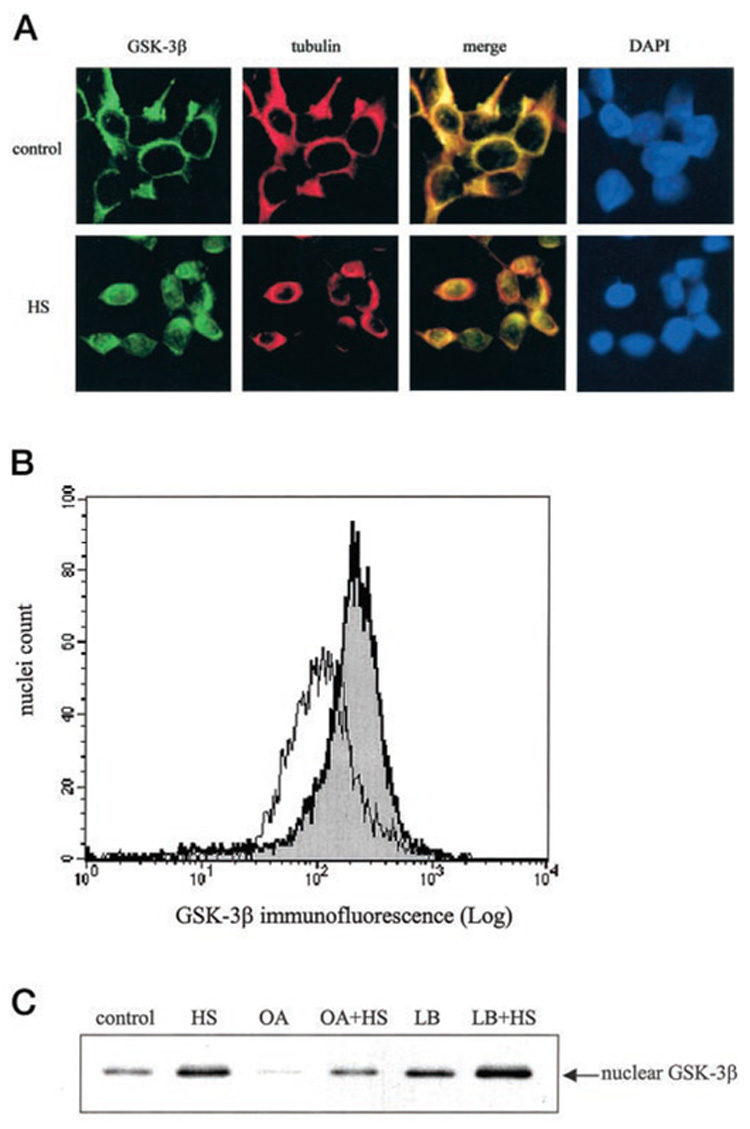
A, cells cultured on glass coverslips were either maintained in a 37 °C chamber as controls or subjected to heat shock (HS) at 45 °C for 30 min and then transferred to a 37 °C chamber for an additional 30 min. Cells were fixed, permeabilized, and immunofluorescently labeled as described under “Experimental Procedures.” Cells immunofluorescently labeled with GSK-3β-fluorescein isothiocyanate-conjugated antibody fluoresce green, and cells labeled with tubulin-Texas Red-conjugated antibody fluoresce red. Colocalization of GSK-3β and tubulin appears yellow in the merged image. Nuclei were visualized by 4,6-diamino-2-phenylindole (DAPI) staining. A representative section is shown at 400× magnification. B, isolated nuclei from control cells (white peak) and heat shock-treated cells (gray peak) were fixed, immunofluorescently stained with GSK-3β and stained with Hoechst 33342, and analyzed by flow cytometry as described under “Experimental Procedures.” C, control and heat shock-treated (HS) cells were incubated with 100 nM okadaic acid (OA) 1 h prior to heat shock treatment or with 10 ng/ml leptomycin B (LB) 2.5 h prior to heat shock treatment. The nuclear extracts were immunoblotted for GSK-3β.
To examine the temporal relationship between heat shock-induced accumulation of nuclear GSK-3β and activation of apoptosis, the time-dependent changes in heat shock-induced nuclear GSK-3β accumulation and activation of caspase-9 and caspase-3, and the cleavage of PARP were measured. The nuclear level of GSK-3β was increased within 7.5 min of incubation of cells at 45 °C (Fig. 5, A and B), and a maximum level of 277% of control was attained after 30 min of incubation at 45 °C. Upon incubation at 37 °C, there was a gradual decline in nuclear GSK-3β, which returned to near control levels after 240 min. Heat shock-induced caspase-9 activity was detected 30 min after heat shock treatment, considerably after the accumulation of nuclear GSK-3β had commenced. Similarly caspase-3 activation and PARP proteolysis were also detected after 30 min of heat shock treatment.
FIG. 5. Heat shock- and staurosporine-induced GSK-3β nuclear accumulation precedes activation of caspase-9 and caspase-3.
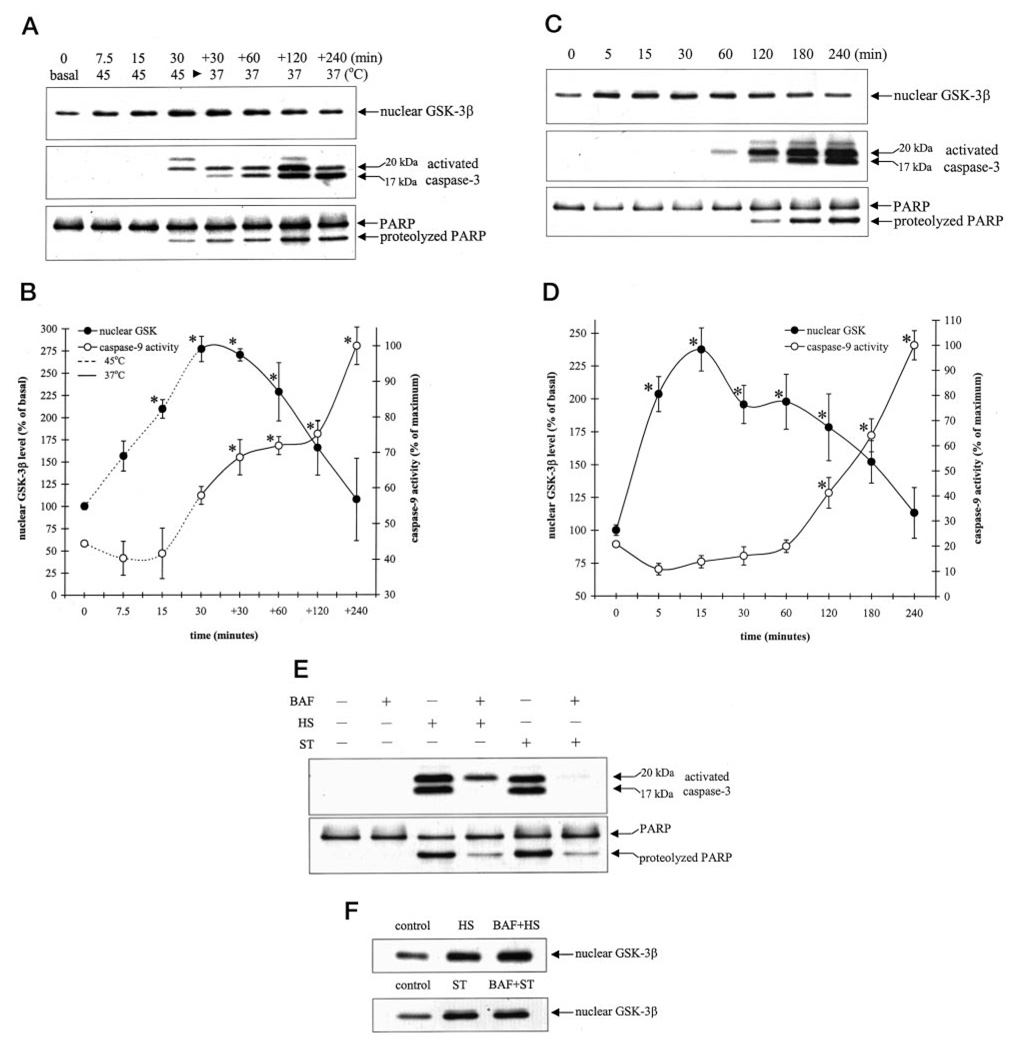
Cells were subjected to heat shock at 45 °C for 7.5, 15, and 30 min or subjected to heat shock for 30 min and then transferred to a 37 °C chamber for an additional (+) 30, 60, 120, and 240 min (A and B) or treated with 0.5 µm staurosporine for 5, 15, 30, 60, 120, 180, and 240 min (C and D). Nuclear extracts were immunoblotted for GSK-3β, and total cell lysates were immunoblotted for activated caspase-3 and PARP proteolysis (A and C) or were used to measure caspase-9 activity (B and D) as described under “Experimental Procedures.” Nuclear GSK-3β protein bands were quantitated by densitometer. Means ± S.E., n = three experiments; *, p < 0.05 (ANOVA) compared with untreated cells. E, cells were treated with 100 µm BAF 1 h prior to treatment with 30-min heat shock (HS), followed by incubation at 37 °C for 4 h or with 0.5 µm staurosporine (ST) for 4 h. Total cell lysates were immunoblotted for activated caspase-3 and PARP. F, cells were treated with 100 µm BAF 1 h prior to treatment with 30 min of HS followed by incubation at 37 °C or with 0.5 µm ST for 15 min. Nuclear extracts were immunoblotted for GSK-3β.
Staurosporine is a widely used agent to induce apoptosis in many types of cells, including SH-SY5Y cells (23, 32, 33). Treatment with staurosporine (0.5 µM) caused a rapid increase in nuclear GSK-3β (Fig. 5, C and D), resulting in a 2-fold increase in nuclear GSK-3β levels within 5 min of treatment and a maximal increase after 15 min of treatment. Whereas increased nuclear GSK-3β was evident within 5 min of staurosporine treatment, the earliest activation of caspase-9 and caspase-3 and proteolysis of PARP was detected between 60 and 120 min of staurosporine treatment. These results demonstrate that the intracellular distribution of GSK-3β is a dynamic process regulated by heat shock and staurosporine and that it precedes activation of caspase-9 and caspase-3.
The results with heat shock and staurosporine indicated that nuclear accumulation of GSK-3β preceded activation of caspases. To test this further, cells were preincubated with the broad-spectrum caspase inhibitor Boc-Asp(OMe)-fluoromethyl ketone (BAF) (100 µM) 1 h prior to treatment with heat shock or staurosporine. Although pretreatment with BAF markedly reduced heat shock- and staurosporine-induced activation of caspase-3 and PARP proteolysis (Fig. 5E), BAF pretreatment did not affect heat shock- or staurosporine-induced accumulation of nuclear GSK-3β (Fig. 5F). These results indicate that the nuclear accumulation of GSK-3β is an early event caused by apoptosis-stimulating agents that occurs upstream of caspase activation.
GSK-3β Nuclear Accumulation Is Independent of Ser9 and Tyr216 Phosphorylation
GSK-3β activity is inhibited by phosphorylation at Ser9. To test whether Ser9 phosphorylation regulates GSK-3β nuclear accumulation, cells were incubated without or with LY294002 and then subjected to serum withdrawal, heat shock, or staurosporine. Serum withdrawal and staurosporine treatment both decreased Ser9 phosphorylation of cytosolic GSK-3β (Fig. 6A), and this phosphorylation was further decreased in LY294002 pretreated cells. There was a concomitant increase in nuclear GSK-3β, and nuclear Ser9- phosphorylated GSK-3β was nearly undetectable after serum withdrawal or staurosporine treatments, suggesting that reduced Ser9 phosphorylation may facilitate nuclear accumulation of GSK-3β. In contrast, heat shock treatment caused a marked increase in the nuclear levels of phospho-Ser9 GSK-3β and total GSK-3β, and although LY294002 treatment reduced the nuclear level of phospho-Ser9 GSK-3β after heat shock treatment, the nuclear level of total GSK-3β was not decreased. These results indicate that Ser9 phosphorylation of GSK-3β is unlikely to regulate the nuclear accumulation of GSK-3β.
FIG. 6. GSK-3β nuclear accumulation is independent of Ser9 and tyrosine phosphorylation.
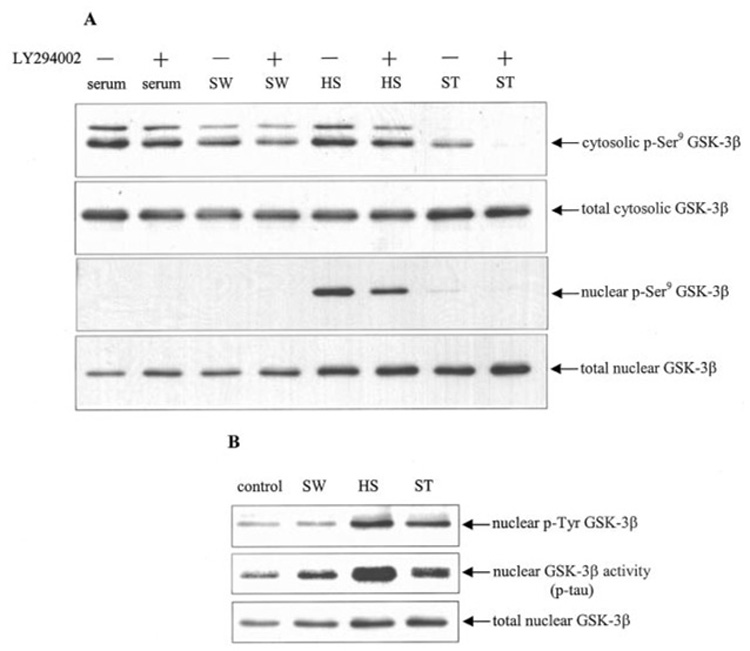
Cells were subjected to 1 h of serum withdrawal (SW), 30 min of heat shock (HS) and incubation at 37 °C for 30 min, or 0.5 µm staurosporine (ST) for 15 min. Where indicated cells were pretreated for 30 min with 20 µm LY294002. A, cytosolic and nuclear extracts were immunoblotted for p-Ser9 GSK-3β and total GSK-3β. B, to measure tyrosine phosphorylation of GSK-3β, nuclear extracts were immunoprecipitated with anti-phosphotyrosine antibody (PY20) and then immunoblotted for GSK-3β to detect tyrosine-phosphorylated (p-Tyr) GSK-3β. For activity measurements, GSK-3β was immunoprecipitated from nuclear extracts, and GSK-3β activity was measured using recombinant tau protein as substrate as described under “Experimental Procedures,” and the immunoprecipitated nuclear GSK-3β was immunoblotted. A representative result from three experiments is shown.
Phosphorylation of Tyr216 increases GSK-3β activity (34), and Tyr216 phosphorylation was reported recently to be increased in the nucleus during staurosporine-induced apoptosis (20). Serum withdrawal increased nuclear GSK-3β levels but did not change the level of phosphotyrosine GSK-3β. However, heat shock and staurosporine treatments caused increases in phosphotyrosine GSK-3β levels (Fig. 6B), equivalent to the nuclear accumulation of GSK-3β. All three treatments increased the activity of GSK-3β in the nucleus equivalent to the nuclear accumulation of GSK-3β. Taken together, these results indicate that nuclear accumulation of GSK-3β is independent of phosphorylations that regulate GSK-3β activity.
Lithium Induces Nuclear Accumulation of Cyclin D1
Nuclear GSK-3β phosphorylates and induces the nuclear export of the cell cycle-associated protein cyclin D1 (12), a protein that regulates the G1/S transition. To examine whether heat shock-induced accumulation of nuclear GSK-3β affected cyclin D1, cells were treated with heat shock and lithium to inhibit GSK-3β activity. Lithium treatment alone did not affect the nuclear level of cyclin D1 (Fig. 7, A and B). However, lithium concentration-dependently increased the heat shock-induced nuclear accumulation of cyclin D1 without affecting the heat shock-induced increase in nuclear GSK-3β level. These results indicate that nuclear accumulation of GSK-3β contributes to the regulation of nuclear cyclin D1 levels.
FIG. 7. Lithium treatment enhances heat shock-induced nuclear accumulation of cyclin D1.
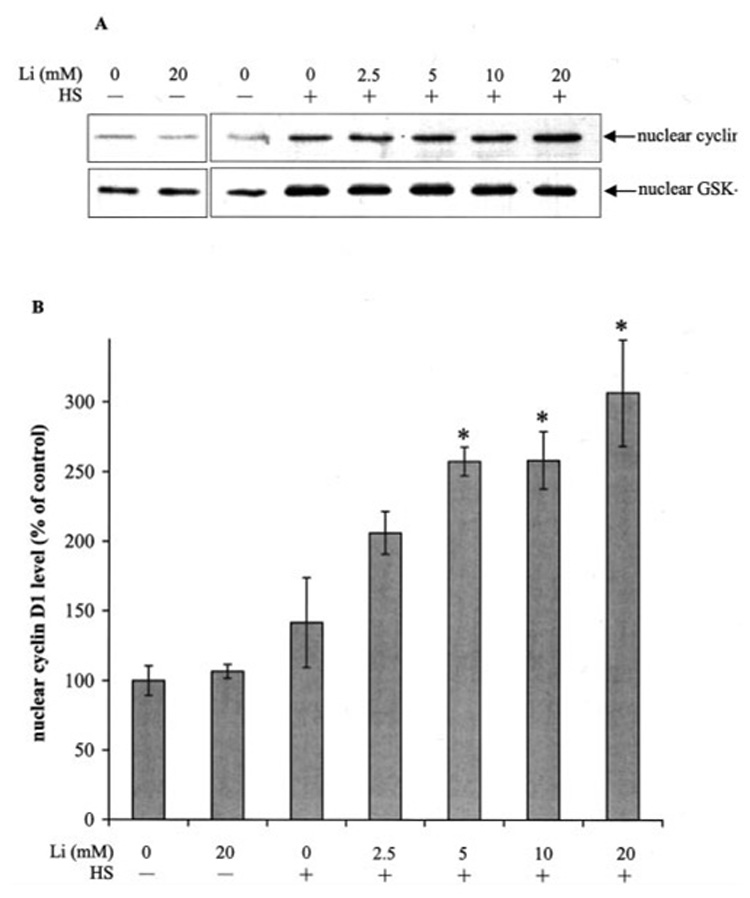
Cells were incubated with 0 or 20 mm lithium for 90 min or pretreated for 30 min with 0, 2.5, 5, 10, and 20 mm lithium and then treated with 30 min heat shock (HS), followed by incubation in a 37 °C chamber for 30 min. Nuclear extracts were immunoblotted for cyclin D1 and GSK-3β (A), and the cyclin D1 protein bands were quantitated by densitometer (B). Means ± S.E., n = three experiments; *, p < 0.05 (ANOVA) compared with cells treated with heat shock alone.
DISCUSSION
In addition to the well known regulation of GSK-3β by Ser9 and Tyr216 phosphorylation, the results of the present study demonstrated that GSK-3β also can be regulated by redistribution within the cell. Three conditions known to induce apoptosis, serum withdrawal, heat shock, and staurosporine, all increased the nuclear accumulation of GSK-3β, and this redistribution appeared to be independent of site-specific phosphorylation of GSK-3β. In addition, the nuclear accumulation of GSK-3β induced by the apoptotic stimuli of heat shock and staurosporine preceded activation of caspase-9 and caspase-3, an observation that may be relevant for apoptotic signaling cascades, because GSK-3β has previously been shown to contribute to the apoptotic signaling pathways induced by these treatments (22, 23, 25). Thus, in addition to the regulation of GSK-3β by phosphorylation, the intracellular distribution of GSK-3β is a dynamic process that also likely contributes to regulating the function of GSK-3β by controlling its accessibility to certain substrates.
In light of recent findings that GSK-3β activity contributes to apoptotic signaling (reviewed in Ref. 1) it was particularly relevant to find that the apoptotic paradigms that are facilitated by increased GSK-3β activity, serum withdrawal, heat shock, and staurosporine, all induced GSK-3β nuclear accumulation. The intranuclear accumulation of GSK-3β was confirmed by inhibition of the nuclear transport machinery, as inhibition of nuclear import mechanisms inhibited the heat shock-induced nuclear accumulation of GSK-3β, and inhibition of the nuclear export-associated protein, CRM1, increased GSK-3β nuclear accumulation. One of the consequences of apoptosis is the disruption of the nuclear membrane by the action of caspases, thereby allowing the diffusion of cytosolic proteins in the nucleus (35). However, GSK-3β nuclear accumulation occurred prior to heat shock- and staurosporine-induced activation of caspase-9 and caspase-3. Furthermore, the broad-spectrum caspase inhibitor, BAF, blocked caspase activation but did not prevent GSK-3β nuclear accumulation. These results indicate that the increase in nuclear GSK-3β levels precedes activation of the caspase cascade and therefore is independent of caspase activity and is an early event in apoptosis. Overall, these results demonstrate that the level of GSK-3β in the nucleus is dynamic and is subject to regulation by treatments that cause apoptosis. Because inhibition of GSK-3β attenuates apoptosis (reviewed in Ref. 28), the rapid accumulation of GSK-3β in the nucleus caused by apoptotic stimuli raises the possibility that phosphorylation of nuclear substrates by GSK-3β contributes, in part, to its facilitory effects on apoptosis. For example, nuclear GSK-3β phosphorylates, and thereby inhibits, the cell survival-promoting transcription factors heat shock factor-1 (14–16), and cAMP response element-binding protein (17, 36). Therefore, it is intriguing to speculate that inhibition of the anti-apoptotic actions of nuclear substrates of GSK-3β may facilitate apoptosis.
Although GSK-3β activity is regulated by Ser9 and Tyr216 phosphorylation, the results presented here indicate that nuclear accumulation of GSK-3β caused by serum withdrawal, heat shock, and staurosporine was independent of phosphorylation at these sites. For example, heat shock and staurosporine had opposite effects on Ser9 phosphorylation of GSK-3β, yet caused similar increases in the nuclear level of GSK-3β. Furthermore, the heat shock-, and staurosporine-induced increases in nuclear tyrosine phosphorylated GSK-3β were equivalent to the increases in total GSK-3β in the nucleus, indicating a lack of preferential nuclear accumulation of phospho-Tyr216-GSK-3β. Additionally, the magnitudes of the increases in nuclear GSK-3β activity were equivalent to the increases in nuclear GSK-3β levels. These results indicate that the recently reported increases in nuclear phospho-Tyr216-GSK-3β following treatments that induce apoptosis (20) likely reflect increased nuclear levels of GSK-3β rather than selective nuclear import of phospho-Tyr216-GSK-3β. Overall, these results suggest that Ser9 and Tyr216 phosphorylation do not regulate GSK-3β nuclear transport. Because these results demonstrate that the intracellular distribution of GSK-3β is a dynamic, regulated process, further investigations will be directed toward identifying the mechanisms responsible for nuclear import and export of GSK-3β.
Nuclear GSK-3β has previously been shown to phosphorylate cyclin D1, which induces the export of cyclin D1 from the nucleus (12). To test whether nuclear GSK-3β accumulation after heat shock contributes to the regulation of nuclear cyclin D1 levels, the effect on cyclin D1 levels of inhibiting nuclear GSK-3β activity was tested using lithium, a selective inhibitor of GSK-3β (24). The paradigm of transiently treating cells with heat shock is particularly well suited for testing the regulation of cell cycle-associated proteins, such as cyclin D1, because short term exposure to heat shock induces a transient cell cycle arrest (reviewed in Ref. 37) and apoptosis. Treatment with lithium concentration-dependently increased nuclear cyclin D1 levels following heat shock, indicating that nuclear GSK-3β phosphorylated and facilitated the export of cyclin D1 from the nucleus in the absence of lithium. This action of lithium may contribute to its reported neuroprotective effects (1) by facilitating cell cycle arrest, thereby supporting recovery from potentially lethal stress.
In summary, the results reported here demonstrate that stimuli that activate apoptotic signaling cascades that are known to be facilitated by GSK-3β induce a redistribution of GSK-3β from the cytosol to the nucleus early in the apoptotic process. These results indicate that besides site-specific phosphorylation, there is a spatial regulatory component associated with GSK-3β activity during apoptosis.
Acknowledgments
We thank Dr. Minoru Yoshida for the kind gift of leptomycin B.
Footnotes
This work was supported by grants from the Alzheimer’s Association and by National Institutes of Health Grant MH38752.
The abbreviations used are: GSK-3β, glycogen synthase kinase-3β; ANOVA, analysis of variance; BAF, Boc-Asp(OMe)-fluoromethyl ketone; IGF-1, insulin-like growth factor-1; PARP, poly(ADP-ribose) polymerase; PBS, phosphate-buffered saline; PI3K, phosphatidylinositol 3-kinase; PIPES, 1,4-piperazinediethanesulfonic acid.
REFERENCES
- 1.Grimes CA, Jope RS. Prog. Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 2.Wang QM, Fiol CJ, DePaoli-Roach AA, Roach PJ. J. Biol. Chem. 1994;269:14566–14574. [PubMed] [Google Scholar]
- 3.Lesort M, Jope RS, Johnson GV. J. Neurochem. 1999;72:576–584. doi: 10.1046/j.1471-4159.1999.0720576.x. [DOI] [PubMed] [Google Scholar]
- 4.Hartigan JA, Johnson GV. J. Biol. Chem. 1999;274:21395–21401. doi: 10.1074/jbc.274.30.21395. [DOI] [PubMed] [Google Scholar]
- 5.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Curr. Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 6.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 7.Goode N, Hughes K, Woodgett JR, Parker PJ. J. Biol. Chem. 1992;267:16878–16882. [PubMed] [Google Scholar]
- 8.Sutherland C, Leighton IA, Cohen P. Biochem. J. 1993;296:15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang X, Yu SX, Lu Y, Bast RC, Jr, Woodgett JR, Mills GB. Proc. Natl. Acad. Sci. U. S. A. 2000;97:11960–11965. doi: 10.1073/pnas.220413597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyle WJ, Smeal T, Defize LH, Angel P, Woodgett JR, Karin M, Hunter T. Cell. 1991;64:573–584. doi: 10.1016/0092-8674(91)90241-p. [DOI] [PubMed] [Google Scholar]
- 11.Hemmings BA, Yellowlees D, Kernohan JC, Cohen P. Eur. J. Biochem. 1981;119:443–451. doi: 10.1111/j.1432-1033.1981.tb05628.x. [DOI] [PubMed] [Google Scholar]
- 12.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Science. 1997;275:1930–1934. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- 14.He B, Meng YH, Mivechi NF. Mol. Cell. Biol. 1998;18:6624–6633. doi: 10.1128/mcb.18.11.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bijur GN, Jope RS. J. Neurochem. 2000;75:2401–2408. doi: 10.1046/j.1471-4159.2000.0752401.x. [DOI] [PubMed] [Google Scholar]
- 16.Xavier IJ, Mercier PA, McLoughlin CM, Ali A, Woodgett JR, Ovsenek N. J. Biol. Chem. 2000;275:29147–29152. doi: 10.1074/jbc.M002169200. [DOI] [PubMed] [Google Scholar]
- 17.Bullock BP, Habener JF. Biochemistry. 1998;37:3795–3809. doi: 10.1021/bi970982t. [DOI] [PubMed] [Google Scholar]
- 18.Ragano-Caracciolo M, Berlin WK, Miller MW, Hanover JA. Biochem. Biophys. Res. Commun. 1998;249:422–427. doi: 10.1006/bbrc.1998.9159. [DOI] [PubMed] [Google Scholar]
- 19.Haq S, Choukroun G, Kang ZB, Ranu H, Matsui T, Rosenzweig A, Molkentin JD, Alessandrini A, Woodgett J, Hajjar R, Michael A, Force T. J. Cell Biol. 2000;151:117–130. doi: 10.1083/jcb.151.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM. Proc. Natl. Acad. Sci. U. S. A. 2000;97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K. Proc. Natl. Acad. Sci. U. S. A. 1993;90:7789–7793. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pap M, Cooper GM. J. Biol. Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 23.Bijur GN, De Sarno P, Jope RS. J. Biol. Chem. 2000;275:7583–7590. doi: 10.1074/jbc.275.11.7583. [DOI] [PubMed] [Google Scholar]
- 24.Klein PS, Melton DA. Proc. Natl. Acad. Sci. U. S. A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hetman M, Cavanaugh JE, Kimelman D, Xia Z. J. Neurosci. 2000;20:2567–2574. doi: 10.1523/JNEUROSCI.20-07-02567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crowder RJ, Freeman RS. J. Biol. Chem. 2000;275:34266–34271. doi: 10.1074/jbc.M006160200. [DOI] [PubMed] [Google Scholar]
- 27.Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S. J. Neurochem. 1999;73:578–586. doi: 10.1046/j.1471-4159.1999.0730578.x. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Bijur GN, Jope RS. Bipolar Disord. 2001 doi: 10.1034/j.1399-5618.2002.40201.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jope RS, Bijur GN. Mol. Psychiatry. 2001 doi: 10.1038/sj.mp.4001017. in press. [DOI] [PubMed] [Google Scholar]
- 30.Kehlenbach RH, Gerace L. J. Biol. Chem. 2000;275:17848–17856. doi: 10.1074/jbc.M001455200. [DOI] [PubMed] [Google Scholar]
- 31.Kudo N, Wolff B, Sekimoto T, Schreiner EP, Yoneda Y, Yanagida M, Horinouchi S, Yoshida M. Exp. Cell Res. 1998;242:540–547. doi: 10.1006/excr.1998.4136. [DOI] [PubMed] [Google Scholar]
- 32.Posmantur R, McGinnis K, Nadimpalli R, Gilbertsen RB, Wang KK. J. Neurochem. 1997;68:2328–2337. doi: 10.1046/j.1471-4159.1997.68062328.x. [DOI] [PubMed] [Google Scholar]
- 33.Chakravarthy BR, Walker T, Rasquinha I, Hill IE, MacManus JP. J. Neurochem. 1999;72:933–942. doi: 10.1046/j.1471-4159.1999.0720933.x. [DOI] [PubMed] [Google Scholar]
- 34.Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR. EMBO J. 1993;12:803–808. doi: 10.1002/j.1460-2075.1993.tb05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faleiro L, Lazebnik Y. J. Cell Biol. 2000;151:951–959. doi: 10.1083/jcb.151.5.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grimes CA, Jope RS. J. Neurochem. 2001 in press. [Google Scholar]
- 37.Kuhl NM, Rensing L. Cell. Mol. Life Sci. 2000;57:450–463. doi: 10.1007/PL00000707. [DOI] [PMC free article] [PubMed] [Google Scholar]