

Abstract
Ca2+-dependent inactivation of Ca2+ currents is a physiological phenomenon widely associated with L-type Ca2+ channels. Although the pore-forming α1C subunit of the channel is the target for Ca2+ binding, the amino acid sequences involved in the binding and/or in the coordination of Ca2+-dependent inactivation are still unclear. Based on previous experiments, we have prepared truncation mutants of a human α1C subunit by systematically deleting an EF-hand motif and sequences in a segment of 80 amino acids in the carboxyl-terminal tail. We found that the rate as well as the Ca2+ dependence of inactivation of currents through these mutated channels were very different. We have identified three amino acid sequences, the presence of which is important for Ca2+-dependent inactivation: (i) a putative Ca2+-binding EF-hand motif, (ii) two hydrophilic residues (asparagine and glutamic acid) 77–78 amino acids downstream of the EF-hand motif, and (iii) a putative IQ calmodulin binding motif. We suggest that Ca2+-dependent inactivation is a cooperative process involving several amino acid sequences in cytoplasmic segments of the α1C subunit.
The voltage-gated L-type Ca2+ channel is an essential part of signal transduction systems in many cell types. The channel is a heteromeric protein complex that occurs in several subunit combinations (1, 2). The cardiac type Ca2+ channel consists of the pore-forming α1C subunit and auxiliary β and α2δ subunits. Several splice variants of the gene for the α1C subunit of human L-type Ca2+ channels have been identified (3–5), and some of them have been expressed in Xenopus oocytes (4, 6, 7). This work has led to the identification of α1C subunits through which currents with very different inactivation properties could be activated. Ba2+ currents through a subunit named α1C,77 exhibited slow inactivation kinetics, whereas those of currents through a chimeric α1C,86 channel were fast (7). Furthermore, with the physiological Ca2+ ions as charge carriers, inactivation of the current through α1C,77 was greatly accelerated, in contrast to α1C,86, where it was very similar in both Ba2+ and Ca2+ solutions. The two α1C subunits differed in a stretch of 80 or 81 amino acids in the carboxyl-terminal tail of the protein (7).
Ca2+-dependent inactivation is a typical feature of many L-type Ca2+ channels (8). It may be a physiological safety mechanism against a harmful Ca2+ overload in the cell, notably during large Ca2+ currents through the channels. It has been shown to be caused by binding of Ca2+ to the pore-forming α1C subunit (9–12), although the location of the Ca2+ binding sites are highly controversial. There is general agreement that the long cytoplasmic, carboxyl-terminal tail of α1C subunits is involved in Ca2+-dependent inactivation. However, although one group (13) found the distal half of the C-terminal tail important, others (7, 14, 15) identified amino acid sequences in the proximal part as being essential for Ca2+-dependent inactivation. An EF-hand motif in this region has been suggested as a crucial Ca2+-binding site for Ca2+-dependent inactivation (14, 16). However, this was not confirmed by Zhou et al. (15). Based on our previous experiments with the α1C,77 and α1C,86 subunits (7), we now have approached the problem by systematically deleting the EF-hand motif and segments within the stretch of 80 amino acids that we had identified as being critical for Ca2+-dependent inactivation (7). By this method we have found that the putative Ca2+-binding EF-hand motif, two hydrophilic amino acid residues (asparagine and glutamic acid) downstream of the EF-hand motif, and a consensus IQ calmodulin binding motif help to support this type of inactivation. However, other sites may well be of additional importance in the coordination of this process (13).
METHODS
Construction and in Vitro RNA Transcription of α1C,77 Deletion Mutants.
Details of the preparation of the plasmid pHLCC77 harboring the cDNA that encodes the subunit α1C,77 have been described elsewhere (6). The NsiI–BamHI fragment of pHLCC77 has been subcloned into the PsiI–BamHI polylinker sites of pBluescript SK(−). This construct (p77NB) was used for all subsequent manipulations of C-terminal sequences of α1C,77. The deletion mutant 77d80 (Fig. 1) has been constructed by using the Seamless Cloning Kit (Stratagene). All other deletions (Fig. 1) have been introduced into p77NB by PCR, taking advantage of the unique feature of Pfu DNA Polymerase (Stratagene) to create blunt-ended amplification products. For each deletion two pairs of phosphorylated primers have been designed to amplify two fragments that flank the sequences to be deleted. After isolation and proper restriction of the fragments, they have been cloned into p77NB by triple ligations and were further subcloned into the full-length pHLCC77. All mutations have been confirmed by sequencing. Plasmids encoding mutated α1C subunits as well as the auxiliary subunits β1 (17, 18) and α2δ (19), kindly provided by V. Flockerzi (University of Saarland, Homburg/Saar) and F. Hofmann (Technical University, Munich), were appropriately linearized before in vitro transcription by using Ambion’s T7 mMessage mMachine Kit.
Figure 1.
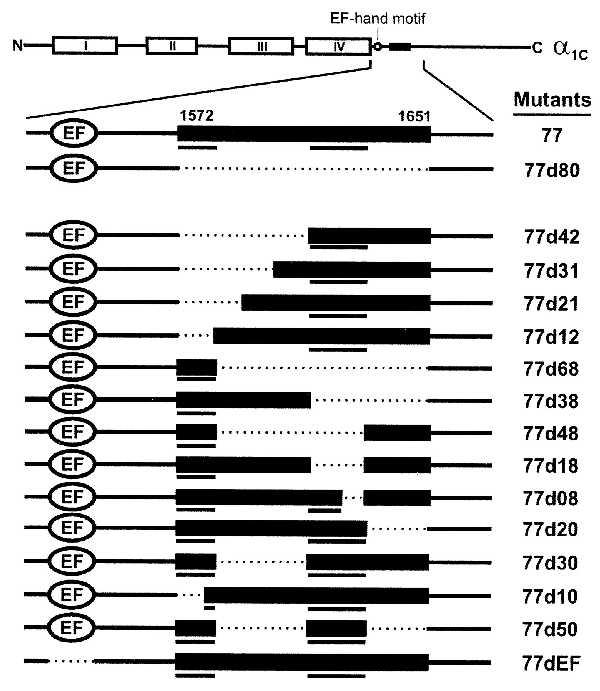
Schematic presentation of the L-type Ca2+ channel α1C,77 subunit (Top) and its deletion constructs used in this study. The pore-forming α1C subunit consists of four homologous, membrane-spanning domains (I–IV). For simplicity, our full-length reference channel α1C,77 is called 77, and deletions derived from 77 are defined as 77dXX, where XX stands for the number of amino acid residues deleted in the C terminus of α1C,77, except for 77dEF, where EF stands for the deleted EF-hand motif. The deletion mutants constructed are (positions of deleted amino acid residues): 77d80 (1572–1651), 77d42 (1572–1613), 77d31 (1572–1602), 77d21 (1572–1592), 77d12 (1572–1583), 77d68 (1584–1651), 77d38 (1614–1651), 77d48 (1584–1631), 77d18 (1614–1631), 77d08 (1624–1631), 77d20 (1632–1651), 77d30 (1584–1613), 77d10 (1572–1581), 77d50 (1584–1613, 1632–1651), and 77dEF (1496–1524). Bars underneath the constructs highlight amino acids sequences important for Ca2+-dependent inactivation.
Functional Expression and Electrophysiological Recordings of Constructs in Xenopus Oocyte.
Xenopus oocytes were isolated according to standard procedures (20). cRNAs for mutated α1C subunits were mixed with those for α2δ and β1 subunits in equimolar ratios at concentrations of 0.1–1.0 ng/nl. About 50 nl of these mixtures were injected into defolliculated Xenopus laevis oocytes. The oocytes were stored at 18°C for 5–8 days before the electrophysiological recordings began. Whole-cell Ba2+ currents (IBa) or Ca2+ currents (ICa) were measured by a conventional two-electrode voltage-clamp method. The glass electrodes were filled with 3 M CsCl and had resistances between 0.2 and 1 MΩ. During IBa recordings, the oocyte was constantly superfused with a solution containing 40 mM Ba(OH)2, 50 mM NaOH, 1 mM KOH, 10 mM Hepes (adjusted to pH 7.4 with methanesulfonic acid). When ICa was measured, Ba(OH)2 was replaced by Ca(NO3)2, and the oocytes were injected beforehand with 50 nl of a 40 mM BAPTA (1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid Na4 salt) solution (pH 7.4) to inhibit Ca2+-activated Cl− currents.
In some instances an endogenous IBa or ICa could be detected in injected oocytes (21). This current is dihydropyridine (DHP)-insensitive and therefore pharmacologically distinct from currents through overexpressed α1C channels. The deletion mutants described in this study expressed peak IBa averaging more than 1 μA, except for 77d48 (0.59 ± 0.17 μA, n = 5), 77d12 (0.44 ± 0.04 μA, n = 7), and 77dEF (0.80 ± 0.32 μA, n = 6). The contaminating fraction of endogenous current for constructs with low expression was determined by bath application of 5 μM (+)-isradipine at the end of an experiment. Experiments showing substantial DHP-insensitive residual current (>10% of peak current) have been discarded. In the case of construct 77d12, ICa could be measured reliably in only one experiment because of contaminating endogenous ICa.
Voltage-clamp protocols, current recordings, and leak subtractions were performed by using the EPC software (Cambridge Electronic Design, Cambridge, UK). Currents were filtered at 1 Hz and sampled at 2 Hz. For the construction of inactivation curves, 2-s prepulses (PPs) to different voltages, from a holding potential (Vh) −90 mV, were followed by a test pulse (TP) to +20 mV. The 2-s PPs were not quite long enough to achieve steady-state inactivation (7). We refer therefore to our inactivation curves with 2-s PPs as “isochronic” instead of steady-state.
RESULTS
Analysis of Currents Through α1C,77 and α1C,77d80.
In a previous paper (7) we have compared electrophysiological properties of the α1C subunit of a human fibroblast L-type Ca2+-channel isoform (α1C,77) with a chimeric α1C,86 homologue. The latter was prepared by incorporation of a partial cDNA clone, isolated from human hippocampus (5), into the nucleotide sequence of the recombinant plasmid for α1C,77. This resulted in the replacement of 80 amino acid residues (1572–1651) in α1C,77 by 81 nonidentical amino acids of α1C,86. This replacement produced a marked acceleration of inactivation and a loss of Ca2+ sensitivity of inactivation of Ca2+ currents (7). Like in the previous study, α1C,77 is our reference channel in this paper. Sequence alignment revealed more than 93% identity with α1C from rabbit cardiac muscle.
We began this study by deleting the 80 amino acids in the α1C,77 subunit (Fig. 1) that previously had been replaced by 81 amino acids in α1C,86 (7). For simplicity, from here on we will use the nomenclature 77 instead of α1C,77, and for the deletion mutants 77d. Thus, 77d80 refers to the α1C,77 subunit where the 80 amino acids 1572–1651 have been deleted (Fig. 1). When either 77 or 77d80 were coinjected with the β1 and α2δ subunits into Xenopus oocytes at a 1:1:1 molar ratio, the resulting currents were very different (Fig. 2A). Whereas Ba2+ currents (IBa) through the 77 channel showed the slow inactivation behavior typical for α1C, inactivation of IBa through 77d80 was as fast as in the previously described chimeric channel α1C,86 (7). Furthermore, Ca2+ currents (ICa) through the 77 channel showed a pronounced acceleration of inactivation (7), whereas the rate of inactivation of currents through 77d80 was very similar with both Ba2+ and Ca2+ as charge carriers (Fig. 2A). These findings suggest that the presence of the 80 amino acids in 77 is important for (i) the slow rate of inactivation of IBa, and (ii) the Ca2+ sensitivity of inactivation in this α1C subunit.
Figure 2.
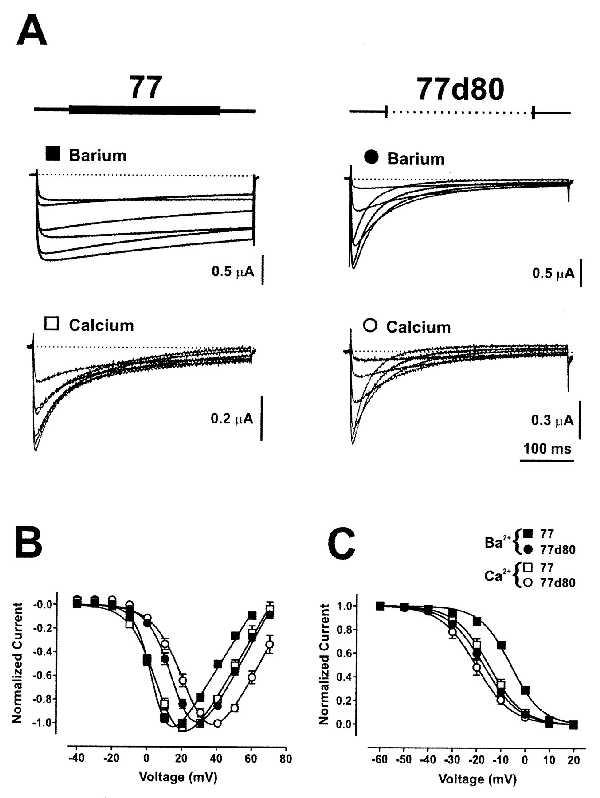
Properties of IBa and ICa through constructs 77 and 77d80 expressed in Xenopus oocytes. (A) C-terminal amino acid sequences between positions 1572 and 1651 (Fig. 1) are shown schematically above the current traces. Ionic currents have been elicited by applying in 30-s intervals a series of 400-ms TPs with 10 mV increments from Vh = −90 mV. Representative families of current traces elicited by TPs between 0 and +50 mV are shown. Bath solutions contained either 40 mM Ba2+ (Upper, filled symbols) or 40 mM Ca2+ (Lower, open symbols) as charge carriers. Normalized I–V curves (B) and isochronic inactivation curves (C) for IBa (filled symbols) and ICa (open symbols) through 77 (squares) and 77d80 (circles) were obtained and plotted according to the procedures described in Table 1. Shown are means ± SEM.
Fig. 2 B and C shows some other more subtle differences between 77 and 77d80. The potential ranges for activation of IBa and ICa through 77 were more negative than for 77d80, whereas those for inactivation were more positive in 77 than in 77d80 (Table 1). These differences are again very similar to those of α1C,77 and α1C,86 in our previous work (7).
Table 1.
Parameters of nonlinear fittings of I-V curves and isochronic (2 s) inactivation curves
| α1C subunit |
I-V curve*
|
Isochronic inactivation curve†
|
||||
|---|---|---|---|---|---|---|
|
V0.5 (mV)
|
V0.5 (mV)
|
|||||
| IBa | ICa | n(IBa/ICa) | IBa | ICa | n(IBa/ICa) | |
| 77 | 4.45 ± 0.77 | 10.30 ± 0.90 | 13/15 | −6.14 ± 0.86 | −15.17 ± 1.66 | 10/6 |
| 77d80 | 18.64 ± 1.08 | 25.46 ± 1.42 | 10/4 | −17.30 ± 1.28 | −20.44 ± 2.02 | 8/4 |
| 77d42 | 20.41 ± 0.42 | 27.25 ± 1.23 | 5/3 | −5.21 ± 0.69 | −10.22 ± 1.31 | 5/3 |
| 77d31 | 10.83 ± 3.05 | 19.26 ± 3.03 | 6/3 | −22.64 ± 1.12 | −25.67 ± 2.23 | 6/3 |
| 77d21 | 8.64 ± 2.51 | 16.40 ± 2.84 | 5/3 | −18.14 ± 0.68 | −22.45 ± 1.01 | 4/3 |
| 77d12 | 18.95 ± 1.15 | 17.03 | 7/1 | −8.23 ± 1.20 | −16.95 | 7/1 |
| 77d68 | 18.22 ± 2.32 | 19.84 ± 2.22 | 6/6 | −16.54 ± 1.68 | −23.21 ± 2.49 | 6/5 |
| 77d38 | 17.73 ± 1.53 | 22.02 ± 1.44 | 7/5 | −10.82 ± 1.30 | −16.03 ± 1.59 | 6/5 |
| 77d48 | 20.15 ± 2.51 | 21.23 ± 1.44 | 5/4 | −13.54 ± 2.39 | −21.39 ± 2.45 | 5/4 |
| 77d18 | 19.42 ± 2.36 | 22.71 ± 1.95 | 6/4 | −9.81 ± 1.93 | −14.98 ± 1.53 | 5/4 |
| 77d08 | 13.57 ± 1.34 | 18.02 ± 1.92 | 10/6 | −14.96 ± 1.65 | −16.36 ± 2.33 | 5/5 |
| 77d20 | −0.72 ± 0.87 | 6.00 ± 1.94 | 7/5 | −17.07 ± 1.22 | −27.52 ± 2.22 | 8/4 |
| 77d30 | 3.41 ± 2.52 | 9.92 ± 1.66 | 5/7 | −14.55 ± 1.33 | −21.64 ± 2.85 | 6/4 |
| 77d10 | 4.69 ± 2.19 | 13.16 ± 2.12 | 9/5 | −20.24 ± 1.28 | −23.93 ± 2.68 | 5/3 |
| 77d50 | 10.34 ± 3.38 | 18.83 ± 1.01 | 4/3 | −15.57 ± 0.07 | −22.58 ± 1.13 | 3/3 |
| 77dEF | 13.35 ± 1.75 | 21.60 ± 1.55 | 6/3 | −10.31 ± 3.43 | −13.61 ± 2.00 | 4/3 |
TPs in the range of −40 to +80 mV were applied in 10 mV increments from Vh = −90 mV. I-V curves were fitted by I = Gmax(V − Erev)/{1 + exp[(V − V0.5)/k]}, where Gmax = maximum conductance; Erev = reversal potential; V0.5 = voltage at 50% of Gmax; k = negative slope factor.
From Vh = −90 mV 2-s conditioning PP were applied in 10 mV increments from −60 up to +20 mV, each followed by a 1-s TP to +20 mV. The interval between TPs was 30 s. Recorded peak current amplitudes were normalized to IBa at −60 mV. Isochronic inactivation curves were fitted by a Boltzmann function: I = 1/{1 + exp[(V − V0.5)/k]}, where V is the conditioning PP voltage, V0.5 is the voltage at half-maximal inactivation, and k is a slope factor. Values are means ± SEM. n = number of tested oocytes.
We also have studied the kinetics of inactivation and of the recovery from inactivation of currents through 77 and 77d80 channels, both in Ba2+ and in Ca2+ solutions. Fig. 3A shows time constants (τ) of the rates of inactivation of IBa and ICa through 77 and 77d80. Whereas the time courses of IBa inactivation in 77 were fitted best by single exponentials, those of ICa required a bi-exponential fit, as did those for IBa and ICa through 77d80 (Table 2). In Fig. 3 A and B only the time constants of the initial exponential decays have been plotted over the potential range from 0 or +10 mV to +50 mV. Fig. 3A shows the slow inactivation kinetics of IBa through 77 (▪), and the large decrease in inactivation time constants with Ca2+ as charge carrier (□). Note also the increase in τ of ICa through 77 at potentials more positive than +20 mV, which is an indication of Ca2+-dependent inactivation (8). The currents through 77d80 showed a very different behavior. Fast-inactivation time constants were strongly voltage-dependent, and those of ICa (○) were slightly larger than those of IBa (•). This result is shown more clearly in Fig. 3B, where all inactivation τs were normalized to those measured for currents at +50 mV.
Figure 3.
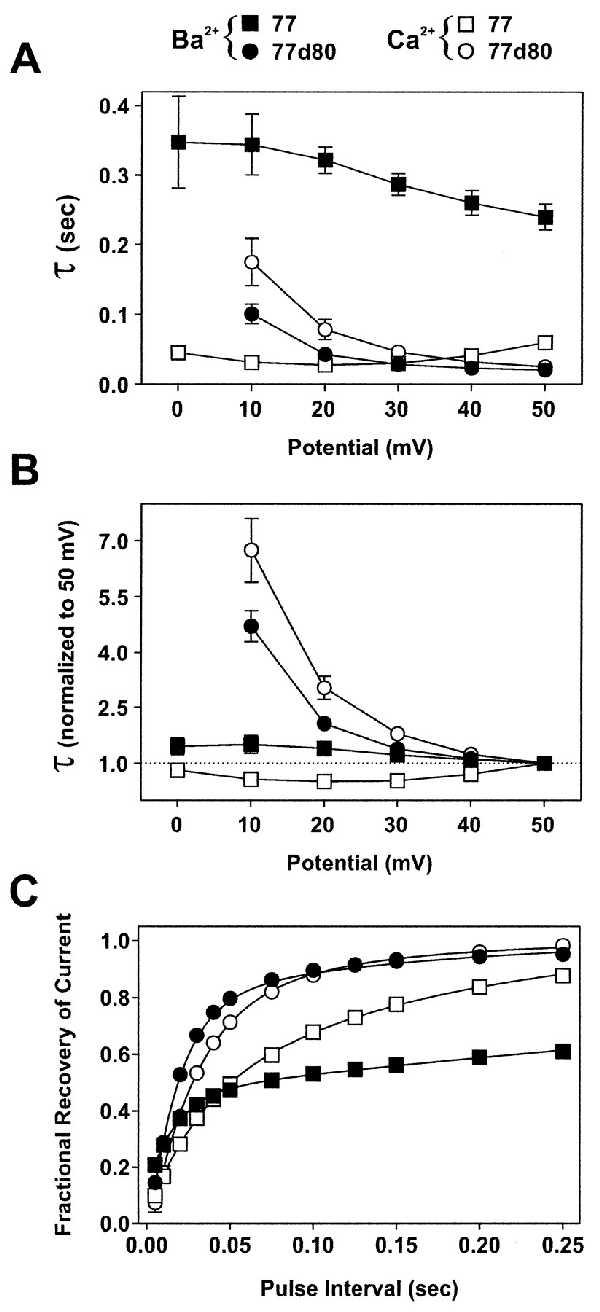
Time constants of inactivation and of recovery from inactivation in constructs 77 and 77d80. (A) IBa (filled symbols) and ICa (open symbols) have been evoked by applying 400-ms pulses to various potentials (abscissa) from Vh = −90 mV. Inactivation time constants (τ) were estimated by exponential fits of IBa and ICa (Table 2). Only τ of the fast component of the bi-exponential fit has been plotted, except for IBa through 77, which was best-fitted, even with 1-s pulses, by a single exponential. The numbers of experiments were: 77 (▪, n = 8; □, n = 10–11), 77d80 (•, n = 8; ○, n = 4). (B) Data from A are normalized to τ at +50 mV. (C) The fractional recovery from inactivation was determined by a two-pulse protocol. A conditioning PP applied from Vh = −90 mV to +20 mV was followed by a TP to +20 mV after variable pulse intervals at Vh. The peak current amplitudes at TPs were plotted against the pulse interval. The PP duration was 3 s for 77 IBa, and 400 ms for all other experiments. Full recovery (>98%) was reached for 77 IBa after 16 s (▪, n = 4) and for ICa after 2 s (□, n = 4); for 77d80 IBa after 1 s (•, n = 6) and for ICa after 250 ms (○, n = 4). Smooth lines are bi-exponential fits of the averaged data between 5 and 250 ms. All data points are means ± SEM (error bars are in most cases smaller than symbols).
Table 2.
Kinetics of IBa and ICa inactivation at +20 mV or +30 mV (according to maximal IBa)
| α1C subunit | Potential at max IBa |
IBa
|
ICa
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| τ1 (ms) | τ2 (ms) | A1 (%) | n | τ1 (ms) | τ2 (ms) | A1 (%) | n | ||
| 77 | +20 mV | 321.7 ± 19.0 | – | – | 8 | 27.8 ± 1.6 | 202.0 ± 9.0 | 68.2 ± 1.9 | 10 |
| 77d80 | +30 mV | 28.8 ± 2.2 | 105.4 ± 7.6 | 82.0 ± 2.0 | 8 | 46.6 ± 8.7 | 127.0 ± 12.1 | 72.8 ± 2.4 | 4 |
| 77d42 | +30 mV | 37.5 ± 1.6 | 140.5 ± 7.1 | 67.1 ± 2.1 | 5 | 45.8 ± 2.8 | 152.8 ± 10.0 | 59.6 ± 1.1 | 3 |
| 77d31 | +20 mV | 44.1 ± 4.3 | 173.3 ± 14.3 | 46.7 ± 2.3 | 6 | 69.9 ± 14.1 | 261.4 ± 63.3 | 60.7 ± 0.8 | 3 |
| 77d21 | +20 mV | 39.2 ± 1.9 | 189.1 ± 7.1 | 45.3 ± 2.2 | 5 | 49.9 ± 7.8 | 171.8 ± 24.3 | 51.1 ± 2.1 | 3 |
| 77d12 | +30 mV | 41.2 ± 2.2 | 177.0 ± 13.5 | 61.2 ± 1.5 | 7 | 38.7 | 139.9 | 41.3 | 1 |
| 77d68 | +30 mV | 32.4 ± 3.1 | 107.6 ± 8.7 | 73.2 ± 2.7 | 5 | 36.6 ± 4.2 | 112.9 ± 12.4 | 67.0 ± 3.7 | 6 |
| 77d38 | +30 mV | 80.1 ± 6.7 | 301.6 ± 50.9 | 61.9 ± 2.7 | 7 | 84.9 ± 4.9 | 286.8 ± 24.2 | 43.5 ± 8.3 | 5 |
| 77d48 | +30 mV | 48.7 ± 9.8 | 199.0 ± 47.7 | 76.3 ± 2.4 | 4 | 65.6 ± 8.3 | 224.9 ± 32.2 | 69.3 ± 1.7 | 4 |
| 77d18 | +30 mV | 96.0 ± 17.7 | 368.4 ± 88.3 | 62.8 ± 2.8 | 6 | 104.3 ± 11.5 | 337.2 ± 56.8 | 53.9 ± 2.9 | 4 |
| 77d08 | +30 mV | 80.3 ± 4.6 | 332.3 ± 46.9 | 60.0 ± 2.5 | 8 | 101.5 ± 7.3 | 318.3 ± 26.5 | 51.2 ± 5.5 | 6 |
| 77d20 | +20 mV | 359.5 ± 21.3 | – | – | 6 | 47.9 ± 6.5 | 199.2 ± 8.4 | 52.6 ± 4.5 | 4 |
| 77d30 | +20 mV | 212.7 ± 7.8 | – | – | 5 | 67.7 ± 6.8 | 239.5 ± 27.8 | 50.2 ± 4.0 | 6 |
| 77d10 | +20 mV | 67.5 ± 4.3 | 362.7 ± 29.9 | 36.1 ± 1.4 | 7 | 71.0 ± 7.4 | 237.1 ± 23.6 | 45.5 ± 3.5 | 5 |
| 77d50 | +20 mV | 52.7 ± 3.3 | 260.3 ± 9.9 | 45.5 ± 3.4 | 4 | 77.5 ± 13.7 | 228.2 ± 41.7 | 35.8 ± 6.0 | 3 |
| 77dEF | +30 mV | 31.0 ± 1.7 | 134.0 ± 10.0 | 68.7 ± 3.0 | 6 | 37.8 ± 0.1 | 139.8 ± 1.0 | 55.4 ± 3.8 | 3 |
During a 400-ms TP to the indicated potential, inactivation time constants were estimated by fitting the inactivating component of the current trace to the following equation: I =Io + I1 exp(−t/τ1) + I2 exp(−t/τ2); Io is the residual current amplitude at equilibrium, I1 and I2 are the amplitudes of the current components. A1 has been calculated as I1/(I1 + I2). IBa of 77, 77d20, and 77d30 were best-fitted by a single-exponential equation. Values are means ± SEM. n = number of tested oocytes.
The time course of recovery from inactivation was measured by a two-pulse protocol. For 77, the first voltage pulse (PP) from a holding potential of −90 mV to +20 mV lasted 3 s, and the second pulse (TP) to +20 mV lasted 400 ms. This pulse was applied after variable intervals at −90 mV. During the PP the currents became inactivated by 80–90%. The recovery of IBa from inactivation during the intervals at −90 mV initially was fast (τ1 = 17.4 ± 0.9 ms; n = 4) and later on very slow (τ2 = 779.4 ± 101 ms; n = 4). The fast and the slow components amounted to about 46% and 54% of the total recovery process, respectively (Fig. 3C). The recovery from inactivation of ICa through 77 was slightly slower than that of IBa in its first component (≈40%; τ1 = 30.9 ± 2.3 ms; n = 4) and considerably faster in its second component (≈60%; τ2 = 157.6 ± 11.3 ms; n = 4). In 77d80 IBa and ICa recovered almost completely during a 250-ms time interval, although the initial time constant of recovery was slightly slower for ICa than for IBa (27.9 ± 3.0 ms vs. 19.5 ± 0.9 ms; n = 4). The slow components of recovery (τ2 = 179.2 ± 24.4 ms and 121.8 ± 33.8 ms, respectively) contributed 17% and 25% to the total time course of ICa and IBa.
The increase in the rate of inactivation when Ca2+ instead of Ba2+ was the charge carrier through the 77 channel (Fig. 3 A and B) points to a Ca2+-dependent inactivation process (8). A crucial test for Ca2+-dependent inactivation is the demonstration that it does depend on the size of ICa through the open channel. This was tested by a standard two-pulse protocol (Fig. 4A). The potentials during the first pulses (400 ms) were changed in 10-mV steps between −40 and +80 mV from a holding potential of −90 mV. During these PPs, current-voltage relationships were established for IBa and subsequently for ICa in the same oocytes. After PPs, the potential was held for 50 ms at −90 mV before a constant TP to +20 mV was applied. The extent of recovery of IBa or ICa after PP during the 50-ms time interval at −90 mV was measured by reactivating the currents at TP. If the current at TP is smallest when the current during PP is largest, a current-dependent inactivation mechanism is suggested. The results of such experiments are shown in Fig. 4B. The peak currents activated by PP (current-voltage relationships) under each conditions were normalized for both channels to the respective maximal current amplitudes. As described above, activation of the currents during PP occurred about 14 mV more positive for 77d80 than for 77 (Fig. 4B, filled symbols). Normalization of the current amplitudes was necessary because of the variability of expression of the channels between different oocytes, and because of the smaller current amplitude of ICa as compared with IBa. The latter is because of the lower permeability of the channels for Ca2+ than for Ba2+ (22). In Ba2+ solution the peak currents through 77 and 77d80 at TP, which were normalized to those measured after a PP to −40 mV, show a slight tendency to become smaller with increasing PP potentials (Fig. 4B, Upper, open symbols). In contrast to Ba2+, with Ca2+ as charge carriers (Fig. 4B, Lower) there is an important difference between the 77 and 77d80 channels in the relative current amplitudes at TP. For 77d80, peak ICa and IBa at TP show a similar tendency to become smaller with increasing PPs (Fig. 4B, ○), whereas ICa through 77 displays a bell-shaped relationship between normalized current at TP and PP potentials (Fig. 4B, Lower, □). Peak ICa at TP is smallest when ICa activated by PPs to +20 or +30 mV is largest (7). Subtraction of IBa from ICa at TP yields the Ca2+-sensitive component of inactivation during PP for both channel types. This result is shown more clearly in Fig. 4C. The open circles show the subtracted currents [1 − (ICa − IBa)] for 77d80, and the open squares show those for 77. The difference between the two curves in Fig. 4C is an indication for the Ca2+ sensitivity of inactivation of ICa in the 77 channel. The larger the Ca2+ influx through the channels during PP, the larger is the extent of Ca2+-dependent inactivation measured at TP.
Figure 4.
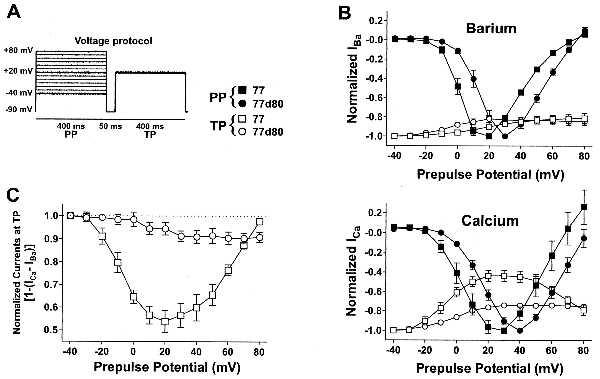
Ca2+-dependent inactivation depends on Ca2+ influx. (A) Two-pulse voltage protocol. Conditioning PPs applied in 10 mV increments from −90 mV to voltages ranging from −40 mV to +80 mV are each followed by a TP to +20 mV after a 50-ms pulse interval at −90 mV. (B) For 77 and 77d80, peak currents at PPs were normalized to the maximal current amplitudes (filled symbols), and those at TPs to the current amplitude obtained after a PP to −40 mV (open symbols). Normalized current amplitudes for IBa (Upper) and ICa (Lower) are plotted against the PP potential. For ICa through 77 (□) the bell-shaped relationship at TP indicates Ca2+-dependent inactivation. IBa and ICa were always recorded from the same Xenopus oocyte. Numbers of experiments: 77 (▪, □, n = 5), 77d80 (•, ○, n = 4). (C) Subtraction of IBa from ICa at TP for both constructs yields a U-shaped relationship for 77 (□) and a more steady decrease for 77d80 (○). The difference between the two curves is a measure of Ca2+-dependent inactivation. Data represent means ± SEM.
Analysis of Currents Through Other Deletion Mutants of α1C,77.
The main task of our study was the attempt to determine sequences within the segment of 80 amino acids in the carboxyl-terminal region of α1C,77 that are involved in Ca2+-dependent inactivation. Therefore, a similar analysis, as described extensively above for the α1C channels 77 and 77d80, has been made for all the constructs shown in Fig. 1. The results for half-maximal voltages (V0.5) of activation and inactivation of IBa and ICa through these channels, as well as time constants of inactivation, and the respective fractional components of inactivation governed by these time constants, are summarized in Tables 1 and 2. The most interesting differences between the various deletion mutants, however, concern their sensitivities to Ca2+-dependent inactivation.
For the analysis of data where IBa and ICa are directly compared, only those experiments have been selected where both currents were measured in the same oocyte. However, the channel 77d12 expressed poorly, and therefore IBa and ICa both could be measured in only one oocyte where no obvious differences in the time courses could be seen (Table 2). In Fig. 5, ICa and IBa through eight α1C constructs are depicted. All current amplitudes have been scaled to the respective peak IBa. The reference channel 77 showed prominent Ca2+-dependent inactivation, as described above. The deletion mutants 77d68, 77d42, and 77d08 showed fast or moderately fast (77d08) inactivation in Ba2+ and Ca2+ solutions and no clear increase in the rates of inactivation by Ca2+. The slight slowing of the initial rate of inactivation in Ca2+ solution also was observed with other deletion mutants (Table 2). Constructs 77d68 and 77d42 include deletions of either the 18 amino acids 1614–1631 or the 12 amino acids 1572–1583, respectively. In construct 77d08 only the eight amino acids 1624–1631 have been deleted. By contrast, the deletion mutants 77d50, 77d30, and 77d20 contain the full amino acid sequences 1572–1583 and 1614–1631, but 50, 30, or 20 amino acids are absent in other parts of the sequences. In 77d10 only the segment 1572–1583 was truncated by the first 10 amino acids. The currents through these four channels all exhibited Ca2+-sensitive inactivation (Fig. 5, lower four traces), though to different extents. The inactivation rates of currents in Ca2+ solution were similar in all four deletion mutants. However, the contribution of Ca2+-dependent inactivation to the total inactivation process was different. The current through channel 77d20 had slow inactivation rates in Ba2+ solution and showed the most prominent Ca2+-dependent inactivation. IBa through 77d50 showed the fastest inactivation rate, whereas that of ICa was only moderately accelerated.
Figure 5.
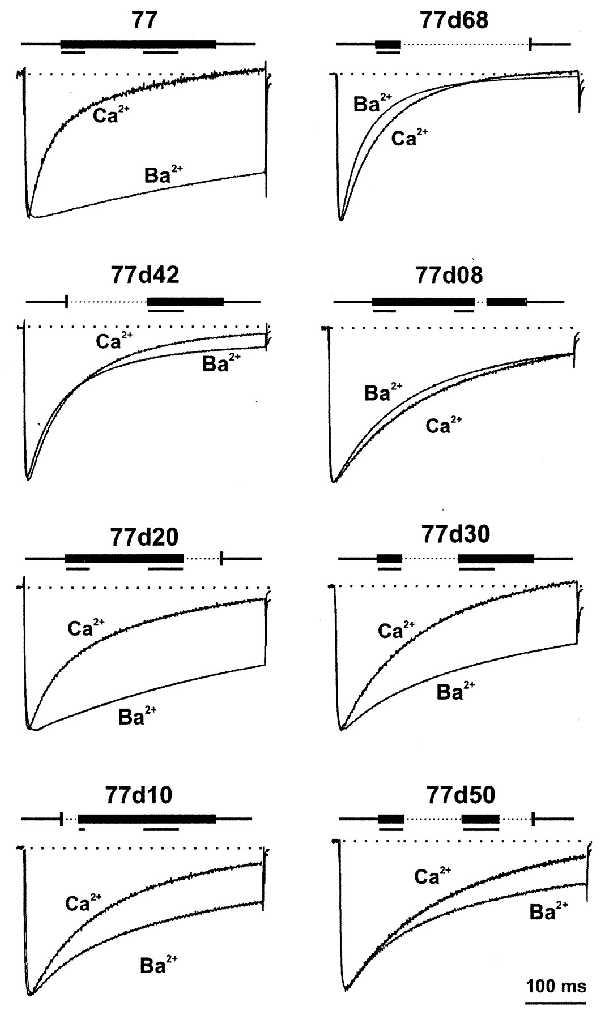
IBa and ICa traces of selected α1C constructs. A 400-ms pulse was applied from Vh = −90 mV to potentials that evoked approximately maximal IBa at +20 mV for 77, 77d20, 77d30, 77d10, 77d50, and at +30 mV for 77d68, 77d42, and 77d08. After switching from a 40 mM Ba2+- to 40 mM Ca2+-containing solution, the same TPs were applied. ICa traces were scaled to the amplitude of the corresponding IBa trace to compare time courses of IBa and ICa. Deletions of C-terminal amino acid residues between positions 1572 and 1651 for each construct are shown above each pair of current traces. Stretches of amino acids in the sequences 1572–1584 and 1614–1631 that were found to be important for Ca2+-dependent inactivation in each construct (Fig. 1) are underlined.
To establish the dependence of the Ca2+ sensitivity of inactivation on Ca2+ influx through all these channel mutants, we have applied the same double-pulse protocols and procedures as described for 77 and 77d80 (Fig. 4 A–C). For constructs 77d10, 77d20, 77d30, and 77d50 the currents at TP became smallest when the preceding ICa at PP was largest. Such a relationship was not found for the other deletion mutants. Normalized peak IBa at TP was subtracted from each normalized ICa at the same potential for every oocyte. The subtracted currents [1 − (ICa − IBa)] are plotted (Fig. 6 A and B) for all constructs shown in Fig. 5. Although clear current minima at TP +20 mV were obtained for the reference channel 77 and for the deletion mutants 77d10, 77d20, 77d30, and 77d50 (Fig. 6A), no such minima could be seen for currents through 77d68, 77d42, and 77d08 (Fig. 6B) and for all other constructs in Tables 1 and 2. Notably the channel 77d08 lacks only the eight amino acids 1624–1631 (Fig. 1). Thus, we conclude that the two amino acids 1582–1583 (channel 77d10) plus the eight amino acids 1624–1631 (channel 77d08) are important sequences involved in Ca2+-dependent inactivation. Moreover, deletion of an EF-hand motif (14–16), 47 amino acids upstream of the stretch of 80 amino acids studied here, also led to a loss of Ca2+-dependent inactivation and to a pronounced acceleration of IBa inactivation (Table 2).
Figure 6.
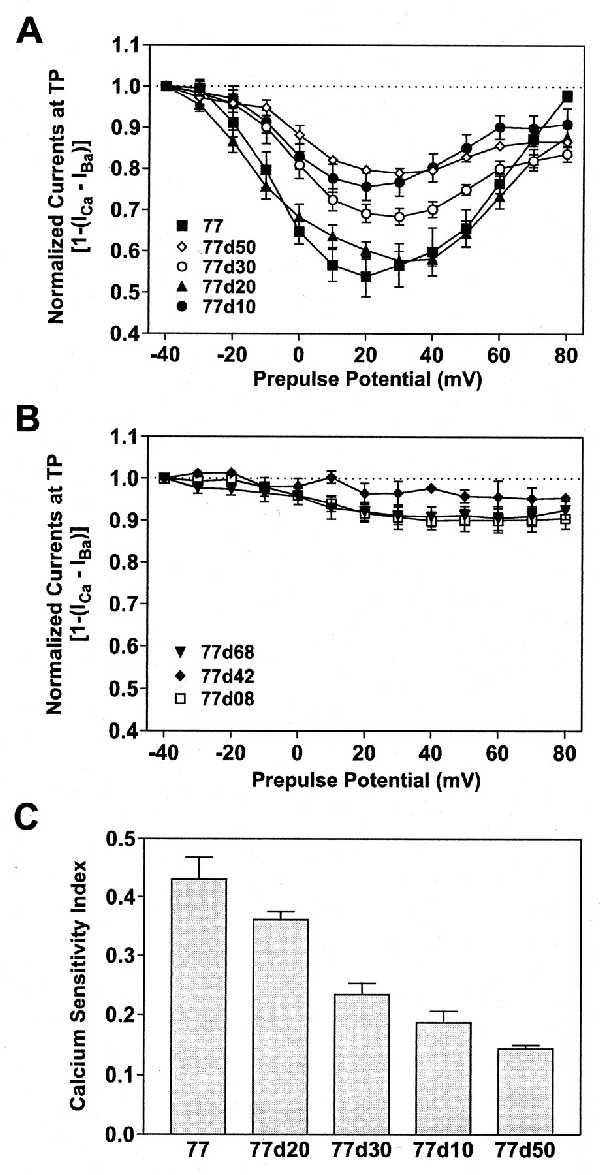
Ca2+ sensitivity of inactivation in selected deletion mutants. By using a double-pulse protocol and following the same procedure as described in Fig. 4, normalized IBa at TP were subtracted from normalized ICa and plotted against the PP potential. (A) Reference channel 77 (▪, n = 5) and its deletion mutants 77d50 (◊, n = 5), 77d30 (○, n = 6), 77d20 (▴, n = 5), and 77d10 (•, n = 5) show U-shaped curves, with minima at +20 to +30 mV. (B) For 77d68 (▾, n = 6), 77d42 (⧫, n = 3), and 77d08 (□, n = 6) no clear minima were seen. (C) A calcium sensitivity index has been calculated and plotted for 77, 77d10, 77d20, 77d30, and 77d50, on the basis of the data presented in A. Normalized currents at TP elicited after a PP to −40 mV (I−40) and +80 mV (I+80) were averaged and subtracted from the normalized current minima at TP (Imin) for each construct: [(1 − Imin) − (I−40 − I+80)/2]. For all other deletion mutants the calcium sensitivity index was smaller than 0.1. Shown are means ± SEM.
To provide a more quantitative picture of Ca2+-dependent inactivation of currents through the α1C constructs 77, 77d10, 77d20, 77d30, and 77d50, we have calculated a “calcium sensitivity index.” The index is based on the data shown in Fig. 6A. The average of the normalized currents at TP elicited after PPs to −40 mV and +80 mV (I−40 and I+80) have been subtracted from the normalized minimal currents (Imin) at TP for each construct [(1 − Imin) − (I−40 − I+80)/2]. The difference is a measure of the Ca2+-dependent fraction of inactivation. This calcium sensitivity index has been plotted in Fig. 6C and shows that Ca2+-dependent inactivation is strongest in 77 and weakest in 77d50. All other constructs had an index of less than 0.1, with Imin after a PP to +30 mV. Other sequences in the carboxyl-terminal cytoplasmic stretch of 80 amino acids, notably within the segment 1584–1623, are significant in determining the rates of voltage-dependent inactivation (Table 2; see also ref. 23).
A summary of our conclusions is provided in Fig. 7. The stretch of 80 amino acids in the carboxyl-terminal end of α1C,77 is shown together with the deletions. The bars above the amino acids correspond to the underlined sequences in the upper scheme of mutations in Fig. 7. Elimination of either one of these sequences abolished Ca2+-dependent inactivation. The dashes above the bars indicate amino acid sequences that were essential for the conservation of Ca2+-dependent inactivation in our experiments with channels 77d10 and 77d08. Inactivation properties of the currents through all channel mutants are indicated on the right. The list provides a qualitative survey of inactivation rates of the currents in Ba2+ solutions and of their Ca2+ sensitivity in Ca2+ solutions.
Figure 7.

Summary of inactivation characteristics for all deletion mutants studied. The schematic presentation of α1C and its deletion mutants (Top and Left) is as in Fig. 1. The two columns to the right qualitatively assess the inactivation rates of IBa (Table 2) and the calcium sensitivity of each construct. Calcium sensitivities are based on calcium sensitivity indices as shown in Fig. 6C. (Bottom) The amino acid sequence between positions 1572 and 1651 of α1C,77 is shown. The heavy bars above the sequence correspond to the underlined sequences in the upper schemes of mutants. The dashed lines above the bars indicate amino acid residues that were found to be essential for the conservation of Ca2+-dependent inactivation.
DISCUSSION
Recently, several articles have been published indicating an important role for Ca2+-dependent inactivation of the carboxyl-terminal cytoplasmic tail in the pore-forming α1C subunit of L-type Ca2+ channels (7, 13–15). However, the exact location of this regulation is still very controversial. A putative Ca2+-binding EF-hand motif near the transmembrane region IVS6 has been shown by one group (14) to be essential for Ca2+-dependent inactivation. But others (15) could not confirm this finding. Disruption of possible Ca2+-coordination sites, by replacement of charged amino acids with alanine in the EF-hand motif, had little effect on Ca2+-dependent inactivation. Deletion of the EF-hand sequence prevented functional expression of the α1C channel in the surface membrane of the oocytes. Those authors (15), however, found that replacement of 134 amino acids in α1E by the homologous 142 amino acid sequence of α1C, downstream of the EF-hand motif, conferred Ca2+-dependent inhibition to the normally Ca2+-insensitive α1E subunit. In our 77 channel, deletions have been made within the same sequence. Although our previous (7) and the present results agree with those by Zhou et al. (15) in showing the importance of sequences downstream of the EF-hand motif, in our hands deletion of this motif did not impede expression of the protein, and it eliminated Ca2+-dependent inactivation. These findings indicate that multiple amino acid sequences in the first half of the putative cytoplasmic carboxyl-terminal region of α1C subunits can participate in the coordination of Ca2+ binding that is required for Ca2+-dependent inactivation. Further sequences that have been described to be important for Ca2+-dependent inactivation are the putative cytoplasmic loops between transmembrane domains I-II and II-III (13).
Recently, Soldatov et al. (23) have shown that replacement of amino acid segments 1572–1598 or 1595–1652 in the carboxyl terminus of α1C,77 by the corresponding sequences of α1C,86 converts the slowly inactivating α1C,77 into the fast-inactivating α1C,86 channel. This finding agrees with our results where deletion of segments 1572–1583 or 1614–1631 in α1C,77 produced fast Ca2+-independent inactivation. They also showed that replacement of the amino acids 1572–1576 in α1C,77 produced a moderately fast, but still Ca2+-sensitive inactivation. This sequence is part of our 77d10 deletion mutant that showed a similar behavior. However, Soldatov et al. (23) could further accelerate inactivation and remove its Ca2+ dependence by the additional replacement of amino acids 1600–1604. Although we have not tried a corresponding deletion in our experiments, we found that deletion of the more distal sequence 1624–1631 converts the slow Ca2+-sensitive inactivation of channel 77 into the faster Ca2+-insensitive inactivation of channel 77d08.
As pointed out to us by R. W. Tsien and K. Deisseroth (personal communication), the sequence 1624–1631 (see Fig. 7) is the initial part of a stretch of 12 amino acids that appears to be a consensus IQ motif for calmodulin binding. This is of special interest not only because of our finding that this motif is involved in Ca2+-dependent inactivation, but also in view of its possible importance for intracellular signaling mechanisms (24). The conserved positions in the consensus sequence of several Ca2+-independent calmodulin-binding motifs are IQXXXRG (or K)XXXRX (25). If Ca2+ binds to calmodulin attached to this motif, this reaction could become part of the possible cooperativity between multiple sites in the α1C subunits that are involved in Ca2+-dependent inactivation. Such a hypothesis appears to be incompatible with the finding that the calmodulin inhibitor calmidazolium has no effect on Ca2+-dependent inactivation (9, 26). We have confirmed this result (unpublished observation). However, it is possible that calmidazolium has a very low affinity to calmodulin when it is bound in a Ca2+-independent way to the IQ motif. The IQ consensus motif also is conserved in α1D- and α1S subunits of L-type Ca2+ channels (27). Although Ca2+-dependent inactivation of ICa seems to be present in α1D of neuroendocrine cells (26), this is not the case for α1S (13, 28). Because, however, other molecular structures that are involved in Ca2+-dependent inactivation in α1C are absent in α1S (13), this may explain the difference between the two channels. The IQ motif is absent and the EF-hand motif scores low in the Tufty–Kretsinger test (14) in α1E, which does not exhibit Ca2+-dependent inactivation (14, 15). Another puzzling result concerns the Hill coefficient near 1 for Ca2+-induced inactivation (12). This result could be explained, however, by the sum of several steps with low cooperativity involved in Ca2+-dependent inactivation.
In conclusion, we favor a hypothesis for Ca2+-dependent inactivation that requires multiple amino acid sequences in the cytoplasmic regions of the α1C subunit of L-type Ca2+ channels. Two possible Ca2+-binding sites that may be directly involved in the cooperative interaction between several sequences in α1C could be the putative Ca2+-binding EF-hand and the calmodulin-binding IQ motifs. We imagine coiling and interaction of the flexible cytoplasmic sequences and, thereby, plugging of the pore. Further studies involving extensive site-directed mutagenesis are required to substantiate this hypothesis.
Acknowledgments
We thank Heleen Van Hees for excellent technical assistance, Andreas Buhr for suggesting the triple ligation technique for mutagenesis, Hartmut Porzig and Kurt Baltensperger for reading the manuscript, and Richard W. Tsien and Karl Deisseroth (Stanford) for helpful discussions. This work has been supported by Swiss National Science Foundation Grant 31-29862.90 (H.R.).
ABBREVIATIONS
- IBa
whole-cell Ba2+ currents
- ICa
whole-cell Ca2+ currents
- PP
prepulse
- TP
test pulse
References
- 1.Hofmann F, Biel M, Flockerzi V. Annu Rev Neurosci. 1994;17:399–418. doi: 10.1146/annurev.ne.17.030194.002151. [DOI] [PubMed] [Google Scholar]
- 2.Catterall W A. Annu Rev Biochem. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- 3.Soldatov N M. Proc Natl Acad Sci USA. 1992;89:4628–4632. doi: 10.1073/pnas.89.10.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schultz D, Mikala G, Yatani A, Engle D B, Iles D E, Segers B, Sinke R J, Weghuis D O, Klöckner U, Wakamori M, et al. Proc Natl Acad Sci USA. 1993;90:6228–6232. doi: 10.1073/pnas.90.13.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soldatov N M. Genomics. 1994;22:77–87. doi: 10.1006/geno.1994.1347. [DOI] [PubMed] [Google Scholar]
- 6.Soldatov N M, Bouron A, Reuter H. J Biol Chem. 1995;270:10540–10543. doi: 10.1074/jbc.270.18.10540. [DOI] [PubMed] [Google Scholar]
- 7.Soldatov N M, Zühlke R D, Bouron A, Reuter H. J Biol Chem. 1997;272:3560–3566. doi: 10.1074/jbc.272.6.3560. [DOI] [PubMed] [Google Scholar]
- 8.Eckert R, Chad J E. Prog Biophys Mol Biol. 1984;44:215–267. doi: 10.1016/0079-6107(84)90009-9. [DOI] [PubMed] [Google Scholar]
- 9.Imredy J P, Yue D T. Neuron. 1994;12:1301–1318. doi: 10.1016/0896-6273(94)90446-4. [DOI] [PubMed] [Google Scholar]
- 10.Neely A, Olcese R, Wei X, Birnbaumer L, Stefani E. Biophys J. 1994;66:1895–1903. doi: 10.1016/S0006-3495(94)80983-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zong X G, Hofmann F. FEBS Lett. 1996;378:121–125. doi: 10.1016/0014-5793(95)01434-9. [DOI] [PubMed] [Google Scholar]
- 12.Höfer G F, Hohenthanner K, Baumgartner W, Groschner K, Klugbauer N, Hofmann F, Romanin C. Biophys J. 1997;73:1857–1865. doi: 10.1016/S0006-3495(97)78216-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adams B, Tanabe T. J Gen Physiol. 1997;110:379–389. doi: 10.1085/jgp.110.4.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Leon M, Wang Y, Jones L, Perez-Reyes E, Wei X Y, Soong T W, Snutch T P, Yue D T. Science. 1995;270:1502–1506. doi: 10.1126/science.270.5241.1502. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J M, Olcese R, Qin N, Noceti F, Birnbaumer L, Stefani E. Proc Natl Acad Sci USA. 1997;94:2301–2305. doi: 10.1073/pnas.94.6.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babitch J. Nature (London) 1990;346:321–322. doi: 10.1038/346321b0. [DOI] [PubMed] [Google Scholar]
- 17.Mori Y, Friedrich T, Kim M, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuchi T, et al. Nature (London) 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- 18.Ruth P, Röhrkasten A, Biel M, Bosse E, Regulla S, Meyer H E, Flockerzi V, Hofmann F. Science. 1989;245:1115–1118. doi: 10.1126/science.2549640. [DOI] [PubMed] [Google Scholar]
- 19.Singer D, Biel M, Lotan I, Flockerzi V, Hofmann F, Dascal N. Science. 1991;253:1553–1557. doi: 10.1126/science.1716787. [DOI] [PubMed] [Google Scholar]
- 20.Sigel E. J Physiol (London) 1987;386:73–90. doi: 10.1113/jphysiol.1987.sp016523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouron A, Soldatov N M, Reuter H. FEBS Lett. 1995;377:159–162. doi: 10.1016/0014-5793(95)01327-x. [DOI] [PubMed] [Google Scholar]
- 22.Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer; 1992. pp. 105–108. [Google Scholar]
- 23.Soldatov N M, Oz M, O’Brien K A, Abernethy D R, Morad M. J Biol Chem. 1998;273:957–963. doi: 10.1074/jbc.273.2.957. [DOI] [PubMed] [Google Scholar]
- 24.Deisseroth K, Tsien R W. Soc Neurosci Abstr. 1997;23:310. [Google Scholar]
- 25.Rhoads A R, Friedberg F. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- 26.Victor R G, Rusnak F, Sikkink R, Marban E, O’Rourke B. J Membr Biol. 1997;156:53–61. doi: 10.1007/s002329900187. [DOI] [PubMed] [Google Scholar]
- 27.Perez-Reyes E, Schneider T. Drug Dev Res. 1994;33:295–318. [Google Scholar]
- 28.Beam K G, Knudson C M. J Gen Physiol. 1988;91:781–798. doi: 10.1085/jgp.91.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]