

Summary
Beta-site APP-cleaving enzyme (BACE) is required for production of the Alzheimer's disease (AD)-associated Aβ protein. BACE levels are elevated in AD brain, and increasing evidence reveals BACE as a stress-related protease that is upregulated following cerebral ischemia. However, the molecular mechanism responsible is unknown. We show that increases in BACE and β-secretase activity are due to post-translational stabilization following caspase activation. We also found that during cerebral ischemia, levels of GGA3, an adaptor protein involved in BACE trafficking, are reduced, while BACE levels are increased. RNAi silencing of GGA3 also elevated levels of BACE and Aβ. Finally, in AD brain samples, GGA3 protein levels were significantly decreased and inversely correlated with increased levels of BACE. In summary, we have elucidated a novel GGA3-dependent mechanism regulating BACE levels and β-secretase activity. This mechanism may explain increased cerebral levels of BACE and Aβ following cerebral ischemia and in AD.
Introduction
A key neuropathological event in Alzheimer's disease (AD) is the cerebral accumulation of an ∼4kDa peptide termed Aβ, the principle component of senile plaques. The Aβ peptide is derived by serial proteolysis of APP by β-secretase at the N-terminus followed by γ-secretase at the C-terminus (De Strooper and Annaert, 2000). β-secretase has been identified as a novel membrane-tethered member of the aspartyl proteases, termed BACE (Sinha et al., 1999; Vassar et al., 1999). BACE is an N-glycosylated type 1 transmembrane protein that undergoes constitutive N-terminal processing in the Golgi apparatus. The ectodomain contains four glycosylation sites and two signature sequences typically associated with aspartyl proteases (DT/SGT/S). BACE is targeted through the secretory pathway to the plasma membrane where it can be internalized to endosomes (Citron, 2004). The BACE- C-terminal fragment (CTF) contains a specific di-leucine (DXXLL) sorting signal that is present in several transmembrane proteins (e.g. cation-dependent and cation-independent mannose-6-phosphate receptor, CD- and CI-MRP) and that regulates endocytosis and ultimately lysosomal degradation (Bonifacino and Traub, 2003). Mutagenesis of LL to AA results in retention of BACE at the plasma membrane (Huse et al., 2000; Pastorino et al., 2002). Furthermore, the di-leucine motif may play a role in BACE degradation since BACELL/AA mutations increase protein levels of BACE (Pastorino et al., 2002). More recently, we reported that BACE is normally degraded in lysosomes, and that mutagenesis of the di-leucine motif in the BACE CTF prevents accumulation of BACE in the lysosomes following inhibition of lysosomal hydrolases (Koh et al., 2005). The BACE acidic di-leucine motif has been shown to bind GGA1, 2, and 3 (Golgi-localized γ-ear-containing ARF binding proteins) and phosphorylation of BACE-S498 appears to increase their binding (He et al., 2002; He et al., 2003; Shiba et al., 2004; von Arnim et al., 2004; Wahle et al., 2005). GGA1, 2, and 3 are monomeric adaptors that are recruited to the trans-Golgi network by the Arf1-GTPase. They consist of four distinct segments: a VHS (VPS27, Hrs, and STAM) domain that binds the acidic di-leucine sorting signal, DXXLL; a GAT (GGA and Tom1) domain which binds Arf:GTP; a hinge region which recruits clathrin; and a GAE (gamma-adaptin ear homology) domain which exhibits sequence similarity to the ear region of γ-adaptin and recruits a number of accessory proteins. GGAs are necessary for the sorting of acid hydrolases to the lysosomes. Newly synthesized acid hydrolases modified with mannose 6-phosphate groups bind to mannose 6-phosphate receptors (MPRs). MPRs bind to the VHS domain of GGAs via the DXXLL motif (Bonifacino, 2004). GGAs are likely involved in the transport from the Golgi complex to the endosome of proteins containing the DXXLL signal. However, Puertollano et al. (Puertollano and Bonifacino, 2004) have recently reported that the GGA3 GAT domain binds ubiquitin, and that ubiquinated GGA3 is necessary for the delivery of activated epithelial growth factor receptor (EGFR) to lysosomes. RNAi silencing of GGA3, but not GGA1 or GGA2, resulted in the accumulation of EGFRs in enlarged early endosomes and partially blocked their delivery to lysosomes where they are normally degraded. These findings indicate that GGAs may be involved in the delivery of endosomal cargoes to lysosomes. Recently, it has also been shown (He et al., 2005) that RNAi silencing of GGA1, 2, and, 3 significantly increases the levels of BACE in endosomes.
Increasing evidence suggest that BACE is a stress-induced protease. BACE levels increase in cells exposed to oxidative stress (Tamagno et al., 2002; Tamagno et al., 2003; Tamagno et al., 2005; Tong et al., 2005), in in vivo animal models following traumatic brain injury (TBI) (Blasko et al., 2004), cerebral ischemia (Wen et al., 2004) and impaired energy metabolism (Velliquette et al., 2005), and in AD brains (Fukumoto et al., 2002; Holsinger et al., 2002; Li et al., 2004a; Tyler et al., 2002; Yang et al., 2003). The mechanisms underlying such increases remain unknown. In the current study, we report that BACE protein levels and consequently, β-secretase cleavage of APP, are potentiated during apoptosis. In exploring the mechanism underlying this novel apoptotic event, we found that the elevation in BACE following caspase activation is due to post-translational stabilization owing to a significant impairment in the degradation and turnover of BACE. Given our previous findings that BACE is normally degraded in the lysosomes and since the C-terminal di-leucine motif is required for trafficking to lysosomes, we next investigated the fate of GGA3 during apoptosis. We discovered that GGA3 is a novel caspase 3 substrate that is cleaved during apoptosis. This was shown both in vitro, in cell cultures, and in vivo, in a rat model of cerebral ischemia. Moreover, we observed that as GGA3 is removed by caspase cleavage, BACE is stabilized leading to elevated β-secretase cleavage of APP. In further support of our hypothesis that GGA3 plays a key role in BACE degradation, we show that RNAi silencing of GGA3 increases levels of BACE, APP-C99, and Aβ. Finally, to begin to investigate whether decreased GGA3 protein levels may account for increased BACE levels and β-secretase activity in AD brains (Fukumoto et al., 2002; Holsinger et al., 2002; Li et al., 2004a; Tyler et al., 2002; Yang et al., 2003), we measured levels of BACE and GGA3 in a series of AD and non-demented (ND) control brain samples. While BACE protein levels were significantly increased in the AD temporal cortex, in contrast, GGA3 protein levels were significantly decreased and were inversely correlated with BACE levels in the AD group, but not in the ND control group. These data suggest that depletion of GGA3 is responsible for enhanced BACE levels and β-secretase activity during ischemia and in AD brain.
Results
Caspase activation increases BACE and APP-C99 levels
Apoptosis has been shown to enhance Aβ levels in neuronal and non-neuronal cells (Barnes et al., 1998; Galli et al., 1998; Gervais et al., 1999; Guo et al., 2001; LeBlanc, 1995; Sodhi et al., 2004; Tesco et al., 2003). Gervais and colleagues (Gervais et al., 1999) proposed that caspase-mediated cleavage of APP at APPD720 was responsible for increased Aβ generation associated with apoptosis. We subsequently showed that increased levels of Aβ following induction of apoptosis in CHO cells, did not require caspase-mediated cleavage of APP at either its C-terminal (D720) and/or N-terminal caspase sites (Tesco et al., 2003) (Fig. 1A). In that same study we reported that 6 hr treatment with STS significantly increased Aβ1-total production compared to untreated cells. The Aβ ELISA measured Aβ1-total using biotinylated 3D6, which specifically recognizes the aspartate in position 1 of Aβ, as a reporter antibody. The ELISA data demonstrated that the increase in Aβ following caspase activation is exclusively due to β-secretase cleavage at position 1 and does not involve other Aβ species starting at other residues (e.g. at position 2 as expected from a caspase cleavage in the β-secretase region). Here, we investigated whether apoptosis-mediated increases in Aβ levels may be due to enhanced cleavage of APP by BACE, the protease responsible for β-secretase cleavage of APP (Vassar et al., 1999). For this purpose, we tested whether apoptosis/caspase activation leads to increased levels of APP-C99, the C-terminal fragment generated by β-secretase cleavage of APP. Western blot (WB) analysis with the antibody WO2, (raised against amino acids 1-17 of Aβ region), revealed that apoptosis induced by staurosporine (STS) resulted in increased levels of C99 in H4 human neuroglioma cell lines expressing APP751 (H4-APP751) (Fig. 1B). Western blot analysis with an antibody recognizing the caspase 3 active fragment showed that caspase 3 activation occurs as early as 6 hr following treatment with STS. It should be noted that we also observed a small increase of APP-C99 and activated caspase fragment in untreated cells after 12 and 24 hr, most likely due to marginal caspase activity triggered by serum deprivation, which is known to activate caspases in cells of neuronal origin. The loading control for caspase 3 and WO2 blots is the same as that shown in Fig. 1D.
Fig. 1. Caspase activation increases BACE and APP-C99 levels.
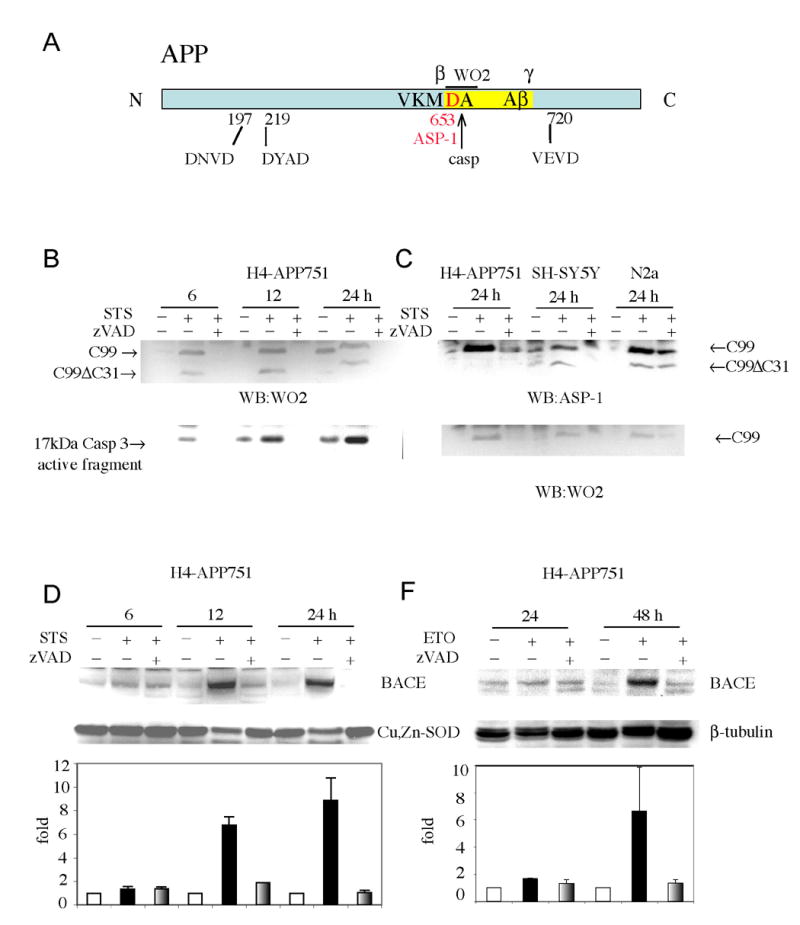
A: Schematic representation of APP caspase sites, and antibody recognition sites. B: Apoptosis was induced in human H4 neuroglioma cells expressing APP751 by STS treatment. Western blot analysis performed with WO2 antibody revealed APP-C99 and APP-C99ΔC31 during time-course experiments. Western blot analysis with anti-caspase 3 active fragment antibody showed caspase 3 activation as early as 6 hr during STS treatment. Limited caspase activity and a small increase of APP-C99 in the untreated cells at time point 12 (horizontally compressed band) and 24 hr was also detected in the control sample owing to the fact that the control cells were grown in serum-free media. C: WB analysis with an antibody, ASP-1 (Oncogene), that recognizes only the first aspartyl residue of Aβ region confirmed that the 12kDa APP fragment increasing following caspase activation is APP-C99 in the cells H4-APP751, and also in human SH-SY5Y and murine N2A cells expressing only endogenous APP. D-F: Western blot analysis with anti-BACE antibody revealed increased BACE protein levels following apoptosis induced by STS or etoposide treatment, respectively. Cu,Zn-SOD or β-tubulin were used as a loading control. Densitometry analysis was performed using NIH image software. The graphs represent BACE levels expressed as percentage increase versus BACE levels in control cells (100%). Each bar represents the mean ± SEM of at least three experiments.
A 6.5 kDa fragment corresponding to caspase cleavage at D720 (APP-C99ΔC31) was also detected in apoptotic H4-APP751 as previously described (Tesco et al., 2003). These data agree with those of LeBlanc and colleagues (LeBlanc et al., 1999) who reported that the generation of a 6.5 kDa fragment precedes the increased production of both secreted and intracellular Aβ in apoptotic neurons. However, LeBlanc et al. (1999) proposed that the 6.5 kDa fragment was generated by caspase-mediated cleavage at D653 (Fig. 1A). A caspase 6-like site (VKMD) has been identified at D653 in the β-secretase region of APP (Gervais et al., 1999; LeBlanc et al., 1999) and it has been reported that the APP Swedish mutation KM/NL improves the likelihood that caspase 6 can cleave at this site (Gervais et al., 1999). Caspase cleavage between D653 and A654 is expected to generate a novel APP-CTF that could be termed C98 starting with A654. In contrast, β-secretase generates APP-C99 starting with D653. Western Blot analysis with an antibody, ASP-1 (Oncogene), that recognizes only the first aspartyl residue of Aβ region confirmed that the 12 kDa APP fragment increasing following caspase activation is APP-C99. This was shown to be the case in the cells H4-APP751, and also in human SH-SY5Y and murine N2A cells expressing only endogenous APP (Fig. 1C).
We next asked whether the increased levels of APP-C99 was due to increased levels of endogenous BACE in the H4-APP751 cells. We found that BACE protein levels were increased ∼6-fold and ∼9 fold after 12 or 24 hr of STS treatment, respectively (Fig. 1D). Next, to confirm that increased BACE protein levels were the result of caspase activation, we used a different apoptotic inducer, etoposide. Apoptosis induced by etoposide also increased BACE protein levels, albeit with a different time course (Fig. 1F). Inhibition of caspase activity by zVAD prevented BACE accumulation in both treatments (Fig. 1D-F). As a control for protein loading, the same blots were reprobed with anti-Cu, Zn-SOD and anti-β-tubulin antibodies, respectively. We chose to use unrelated proteins for which sensitive antibodies were available (and worked best), as loading controls, for each set of experiments. The use of two different loading controls serves to confirm our findings. Using the anti-BACE antibody currently available, detection of steady state levels of BACE is not as robust as detection of steady state levels of APP-C99. Thus, the apparent discrepancy between the increase in C99 levels at 6 hr and absence of increase of BACE levels is most likely due to the difference in the sensitivity of the antibodies used for detection of BACE versus APP-C99. However, we cannot rule out the possibility that generation of the C99 fragment earlier (6 hr) in the STS model is BACE-independent, but caspase-dependent. These findings were also confirmed in CHO cells expressing BACE and APP 751 and in primary neuronal cultures obtained from E15 embryos of transgenic mice expressing human APP with the “Swedish” mutation (KM670/671NL) (SWE) (data not shown). These results indicate that caspase activation increases protein levels of BACE and potentiates β-secretase-mediated processing of APP in neuronal and non-neuronal cell cultures.
Caspase activation increases the half-life of BACE
We next asked whether apoptosis increases protein levels of BACE via increased gene expression. BACE mRNA levels were not increased, but were virtually undetectable after only 6 hr of STS treatment in the H4-APP751 cells (Fig. 2A). The preservation of 28S and 18S ribosomal RNA ruled out non-specific RNA degradation (Fig. 2A). This led us to ask whether BACE protein levels were increased during apoptosis due to decreased degradation.
Fig. 2. Caspase activation increases the half-life of BACE.
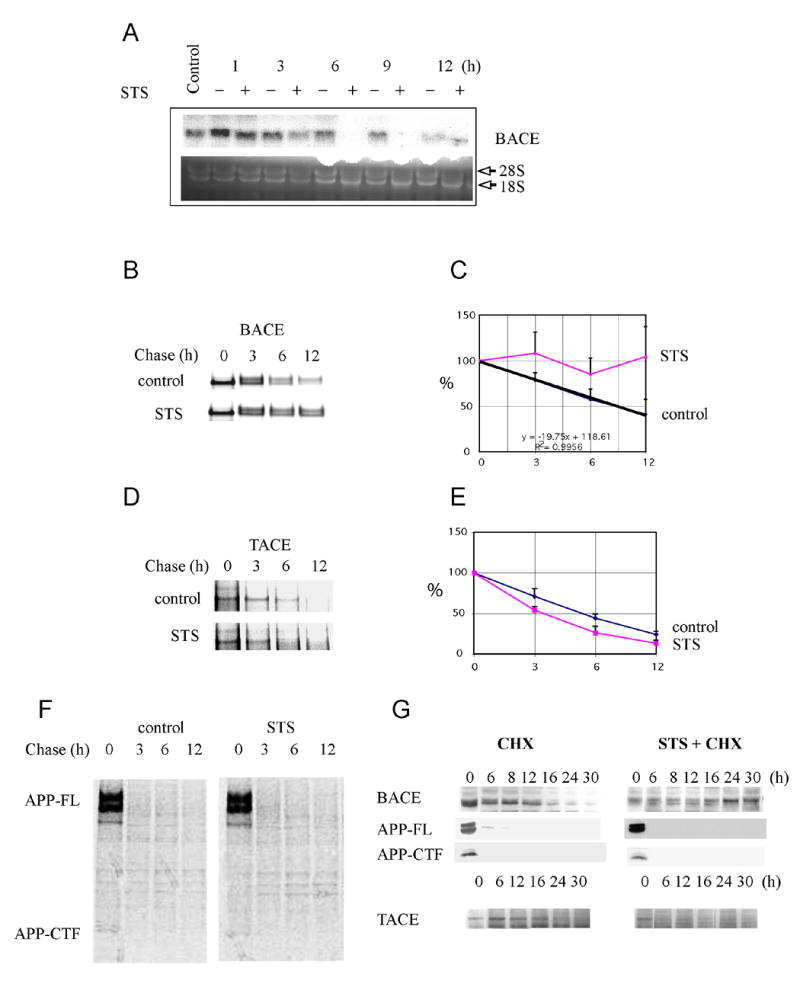
A: Northern Blot analysis: H4-APP751 cells were treated with STS during time course experiments. Total RNA was extracted, and 20 μg of the resulting RNA were analyzed by northern blotting with BACE cDNA. Ethidium staining of the gel (lower panel) confirmed equal RNA loading and absence of non-specific degradation. B: Pulse-chase analysis. H4-APP751 cells were transiently transfected with BACE-Myc cDNA and were metabolically labeled after 24 hr. Lysates from each time point were immunoprecipitated with anti-Myc antibody. C: Protein amounts were quantified by phosphorimager and represented in the graph. The mean ± SEM of at least three experiments is shown. D-E-F: For the TACE and APP pulse-chase, H4-APP751 cells were metabolically labeled. Lysates from each time point were immunoprecipitated wither with anti-TACE or anti-APP (A8717) antibody. Protein amounts were quantified by phosphorimager and represented in the graph. The mean ± SEM of at least three experiments is shown. D: Cycloheximide degradation time-course: BACE, TACE, and APP proteins were detected by Western blot at various times after addition of CHX (40 μg/ml) only or STS + CHX in H4-APP751 cells. The degradation of BACE was decreased while the degradation of TACE and APP was unchanged during the apoptosis.
Pulse-chase analyses revealed that the approximate half-life of BACE was ∼9 hr under normal conditions. However, following caspase activation, BACE levels did not significantly decrease (Fig. 2B-C). In contrast, the half-lives of tumor necrosis factor-alpha (TNF) converting enzyme (TACE) (Fig. 2D-E) and full-length APP (and APP-CTFs) (Fig. 2F) were not increased during apoptosis. In confirmation of these findings, the approximate half-life of endogenous BACE was roughly 10 hr under normal conditions in cycloheximide (CHX) time course experiments. However, following caspase activation, BACE levels did not significantly decrease even after 30 hr into the time-course. Given that BACE is stabilized during apoptosis, while many other proteins are degraded at normal or accelerated rates (e.g., APP and TACE), the apparent increase of BACE protein levels at the latest time points of the CHX+STS time course was most likely due to a relative increase of stabilized BACE when normalized for equal amounts of total protein. The half-life of the negative control, TACE, and APP were somewhat decreased during apoptosis (Fig. 2G). A possible explanation for the apparent discrepancy between BACE half-life (9-10 hr) and a 6 fold increase in BACE protein levels after 12 hr treatment with STS is that BACE levels in these studies are relative to total protein during the apoptotic time-course. As measured, BACE levels are determined relative to equal amounts of total protein loaded throughout the time course. Thus, absolute BACE levels are not increasing during apoptosis, BACE levels are only increasing as a percentage of total cellular protein. Since BACE is being stabilized during apoptosis, while many other proteins are succumbing to caspase cleavage, e.g. SOD and GGA3, BACE levels remain fairly constant in the cell during apoptosis due to a significantly longer half-life. This translates into the detection of increased levels of BACE/total cell protein during the apoptosis time course.
The BACE trafficking molecule GGA3 is cleaved by caspase 3 during apoptosis
We have previously reported that endogenous BACE is normally degraded by lysosomal hydrolases (Koh et al., 2005). Thus, we hypothesized that caspase activation leads to BACE stabilization by interfering with its degradation in the lysosomes. We previously reported that the BACE di-leucine motif, the binding site for GGAs, plays a role in targeting BACE to the lysosomes (Koh et al., 2005). Since GGA3 has been shown to be necessary for lysosomal degradation of EGFR (Puertollano and Bonifacino, 2004), we hypothesized that GGA3 may play a role in the sorting of BACE to the lysosomes and that caspase cleavage of GGA3 during apoptosis may impair the degradation of BACE.
We first asked whether GGA3 is cleaved by caspases during apoptosis. For this purpose, H4-APP751 cells were treated with STS during time course experiments. Western Blot analysis with an anti-GGA3 antibody, targeted to a portion (amino acids 424-542) of the hinge domain of GGA3 (Fig. 3F), revealed that full length GGA3 is cleaved into two major fragments of ∼48 and ∼37 kDa during apoptosis. β-catenin, which is a known caspase 3 substrate (Brancolini et al., 1998), was cleaved in the H4-APP751 cells with a temporal pattern similar to that observed for GGA3 (Fig. 3A). It is important to note that the fragments were not detected at the 24 hr time point. The latter is most likely due to their degradation during the progression of apoptosis. Since many proteases are activated during apoptosis, we next employed an in vitro cell-free assay to determine whether caspase 3 is capable of cleaving of GGA3 to produce ∼48 and ∼37 kDa fragments. Recombinant caspase 3 cleaved in vitro translated [35S]-labeled GGA3 into several fragments (Fig. 3B). To determine which of these fragments corresponded to the ones detected by WB analysis of the apoptotic lysates, GGA3 was in vitro translated in the presence of cold methionine and then incubated with recombinant caspase 3. Western Blot analysis with anti-GGA3 antibody revealed that recombinant caspase 3 cleaves in vitro translated GGA3 (cold methionine) into three major fragments, the ∼48 and ∼37 kDa bands along with a third band at ∼50 kDa (Fig. 3C). Meanwhile, the additional caspase 3-derived fragments detected in the autoradiography are most likely N-terminal fragments based on the epitope of the anti-GGA3 antibody (Fig. 3F).
Fig. 3. The BACE trafficking molecule GGA3 is cleaved by caspase 3 during apoptosis.
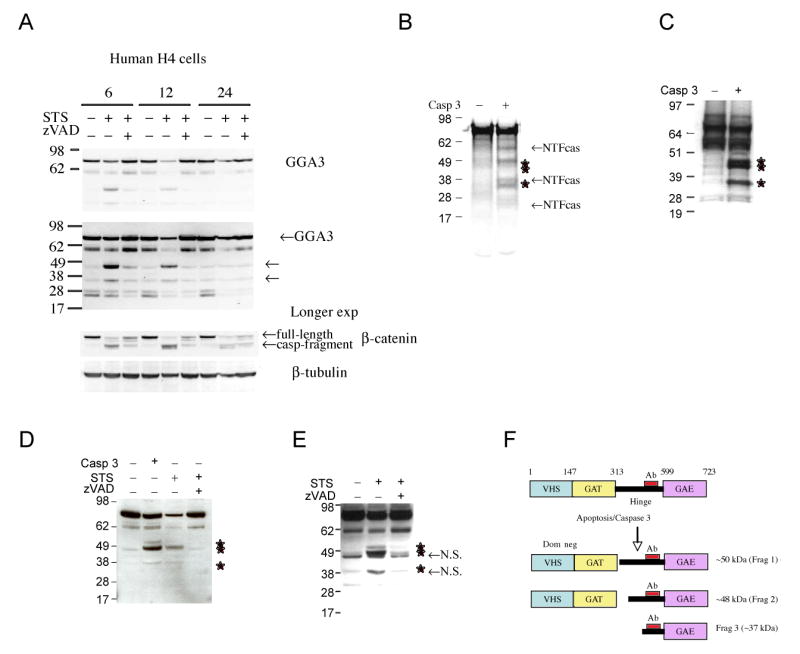
A: Apoptosis was induced in H4-APP751 cells by STS treatment during time course experiments. Western blot analysis with an anti-GGA3 antibody revealed cleavage of GGA3 in two major fragments of ∼48 and ∼37 kDa (indicated by the arrows) in apoptotic H4 cells, which was prevented by caspase inhibition (zVAD). A longer exposure (Long exp) better evidentiates the caspase-derived fragments. Western Blot analysis with anti-β-catenin antibody revealed that β-catenin was cleaved by caspase (casp-fragment) in the H4-APP751 cells with a temporal pattern similar to that observed for GGA3. B: recombinant caspase 3 cleaves in vitro translated GGA3 (labeled with [35S] methionine) in several fragments. The stars indicate the fragments detected by WB in Fig. 3A. The additional fragments most likely are caspase-derived N-terminal fragments (NTFca). C: recombinant caspase 3 cleaves in vitro translated GGA3 (cold methionine) in three fragments detected in WB by anti-GGA3 antibody (stars indicate the fragments). D: recombinant caspase 3 cleaves endogenous GGA3 from lysates of control cells with a pattern similar to the one observed in H4 apoptotic lysates (stars indicate the fragments). E: apoptosis induced in H4 cells stably overexpressing GGA3 produces a cleavage of GGA3 identical to the one produced by recombinant caspase 3 (stars indicate the fragments). Non-specific bands are also detected just below the ∼48 kDa fragment and in correspondence of the ∼37 kDa fragment (N.S.). F: Schematic representation of GGA3 caspase cleavage.
Then we assessed whether recombinant caspase 3 cleaves endogenous GGA3 from normal cell lysates and in vitro translated GGA3 to produce similar sets of fragments. Lysates of control cells were incubated with recombinant caspase 3 at 37° for 2 hr. WB analysis with the anti-GGA3 antibody revealed that recombinant caspase 3 cleaves endogenous GGA3 to generate a pattern of fragments similar to that observed in the in vitro assay (three fragments). Treatment of cell lysates with recombinant caspase 3 generated an ∼50 kDa GGA3 fragment (Fig. 3D), which was not observed in the H4 apoptotic lysates (Fig. 3A). One possible explanation is that its levels were below the levels of detection in the apoptotic cells. To address this possibility, H4 cells stably overexpressing GGA3 were treated with STS to induce caspase activation. This led to cleavage of GGA3 into a pattern of fragments (Fig. 3E) that was identical to that produced by recombinant caspase 3 cleavage of both in vitro translated GGA3 (Fig. 3C) and endogenous GGA3 in cell lysates (Fig. 3D). Collectively, these data indicate that caspase 3 cleaves GGA3 at three major sites. Given that the anti-GGA3 antibody epitope and the size of the fragments generated during apoptosis, we predicted that caspase-mediated cleavage of GGA3 occurs within the hinge domain (Fig. 3F) to produce three fragments of ∼50 kDa (Frag. 1), ∼48 kDa (Frag. 2), and ∼37 kDa (Frag. 3).
Site-directed mutagenesis of GGA3 at D313/D328/D333/D428 prevents the generation of the three major caspase-derived fragments
Several putative caspase consensus sites, conserved in the GGA3 sequence of mammals, can be identified in the hinge domain (Fig. 4A). Since the motif 310TLPD313 in GGA3 has previously been shown to serve as a caspase site in the Golgi resident protein, GRASP65 (Lane et al., 2002), we performed site-directed mutagenesis of D313 to alanine. Next, wild type GGA3 (w.t.) and mutated GGA3 (D313A) were in vitro translated in the presence of cold methionine and subjected to the in vitro caspase 3 cleavage assay as described above. Western Blot analysis with anti-GGA3 antibody revealed that the D313A mutation prevented the generation of the caspase-derived ∼50 kDa fragment 1 (Fig. 4B).
Fig. 4. Site-directed mutagenesis of GGA3 at D313/D328/D333/D428 prevents the generation of the three major caspase-derived fragments.
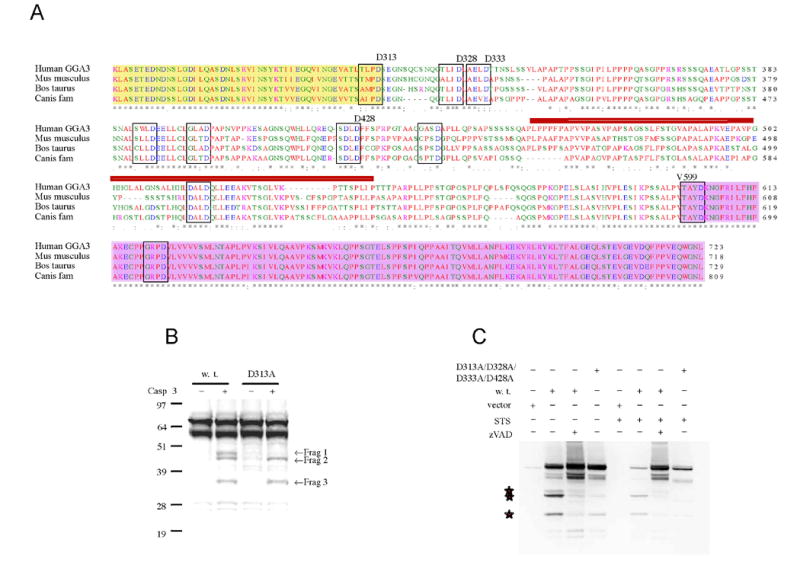
A: Multiple sequence alignment of human (NP_619525), mus musculus (NP_766636), Bos taurus (XP_587687) and canis familiaris (XP_540429) GGA3. Putative caspase consensus sequence in the hinge and GAE domain are in box. The red bar indicates the epitope recognized by the anti GGA3 antibody. The Hinge domain starts at D313 and ends at V599. The yellow and pink background indicates GAT and GAE domain, respectively. B: Site-directed mutagenesis of D313 to alanine prevents the caspase 3-mediated cleavage of in vitro translated GGA3 (cold) at one of the three major sites (WB with anti-GGA3 Antibody). C: H4 cells were transiently transfected with empty vector, GGA3 wild type (w.t.), and GGA3D313A/D328A/D333A/D428A. The overexpression of GGA3 w.t. induced an artefactual caspase-mediated cleavage (stars indicate the fragments). The cleavage was inhibited when cells were treated with zVAD during the transfection. The caspase-derived fragments were not detected in the GGA3D313A/D328A/D333A/D428A when cells where treated with STS. However zVAD treatment, but not GGA3D313A/D328A/D333A/D428A, preserved full-length GGA3.
Subsequently, we mutagenized aspartates downstream of D313 and found that D328A/D333A prevented the generation of fragment 2, while D428A prevented the generation of fragment 3 in the in vitro caspase 3-cleavage assay as described above (data not shown). We next tested whether all four mutations D313A/D333A/D328A/D428A could prevent caspase cleavage of GGA3 during apoptosis. H4 cells were transiently transfected with GGA3 w.t. and GGA3- D313A/D328A/D333A/D428A, and then apoptosis was induced by STS treatment. Overexpression of GGA3 w.t. in H4 cells artefactually induced GGA3 caspase cleavage, which was prevented by treatment with zVAD (Fig. 3I). As expected, the GGA3-D313A/D328A/D333A/D428A did not produce fragments 1-3 following caspase activation (Fig. 4C). However, levels of full-length GGA3-D313A/D328A/D333A/D428A were decreased under these conditions, suggesting that GGA3 hosts additional cleavage sites that render it susceptible to its degradation during apoptosis.
Caspase-mediated cleavage of GGA3 at D313 generates a dominant negative molecule
It has been previously shown that the moderate expression of a truncated GGA construct lacking the Hinge and GAE domain (GGA1 VHS+GAT) operates as a dominant negative (DN), that blocks the clathrin-dependent transport of the cation-independent MPR from the TGN to the endosomes (Puertollano et al., 2001). Thus, cleavage of GGA3 D313 at the border of the hinge domain would not only reduce active GGA3 molecules during apoptosis, but also potentially generate the equivalent of a dominant negative form of GGA3.
To address this possibility we engineered a HA-tagged GGA3 N-terminal fragment 1-313 mimicking the N-terminal caspase-derived fragment generated by cleavage at D313 (GGA3DN). Then we co-transfected GGA3DN, HA-tagged GGA3 w.t. or vector alone with myc-tagged BACE in H4-APP751 cells (Fig. 5A) and found that BACE levels were increased in the cells expressing GGA3DN compared to cells expressing vector alone or GGA3 (Fig. 5A-B). Levels of secreted Aβ40 also increased in cells expressing GGA3DN (Fig. 5C). However, the increase was not statistically significant because the GGA3DN most likely did not completely inhibit the endogenous GGA3. During apoptosis, however, caspase-mediated cleavage of GGA3 results not only in the production of GGA3DN but also the degradation of GGA3.
Fig. 5. Caspase-mediated cleavage of GGA3 at D313 generates a dominant negative molecule.
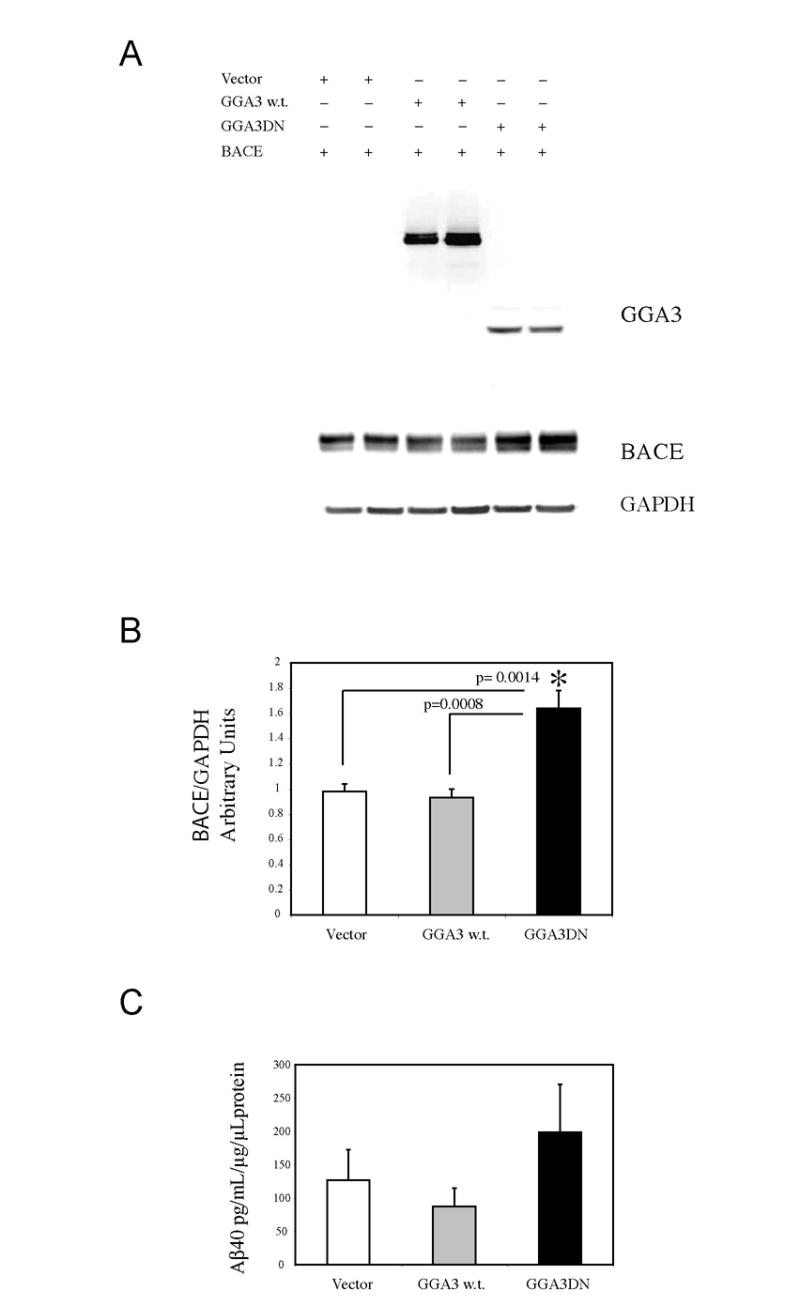
A: HA-tagged GGA3DN, HA-tagged GGA3 w.t. or vector alone with myc-tagged BACE in H4-APP751 cells. WB analysis performed with anti-HA antibody (Cell Signaling) revealed the expression of GGA3 construct. WB analysis performed with anti-myc (Cell Signaling) revealed that BACE levels were increased in the cells expressing GGA3DN compared to cells expressing vector alone or GGA3. B: The graph represents mean ± SEM of at least 6 BACE levels measurements. Densitometry was performed using Versadoc Imager and QuantityOne software (Bio-Rad). BACE densitometry values were normalized against GAPDH values. Unpaired T-test with Welch correction was used for statistical analysis. p = 0.0014 vector vs GGA3DN. p = 0.0008 GGA3 w.t vs GGA#DN. C: The graph represents mean ± SEM of at least 6 Aβ0 measurements by ELISA. Aβ concentration was normalized against the concentration of protein in cell lysates.
RNAi silencing of GGA3 increases levels of BACE, APP-C99, and Aβ
To determine whether GGA3 depletion plays a role in BACE degradation, we assessed the effects of RNAi silencing of GGA3 on BACE and Aβ levels (as measure of β-secretase activity). H4 cells stably expressing wild type APP751 or APP containing the Swedish FAD mutation (APPSwe) were transfected with 200 nM siGGA3 to deplete GGA3. A 19 bp scrambled sequence was used as negative control for siRNA (siNeg). 72 hr later, we measured GGA3 levels and found that endogenous GGA3 was decreased by 50% (Fig. 6A; Suppl Fig.1). At this same time point, a sister plate of the same cells was co-transfected with myc-tagged BACE and either siGGA3 or siNeg. The media was changed after 48 hr. Cells and media were collected 72 hr after co-transfection, and BACE levels were determined by WB using anti-myc antibody. BACE levels were increased by ∼ 7 and ∼4 fold in H4-APP751 and H4 APPSWE, respectively (Fig. 6 B-C-D). Since degradation of EGFR has previously been shown to be impaired in cells depleted of GGA3, as a positive control, we determined the effect of siRNA silencing of GGA3 on EGFR in these same cells. As expected, decreased levels of GGA3 resulted in increased levels of EGFR protein (Fig. 6B; Suppl Fig. 1B). Meanwhile, as a negative control, downregulation of GGA3 had no effect on levels of GAPDH, which was also used as a loading control (Fig. 6B). Similar results were observed in H4 APPSWE cells (data not shown). In accord with these data, levels of APP-C99, detected by WO2 antibody, were increased while levels of full length APP were unchanged in H4-APPSWE cells depleted of GGA3 (Fig. 5E). These data indicate enhanced β-secretase activity. Next, we measured Aβ40 levels in the media (conditioned for 24 hr) by ELISA. Downregulation of GGA3 increased Aβ40 levels by >2 fold in H4-APP751 (Fig. 5F) and by 50% in H4 APPSWE cells (Fig. 6G). As previously reported (Vassar et al., 1999), increased levels of BACE enhance Aβ production to a lesser extent in cells expressing APPSWE than in cells expressing APP wild type. However, increased levels of Aβ corresponded well with the increase in BACE levels in these experiments (Fig. 6B). Similar results were observed when H4 cells stably expressing myc-tagged BACE were co-transfected with siGGA3 and either w.t. APP or APPSWE (data not shown). Our aim was to assess β-secretase activity, and since increases in BACE activity have previously been shown to increase both Aβ40 and Aβ42 (Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999), we did not measure Aβ42 in these experiments. We have confirmed that downregulation of GGA3 increases BACE levels in murine N2A cells using lentiviral vectors expressing shRNA for mouse GGA3. Three shRNA lentiviral vectors downregulated GGA3 to different degree (Suppl Fig. 2A-B). Levels of ectopically expressed BACE inversely correlated with GGA3 levels (Suppl Fig. 2A-C). Moreover, endogenous BACE also increased upon downregulation of GGA3 (Suppl Fig. 2D-E). To determine whether downregulation of GGA3 also affects γ-secretase, GGA3 RNAi has been performed in H4 cells expressing either APP-751 or the APP-CTF (APP105) using lentiviral vectors expressing shRNA for human GGA3 (Suppl Fig. 3A). We have found that downregulation of GGA3 produces an increase in Aβ40 generation in full-length APP-751-expressing cell lines but not in the APP-C105-expressing cell lines (Suppl Fig.3B and C, respectively). These results indicate that depletion of GGA3 affects APP processing independently of γ-secretase activity. It is important to note that BACE levels increase when depletion of GGA3 is achieved with both synthetic RNAi duplex and lentiviral vector expressing shRNA targeting different region of GGA3 gene. Collectively, these findings show that downregulation of GGA3 serves to increase levels of BACE, APP-C99, and Aβ. These data also support the hypothesis that GGA3 normally plays a role in modulating BACE turnover and stability most likely by sorting BACE to lysosomes (based on our earlier findings; (Koh et al., 2005)).
Fig. 6. RNAi silencing of GGA3 increases levels of BACE, APP-C99 and Aβ.

A: H4-APP751 cells were transfected with 200 nM siRNA GGA3 or 200 nM siNeg control. After 72 hr, GGA3 protein levels were determined by WB with anti-GGA3 antibody (Transduction Laboratories). GAPDH was used as loading control. B: After 72 hr, a sister plate of the same cells was co-transfected with myc-tagged BACE and siGGA3 or siNeg control. EGFR levels detected by WB with anti-EGFR antibody (Cell Signaling) were increased in the siGGA3 treated cells. BACE protein levels were detected by WB using anti-myc polyclonal antibody (Cell signaling) were also increased in the siGGA3 treated cells. Levels of GAPDH were unchanged. C-D: the graph represents mean ± SEM of 6 or 7 BACE levels measurements for H4-APP751 and H4 APPSwe, respectively. Densitometry was performed using Versadoc Imager and QuantityOne software (Bio-Rad). BACE densitometry values were normalized against GAPDH values. Unpaired T-test with Welch correction was used for statistical analysis. E: Full-length APP and APP-C99 levels were detected by WB with WO2 antibody in H4-APPSWE cells. F-G: the graph represents mean ± SEM of 6 or 9 Aβ0 measurements by ELISA in H4-APP751 or H4 APPSWE, respectively. Aβ concentration was normalized against the concentration of protein in cell lysates. Unpaired T-test with Welch correction was used for statistical analysis.
GGA3 is degraded during cerebral ischemia concurrently with caspase activation and increases in BACE levels
BACE protein levels and activity have previously been shown to be elevated following cerebral ischemia in a rat model (Wen et al., 2004). Previous studies have also shown that caspase 3 activation can be detected following cerebral ischemia from 1 to 24 hr after reperfusion in both mice and in rats (Davoli et al., 2002; Namura et al., 1998). We thus asked whether caspase activation induced by cerebral ischemia leads to degradation of GGA3 and increased levels of BACE protein in the same rat model of cerebral ischemia used by Wen et al. (2004). Ischemic stroke was induced by middle cerebral artery occlusion (MCAO) in rats as previously described (Liu et al., 2002). At 48 hr of reperfusion following induction of cerebral ischemia, BACE protein levels were significantly increased (> 2-fold) in the ischemic (ipsilateral) cortical and sub-cortical hemispheres, but not in the contralateral hemispheres (Fig. 7A-B). Increased levels of BACE were detected as early as 12 hr post-reperfusion (data not shown). Unfortunately, we were unable to detect caspases in the rat homogenates by WB. One possible explanation is that the anti-caspase antibodies are human specific and thus unable to detect rat caspases. It is also likely that caspase-3 activation occurs much earlier than the 48 hr time point. Since APP is also a substrate for caspase cleavage (Barnes et al., 1998; Gervais et al., 1999; LeBlanc et al., 1999; Pellegrini et al., 1999; Weidemann et al., 1999), we tested whether APP undergoes caspase-mediated cleavage in the rat brain following ischemia. At 48 hr of reperfusion following induction of cerebral ischemia, full-length APP protein levels were slightly decreased owing to the processing of full-length APP into a fragment of roughly 90kDa (Fig. 7A), that we have previously reported to be the N-terminal APP caspase fragment in cells undergoing apoptosis (Tesco et al., 2003). Based on APP, caspase activation occurred concomitant with the increase in BACE levels.
Fig. 7. GGA3 is degraded during cerebral ischemia concurrently with caspase activation and increased BACE levels.
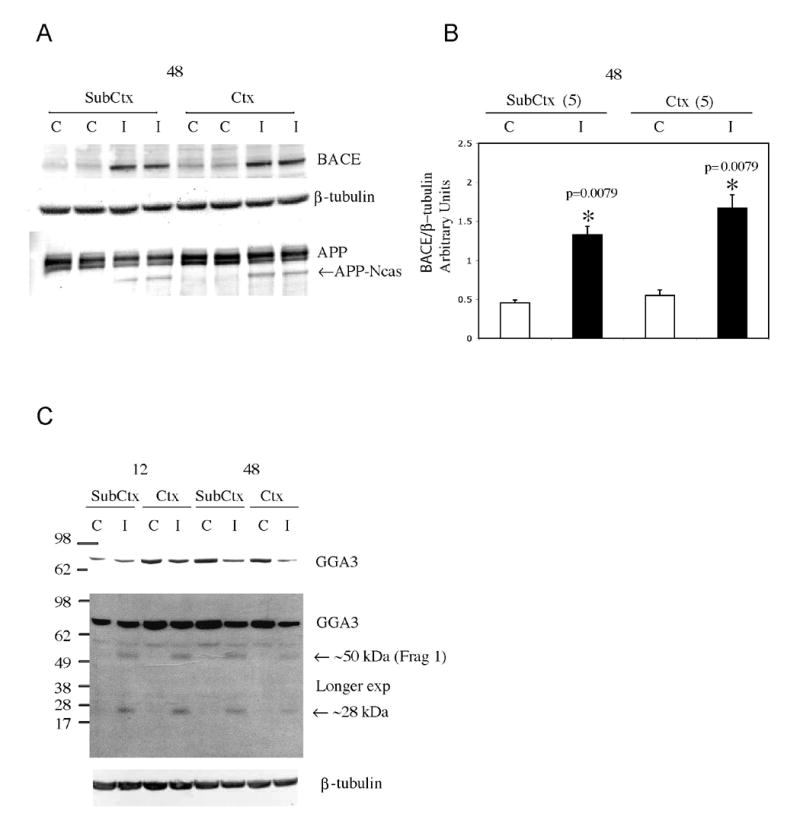
A: Cerebral ischemia was induced by middle cerebral artery occlusion for 1 hr in female rats. After desired times of reperfusion, the animals were sacrificed. The brains were dissected in the ischemic (ipsilateral, I) and not-ischemic (contralateral, C) hemispheres and each hemisphere in cortex (Ctx) and sub-cortex (Sub-Ctx). A-B: WB analysis with anti-BACE antibody (ABR) revealed an increase in BACE proteins levels in the ischemic hemisphere (samples were run in duplicate) after 48 hr of reperfusion. β-tubulin was used as loading control. The densitometry analysis of BACE after normalization against β-tubulin levels is represented in the graph. Mean ± SEM of 5 rats. Statistical analysis was performed using Mann-Whitney test. WB with an anti-APP-CTF antibody (A8717, Sigma) revealed an APP caspase-derived fragment in the rat ischemic hemisphere (samples were run in duplicate) after 48hr of reperfusion. C: WB analysis with anti-GGA3 antibody revealed a decrease in GGA3 full-length and a longer exposure two fragments of GGA3 generated during ischemia after 12 and 48 hrs of reperfusion (C=contralateral hemisphere; I=ipsilateral hemisphere; SubCtx= subcortex; Ctx=cortex).
We next determined whether GGA3 undergoes caspase-mediated cleavage during cerebral ischemia. At 12 and 48 hr of reperfusion following induction of cerebral ischemia, GGA3 was cleaved with a temporal pattern similar to that observed for the increase in BACE protein levels (Fig. 7C). GGA3 full length was decreased in the rat ischemic hemisphere samples and two cleavage fragments were detected in the ischemic rat hemisphere: a fragment of ∼50 kDa most likely corresponding to fragment 1 generated by cleavage at D313 (Fig. 3C) and a second fragment of ∼ 28kDa. The origin of the second fragment is not clear; however, one possible explanation is that the 28kDa fragment may be the result of degradation of the larger fragments detected in the in vitro experiments (Fig. 2 and 3). In any event, these data indicate that full-length GGA3 is cleaved and depleted following ischemia. Moreover, depletion of GGA3 occurs concurrently with caspase activation (based on caspase cleavage of APP) and increasing levels of BACE in the rat model of cerebral ischemia.
Levels of GGA3 are decreased and inversely correlated with increased levels of BACE in AD temporal cortex
Several studies have shown that BACE protein levels and β-secretase activity are increased in AD brains (Fukumoto et al., 2002; Holsinger et al., 2002; Li et al., 2004a; Tyler et al., 2002; Yang et al., 2003). However, the molecular mechanism underlying these increases remains unresolved. The data presented in this study raise the possibility that increased BACE levels in AD brains may be at least partly due to decreased levels of GGA3. To test this hypothesis, we measured GGA3 and BACE protein levels in 19 non-demented (ND) versus 20 AD brain (temporal cortex) samples that have already been shown to possess increased BACE levels and β-secretase activity (Li et al., 2004a; Tyler et al., 2002). We first confirmed that BACE protein levels are significantly increased in the AD temporal cortex (Fig. 8A-B) as shown by WB analysis using the SECB1 antibody (Li et al., 2004a; Tyler et al., 2002). Next, we found that GGA3 protein levels were significantly decreased in the same set of AD brains (Fig. 8A-C). Moreover, the decreased levels of GGA3 were inversely correlated with the increased BACE levels in the AD group, but not in the ND group (Fig. 8D-E). Quantification of GGA3 mRNA levels by real-time PCR showed that the decrease in GGA3 protein levels is not due to reduced gene expression (Fig. 8F) but most likely occurs at the translational or post-translational level. We next asked whether reduced levels of GGA3 in the AD brains could be due to degradation by caspase 3. Levels of full-length caspase 3 were decreased in the AD (but not ND) brains, most likely because of increased activation as shown to occur in apoptotic H4 cells (STS) (Fig. 8G). However, we were unable to detect either the active caspase 3 fragment or the caspase-derived GGA3 fragments in these brains. One possible explanation is that the caspase-generated fragments were readily degraded in these post-mortem tissues. On the other hand, the GGA3 caspase-derived fragments were also not detectable in cells during advanced apoptosis (Fig. 3A). To further investigate the mechanism underlying our observed depletion of GGA3 in the AD brains, we have analyzed BACE and GGA3 levels not only in the temporal cortex, but also in the cerebellum, which is usually spared of AD pathology (Suppl Fig. 4). GGA3 levels were decreased in the cerebellum of AD as compared to control subjects by 40% (Suppl Fig. 4C). However, the decrease was more pronounced in the temporal cortex (55%) (Fig. 8). Moreover, while decreased levels of GGA3 inversely correlated with BACE levels in the temporal cortex, BACE levels were not increased in the cerebellum of AD as compared to control subjects (Suppl Fig. 4A-B), suggesting that the levels of GGA3 were sufficient for a normal degradation of BACE. The generation of GGA3 null mice, currently not available, will be useful for determining the threshold level of GGA3 depletion required to impair BACE degradation in vivo. While GGA3 levels in the TC correlate with GGA3 levels in the cerebellum in ND (Suppl Fig. 4D), such correlation is lost in the AD patients in which the levels of GGA3 are further reduced in the TC (Suppl Fig. 4E). These data suggest that subjects with lower levels of GGA3 (Suppl Fig. 4F) could be at risk of developing AD in conditions associated with caspase activation e.g cerebral ischemia or Aβ toxicity, now, selectively in affected brain regions. These findings lend further support to the hypothesis that that GGA3 normally plays a role in modulating BACE turnover and stability, and that pathological conditions in which GGA3 levels are reduced lead to increased levels of BACE and β-secretase activity.
Fig. 8. Levels of GGA3 are decreased and are inversely correlated with increased levels of BACE in AD brains.
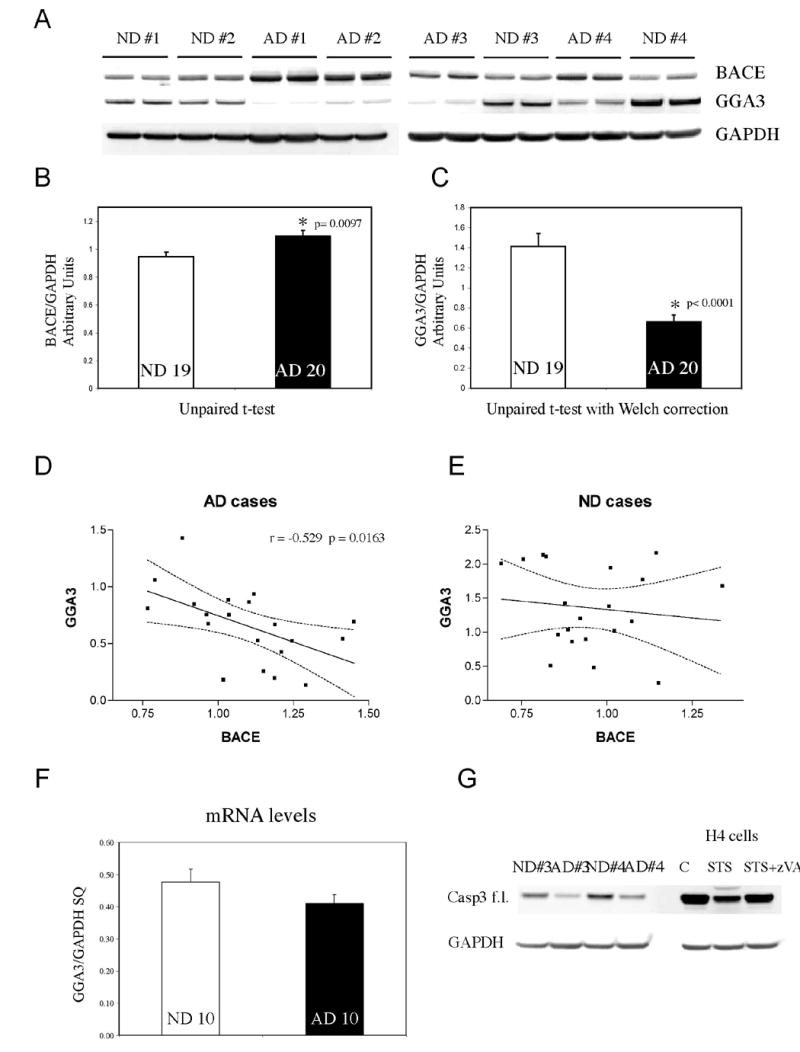
A. Western Blot analysis of temporal cortex of human brains. AD= Alzheimer's disease. ND= non-demented control. BACE was detected by SECB1. GGA3 was detected by anti-GGA3 antibody. GAPDH was used as loading control. B-C. BACE and GGA3 densitometry values were normalized against GAPDH values. At least triplicate of each samples were analyzed. The graphs represent mean ± SEM of 19 ND and 20 AD. Unpaired t-test and unpaired t-test with Welch correction ware used to perform statistical analysis of BACE and GGA3 levels, respectively. D-E. Linear correlation analysis between BACE and GGA3 levels in AD and ND, respectively. The dotted line indicates the 95% confidence interval. F. GGA3 mRNA was quantified by real time PCR. The graph represents mean ± SEM of 10 ND and 10 AD. SQ=starting quantity. Please note that the levels of GGA3 protein were significantly decreased in the same samples. G. Western Blot analysis of temporal cortex of human brains. AD= Alzheimer's disease. ND= non-demented control. Full-length caspase 3 was detected with anti-caspase 3 antibody. To show that full-length caspase 3 decrease during apoptosis, lysates from control (C), treated with (STS), and treated with STS+zVAD H4 cells were also included. GAPDH was used as loading control.
Discussion
Recent studies have revealed BACE as a stress-related protease that is upregulated in the brains of AD patients (Fukumoto et al., 2002; Holsinger et al., 2002; Li et al., 2004a; Tyler et al., 2002; Yang et al., 2003). BACE levels have also been shown to increase following ischemia (Wen et al., 2004); however, the molecular mechanism responsible has remained elusive. Here, we show that caspase activation elevates BACE levels and β-secretase activity owing to post-translational stabilization of BACE. Using a rat ischemia model, we found that levels of GGA3, an adaptor molecule implicated in BACE trafficking, are reduced in a temporally coordinated manner with caspase activation and increases in BACE proteins levels. In cell-based studies, RNAi silencing of GGA3 directly led to increased BACE protein levels and β-secretase activity as evidenced by enhanced APP-C99 and Aβ levels. Together, these data suggest a model in which apoptosis, e.g. induced by ischemia, drives the depletion of GGA3, which, in turn, leads to the stabilization of BACE and increased β-secretase activity. We also analyzed brain samples of AD patients in which BACE levels and β-secretase activity have previously been shown to be elevated in the temporal cortx. In further support of our model of GGA3-dependent degradation/stabilization of BACE, in the brains of AD patients, GGA3 protein levels were significantly decreased. Moreover, this decrease was inversely correlated with increased BACE levels in the temporal cortex.
Decreased levels of GGA3 most likely engender an increase in BACE protein levels by interfering with the sorting of BACE to lysosomes where it is degraded (Koh et al., 2005). GGA3 has previously been demonstrated to target cargo (e.g. EGFR) to lysosomes (Puertollano and Bonifacino, 2004). RNAi silencing of GGA3 resulted in the accumulation of EGFRs in enlarged early endosomes and partially blocked their delivery to lysosomes where they are normally degraded. These studies indicate that GGA3 is involved in the delivery of endosomal cargoes to lysosomes. Recently, He et al. (2005) showed that RNAi silencing of GGAs significantly increases the levels of BACE in endosomes. They also proposed that GGAs are necessary for BACE and MPRs to be transported back to TGN. However, BACE turnover was not assessed in that study, and the accumulation of BACE in endosomes could also be due to decreased degradation resembling the effect of GGA3 downregulation on EGFR degradation (Puertollano and Bonifacino, 2004). The mechanism by which GGA3 targets some cargo (e.g. EGFR) to lysosomes has been shown to be ubiquitin-dependent (Puertollano and Bonifacino, 2004). While there is some evidence that BACE is ubiquitinated (Qing et al., 2004), future studies will be required to determine whether GGA3-dependent degradation of BACE requires ubiquitination, or whether it occurs via an alternate mechanism (e.g. the binding to GGA3-VHS domain).
We have shown that caspase-mediated degradation of GGA3 occurs following cerebral ischemia and propose that it may explain our and others' observations of elevated β-secretase levels (and activity) following cerebral ischemia. As shown in the Nun Study (Kalaria, 2000; Nolan et al., 1998; Snowdon et al., 1997), and recently confirmed in additional prospective autopsy studies (Petrovitch et al., 2005; Riekse et al., 2004), individuals with AD and cerebrovascular pathologies show greater cognitive impairment than those exhibiting either pathology alone. These studies indicate that there is an additive or synergistic interaction between AD and cerebrovascular pathologies. Furthermore, evidence is accumulating that stroke and transient ischemic attacks significantly increase the risk of AD in elderly individuals (Honig et al., 2003; Zlokovic, 2002). A recent family-based study has shown that stroke increases the risk of AD to a similar extent as the presence of an APOE-ε4 allele in Latinos (Rippon et al., 2006). Thus, stroke may represent either a precipitating or a triggering event in AD.
While there is an increasing body of knowledge indicating a strong association between cerebrovascular disease and AD (Honig et al., 2003; Rippon et al., 2006), the potential role of apoptosis and cerebral ischemia in AD pathogenesis has remained unclear. Apoptosis has been firmly established to enhance Aβ production in neuronal and non-neuronal cells (Barnes et al., 1998; Galli et al., 1998; Gervais et al., 1999; Guo et al., 2001; LeBlanc, 1995; Sodhi et al., 2004; Tesco et al., 2003). Thus, apoptotic events in the brain, e.g. induced by stroke and ischemia could increase risk for, or trigger AD by driving cerebral Aβ accumulation. Several studies have shown cerebral ischemia to upregulate APP messages containing the Kunitz-type protease inhibitor domain, between 1 and 21 days after reperfusion (Abe et al., 1991; Kim et al., 1998; Koistinaho et al., 1996; Shi et al., 2000). Additionally, APP protein levels were increased between 1 and 10 weeks after reperfusion (Banati et al., 1995; Kalaria et al., 1993; Stephenson et al., 1992; Wakita et al., 1992). BACE protein levels and β-secretase activity have also been shown to be increased in animal models of traumatic brain injury, including cerebral ischemia (Wen et al., 2004) and head injury, which is also a risk factor for AD (Blasko et al., 2004; Chen et al., 2004). More recently, caspase inhibition therapy has been shown to prevent brain trauma-induced increases in Aβ peptide (Abrahamson et al., 2006). Collectively, these findings, taken together with our current data, suggest that accumulative insults to the brain over one's lifetime would progressively increase risk for AD by elevating cerebral Aβ accumulation via BACE stabilization owing to caspase-mediated depletion of GGA3. Furthermore, the effect of BACE stabilization on Aβ levels could also be amplified by other events (e.g. upregulation of APP levels at much later time points, e.g. several days after the ischemic event).
We have also shown that GGA3 levels are reduced both in the temporal cortex and cerebellum of AD patients (versus controls). The decrease in GGA3 levels was more pronounced in the temporal cortex versus cerebellum, which is relatively spared of AD pathology. Importantly, decreased levels of GGA3 were inversely correlated with increased levels of BACE only in the temporal cortex, which is strongly impacted by AD pathology. In contrast, BACE levels were not significantly increased in the cerebellum of AD patients as compared to control subjects. These findings suggest that some subjects have lower levels of GGA3 independently of AD pathology, e.g. in cerebellum. Subjects with lower levels of GGA3 may be at risk of developing AD given that conditions associated with caspase activation e.g stroke, which is a risk factor for AD, may further decrease GGA3 levels triggering or precipitating AD pathology.
The contribution of apoptosis to the etiology and pathogenesis of AD remains unclear largely due to the difficulties involved in identifying classic apoptotic markers in vivo (for review see (Cribbs et al., 2004; LeBlanc, 2005). This is most likely due to the long duration of AD and the very rapid clearance of apoptotic cells. Contradictory results could be at least partially due to the use of post-mortem tissues. Many factors (e.g. length of agonal state, collection of tissue at the end point of the disease and time interval before freezing the tissue) may significantly affect the analysis of enzymatic activities. It is also possible that many senile plaques, which can take many years to form, are no longer surrounded by apoptotic neurons by the time of autopsy. On the other hand genetic evidence for sporadic AD, such as the disease associations with DAPK1 (Li et al., 2006), GAPD (Li et al., 2004b) and LOC439999 (Grupe et al., 2006) variants, also point to apoptosis as a disease-relevant process. While there is increasing evidence for caspase activation in AD brain (for review see (Cribbs et al., 2004; LeBlanc, 2005), we cannot rule out the possibility that genetic factors and/or other post-translational mechanisms (e.g. other proteases) may contribute to GGA3 depletion in AD brain.
In summary, our studies suggest that elevated BACE protein levels found in AD patients and animal models of traumatic brain injury including ischemia and acute head trauma, may be at least partly due to impaired degradation and stabilization of BACE. This would then lead to increased production of the Aβ peptide, thereby contributing to AD pathogenesis. Since Aβ has also been reported to induce apoptosis, this could result in a vicious cycle that autopotentiates Aβ generation and cell death. Finally, our in vivo and in vitro data implicate GGA3 as the key player in regulating degradation of BACE in its capacity as a trafficking molecule that delivers BACE to the endosomal-lysosomal system.
Experimental procedures
Reagent and General methods
The monoclonal antibody, WO2, raised against 1-17 amino acids of Aβ region was a gift of Dr. Beyreuther. The anti-Cu,Zn-SOD antibody was a gift from Dr. Naoyuki Taniguchi. BACE was detected in human brains by SECB1, which recognizes amino acids 296-310 of BACE amino terminus (Li et al., 2004a; Yang et al., 2003). Details of standard methods such as cell cultures, site-directed mutagenesis, transfection, Western blotting, Northern blotting, metabolic labeling and pulse-chase experiments, Real time PCR, RNA extraction and information on other antibodies and reagents can be found in the Supplemental Experimental Procedures.
Aβ ELISA
Secreted Aβ40 was measured in the conditioned media using an Aβ40-specific sandwich ELISA (BioSource International, Camarillo, CA). Aβ concentration was normalized against the concentration of protein in the cell lysates.
Cycloheximide degradation time-course
H4-APP751 cells were treated with CHX (40 μg/ml) only or STS (1 μM) + CHX during a 30 hr time-course. Lysates from each time point were immunoblotted with the specific antibodies, anti-BACE, anti-TACE, and the anti-APP antibody A8717.
In vitro translation and recombinant caspase 3 cleavage assay
The HA-GGA3 pcDNA4 plasmid was a generous gift from Dr. Waguri. GGA3 was in vitro translated (IVT) in the presence of [35S]-labeled methionine using TNT Quick Coupled Transcription/Translation Systems as indicated by the manufacturer (Promega, Madison WI). 5μL of IVT reaction were incubated with or without 200ng of recombinant caspase 3 (Pharmingen, San Diego, CA) in caspase reaction buffer (sucrose 20%, NaCl 100 mM, HEPES (pH 7.4) 20 mM, CHAPS 0.1%, DTT 10 mM, EDTA 1 mM) at 37° for 2 hr. The reactions were separated by SDS-page (4-12% Bis-tricine gel with MES running buffer, Invitrogen Carlsbad, CA). The gel was fixed, dried and exposed to a phosphorimager screen. The images were acquired with a FX phosphorimager (Bio-Rad, Hercules CA). GGA3 in vitro translation was also performed in the presence of cold methionine. Then, 5μL of IVT reaction were incubated with or without 200ng of recombinant caspase 3 overnight at 37°. The reactions were separated by SDS-page. In order to better separate the bands generated by caspase 3 cleavage we used 12% Bis-tricine gel and MOPS running buffer (Invitrogen). Then, the proteins were transferred to PVDF membrane and WB analysis with anti-GGA3 antibody was performed.
GGA3 RNAi silencing
H4 cells stably expressing wild-type APP-751 or APP containing the Swedish FAD mutation (APPSwe) were transfected with 200 nM siGGA3 (ID #36847, Ambion, Austin TX) or a 19 bp scrambled sequence was used as negative control for siRNA (Silencer Negative control #1 siRNA, Ambion) using oligofectamine following manufacturer's instructions (Invitrogen). 72 hr later, GGA3 levels were determined by WB analysis with anti-GGA3 antibody (Transduction Laboratories). At the same time point, a sister plate of the same cells was co-transfected with myc-tagged BACE plasmid and siGGA3 or siNeg. After 48 hr the media was replaced. Cells and media were collected after 72 hr following the co-transfection.
Middle cerebral artery occlusion
Ischemic stroke was induced by occlusion of the middle cerebral artery (MCAO) as described before (Wen et al., 2004). More details can be found in the in the Supplemental Experimental Procedures.
Human brain samples, protein and RNA extraction
20AD and 19ND temporal cortex were obtained from the Brain Donation Program, Sun Health Research Institute, Sun City, Arizona, USA. Human tissue was collected with informed consent of subjects or next of kin and with ethical approval from the Sun Health IRB. Brain Total RNA was obtained by RNA mini column kit (Qiagen) by following the manufactory's instruction. For protein extraction, small piece of temporal cortex was homogenized by 1X RIPA buffer plus PMSF and proteinase inhibitors mix (Sigma), and protein concentration were measured by protein assay kit (Bio-Rad).
Densitometry and Statistical analysis
Densitometry analysis was performed on a Macintosh computer using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/) or using a Versadoc Imager and QuantityOne software (BioRad). Statistical analysis was performed using Instat3 software. Unpaired T-test was employed for data sets that passed normality test. Unpaired T-test with Welch correction was employed for data set that passed normality test but had different standard deviations. Mann-Whitney test was employed for data set that did not pass normality test.
Supplementary Material
Supplemental Data include Supplemental Experimental Procedures, Supplemental References and four Figures.
Acknowledgments
This work is supported by NIH grants 1K12MH069281-01 (to GT) and 1R01AG025952-01A2 (to GT), NIMH grant 1R37MH60009 (to RET), American Health Assistance Foundation (to RET), Cure Alzheimer's Fund (to RET), and the John French Douglas Foundation Fellowship (to YHK). We thank Dr. Steven Wagner for helpful comments on this manuscript.
Abbreviations are
- AD
Alzheimer's disease
- APP
β-amyloid precursor protein
- Aβ
amyloid β-protein
- ELISA
Enzyme-linked immunosorbent assay
- zVAD-fmk
Z-val-Ala-Asp(Ome)-CH2F
- GGA
Golgi-localized γ-ear-containing ARF binding proteins
- BACE
Beta-site APP-cleaving enzyme
- VHS
(VPS27, Hrs, and STAM) domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe K, Tanzi RE, Kogure K. Selective induction of Kunitz-type protease inhibitor domain-containing amyloid precursor protein mRNA after persistent focal ischemia in rat cerebral cortex. Neurosci Lett. 1991;125:172–174. doi: 10.1016/0304-3940(91)90020-t. [DOI] [PubMed] [Google Scholar]
- Blasko I, Beer R, Bigl M, Apelt J, Franz G, Rudzki D, Ransmayr G, Kampfl A, Schliebs R. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer's disease beta-secretase (BACE-1) J Neural Transm. 2004;111:523–536. doi: 10.1007/s00702-003-0095-6. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS. The GGA proteins: adaptors on the move. Nat Rev Mol Cell Biol. 2004;5:23–32. doi: 10.1038/nrm1279. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Citron M. Beta-secretase inhibition for the treatment of Alzheimer's disease--promise and challenge. Trends Pharmacol Sci. 2004;25:92–97. doi: 10.1016/j.tips.2003.12.004. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113(Pt 11):1857–1870. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, et al. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- He X, Chang WP, Koelsch G, Tang J. Memapsin 2 (beta-secretase) cytosolic domain binds to the VHS domains of GGA1 and GGA2: implications on the endocytosis mechanism of memapsin 2. FEBS Lett. 2002;524:183–187. doi: 10.1016/s0014-5793(02)03052-1. [DOI] [PubMed] [Google Scholar]
- He X, Li F, Chang WP, Tang J. GGA proteins mediate the recycling pathway of memapsin 2 (BACE) J Biol Chem. 2005 doi: 10.1074/jbc.M411296200. [DOI] [PubMed] [Google Scholar]
- He X, Zhu G, Koelsch G, Rodgers KK, Zhang XC, Tang J. Biochemical and structural characterization of the interaction of memapsin 2 (beta-secretase) cytosolic domain with the VHS domain of GGA proteins. Biochemistry. 2003;42:12174–12180. doi: 10.1021/bi035199h. [DOI] [PubMed] [Google Scholar]
- Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Ann Neurol. 2002;51:783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- Honig LS, Tang MX, Albert S, Costa R, Luchsinger J, Manly J, Stern Y, Mayeux R. Stroke and the risk of Alzheimer disease. Arch Neurol. 2003;60:1707–1712. doi: 10.1001/archneur.60.12.1707. [DOI] [PubMed] [Google Scholar]
- Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer's disease beta-secretase. J Biol Chem. 2000;275:33729–33737. doi: 10.1074/jbc.M004175200. [DOI] [PubMed] [Google Scholar]
- Kalaria RN. The role of cerebral ischemia in Alzheimer's disease. Neurobiol Aging. 2000;21:321–330. doi: 10.1016/s0197-4580(00)00125-1. [DOI] [PubMed] [Google Scholar]
- Kalaria RN, Bhatti SU, Palatinsky EA, Pennington DH, Shelton ER, Chan HW, Perry G, Lust WD. Accumulation of the beta amyloid precursor protein at sites of ischemic injury in rat brain. Neuroreport. 1993;4:211–214. doi: 10.1097/00001756-199302000-00025. [DOI] [PubMed] [Google Scholar]
- Koh YH, von Arnim CA, Hyman BT, Tanzi RE, Tesco G. BACE is degraded via the lysosomal pathway. J Biol Chem. 2005;280:32499–32504. doi: 10.1074/jbc.M506199200. [DOI] [PubMed] [Google Scholar]
- LeBlanc A. Increased production of 4 kDa amyloid beta peptide in serum deprived human primary neuron cultures: possible involvement of apoptosis. J Neurosci. 1995;15:7837–7846. doi: 10.1523/JNEUROSCI.15-12-07837.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc A, Liu H, Goodyer C, Bergeron C, Hammond J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer's disease. J Biol Chem. 1999;274:23426–23436. doi: 10.1074/jbc.274.33.23426. [DOI] [PubMed] [Google Scholar]
- LeBlanc AC. The role of apoptotic pathways in Alzheimer's disease neurodegeneration and cell death. Curr Alzheimer Res. 2005;2:389–402. doi: 10.2174/156720505774330573. [DOI] [PubMed] [Google Scholar]
- Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R, Beach T, Sue L, Sabbagh M, Cai H, et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer's disease patients. Proc Natl Acad Sci U S A. 2004a;101:3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puertollano R, Bonifacino JS. Interactions of GGA3 with the ubiquitin sorting machinery. Nat Cell Biol. 2004;6:244–251. doi: 10.1038/ncb1106. [DOI] [PubMed] [Google Scholar]
- Puertollano R, Randazzo PA, Presley JF, Hartnell LM, Bonifacino JS. The GGAs promote ARF-dependent recruitment of clathrin to the TGN. Cell. 2001;105:93–102. doi: 10.1016/s0092-8674(01)00299-9. [DOI] [PubMed] [Google Scholar]
- Qing H, Zhou W, Christensen MA, Sun X, Tong Y, Song W. Degradation of BACE by the ubiquitin-proteasome pathway. Faseb J. 2004 doi: 10.1096/fj.04-1994fje. [DOI] [PubMed] [Google Scholar]
- Rippon GA, Tang MX, Lee JH, Lantigua R, Medrano M, Mayeux R. Familial Alzheimer disease in Latinos: interaction between APOE, stroke, and estrogen replacement. Neurology. 2006;66:35–40. doi: 10.1212/01.wnl.0000191300.38571.3e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba T, Kametaka S, Kawasaki M, Shibata M, Waguri S, Uchiyama Y, Wakatsuki S. Insights into the phosphoregulation of beta-secretase sorting signal by the VHS domain of GGA1. Traffic. 2004;5:437–448. doi: 10.1111/j.1600-0854.2004.00188.x. [DOI] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. Jama. 1997;277:813–817. see comments. [PubMed] [Google Scholar]
- Tesco G, Koh YH, Tanzi RE. Caspase activation increases beta-amyloid generation independently of caspase cleavage of the beta-amyloid precursor protein (APP) J Biol Chem. 2003;278:46074–46080. doi: 10.1074/jbc.M307809200. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Velliquette RA, O'Connor T, Vassar R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. J Neurosci. 2005;25:10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Arnim CA, Tangredi MM, Peltan ID, Lee BM, Irizarry MC, Kinoshita A, Hyman BT. Demonstration of BACE (beta-secretase) phosphorylation and its interaction with GGA1 in cells by fluorescence-lifetime imaging microscopy. J Cell Sci. 2004;117:5437–5445. doi: 10.1242/jcs.01422. [DOI] [PubMed] [Google Scholar]
- Wahle T, Prager K, Raffler N, Haass C, Famulok M, Walter J. GGA proteins regulate retrograde transport of BACE1 from endosomes to the trans-Golgi network. Mol Cell Neurosci. 2005;29:453–461. doi: 10.1016/j.mcn.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Wen Y, Onyewuchi O, Yang S, Liu R, Simpkins JW. Increased beta-secretase activity and expression in rats following transient cerebral ischemia. Brain Res. 2004;1009:1–8. doi: 10.1016/j.brainres.2003.09.086. [DOI] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, et al. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Vascular disorder in Alzheimer's disease: role in pathogenesis of dementia and therapeutic targets. Adv Drug Deliv Rev. 2002;54:1553–1559. doi: 10.1016/s0169-409x(02)00150-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data include Supplemental Experimental Procedures, Supplemental References and four Figures.