

Abstract
The use of microorganisms in the open environment would be of less concern if they were endowed with programmed self-destruction mechanisms. Here, we propose a new genetic design to increase the effectiveness of cell suicide systems. It ensures very tight control of the derepression of cell death by the combination of the bacteriophage T7 RNA polymerase-lysozyme system and an inducible synthesis of antisense RNA and the Escherichia coli LacI repressor. Functionality of this regulatory concept was tested by applying it to containment of Gram-negative bacteria, based on the conditional expression of the lethal Streptomyces avidinii streptavidin gene. Toxicity of streptavidin is derived from its exceptionally high binding affinity for an essential prosthetic group, d-biotin. The entire construct was designed to allow the soil bacterium Pseudomonas putida to survive only in the presence of aromatic hydrocarbons and their derivatives which it can degrade. Under favorable growth conditions, clones escaping killing appeared at frequencies of only 10−7–10−8 per cell per generation. The general requirement for biotin through the living world should make streptavidin-based conditional lethal designs applicable to a broad range of containment strategies.
Keywords: heterologous gene expression, bioremediation, Pseudomonas putida
Although genetically engineered microorganisms (GEMs) offer great benefits in environmental applications, it is difficult to predict their behavior in natural ecosystems or how recombinant DNA can spread among indigenous bacterial populations. Potential risk associated with deliberate or unintentional release of GEMs into the open environment can be minimized by the use of debilitated strains. An alternative, and perhaps more appropriate, approach is the introduction of conditional or stochastic maintenance functions into GEMs (1–3). In such a case, the viability of GEMs depends on the expression of an essential gene or on the repression of a lethal gene controlled by a regulatory promoter responding to changes in the chemical or physical constitution of the environment, or by a promoter undergoing recombinational switches. However, the effectiveness of suicide systems is limited by relatively high frequency of their mutational inactivation, resulting in positive selection of uncontained clones. Utilizing several independent killing functions and tightening the regulation of their expression are potential solutions to this problem.
In this report, we address the need both for new killing genes and improved strategies to control their expression. Described here is a potentially universal conditional lethal system based on the tightly regulated derepression of the streptavidin gene (stv) (4) from the actinobacterium Streptomyces avidinii. It targets the metabolism of one-carbon units at the oxidation level of carbon dioxide by depleting an essential prosthetic group, d-biotin (vitamin H); this should complement cell suicide systems for which direct targets are cell membranes and walls, or nucleic acids. Any incompletely repressed expression of the stv gene was eliminated at the level of its transcription, targeting directly the RNA polymerase, as well as at the level of its translation by antisense mRNA. This novel regulatory strategy for containment of GEMs is apparently responsible for the excellent performance of the whole design.
The entire system was tested in the Gram-negative soil rod Pseudomonas putida potentially useful for bioremediation of areas polluted with aromatic hydrocarbon-based organic solvents and petroleum. A containment system for pseudomonads is of particular importance because their nutritional versatility for low molecular weight organic compounds and fast growth rates allow them to rapidly colonize a wide range of habitats and predominate in soil or water microflora. Coupling this system to regulatory elements derived from the P. putida TOL catabolic plasmid (5) conditioned survival of bacteria only in the presence of an aromatic carboxylic acid that they can degrade.
MATERIALS AND METHODS
Bacterial Strains, Plasmids, and Culture Conditions.
Subcloning was performed by using an Escherichia coli K-12 strain XL1-Blue MRF′ (lacIq and Tetr on F′) (Stratagene). Suicide constructs were tested in P. putida mt-2 strain KT2440 {hsdR1} (6). DNA used was a replicative form of a bacteriophage M13 clone, mGP1–2 (7), and plasmids pKK223–3 (Ampr) (8) (Pharmacia), pCC102 (Kanr) (9), pGEM-luc (Ampr) (Promega), pLysE (Cmlr) (7), pRO1614 (Ampr, Tetr) (10), pTSA-13 (Ampr) (11), pUC19 (Ampr), and pVLT33 (Kanr) (12).
Bacteria were grown aerobically in Luria–Bertani (LB) medium at 30°C, unless otherwise stated. Antibiotics were used at the following concentrations: ampicillin, 100 μg/ml (E. coli) or 800 μg/ml (P. putida; optimized for this particular isolate); kanamycin, 25 μg/ml (E. coli) or 75 μg/ml (P. putida); chloramphenicol, 50 μg/ml (E. coli); and tetracycline, 10 μg/ml (E. coli). Isopropyl β-d-thiogalactopyranoside (IPTG) and m-methylbenzoate (3MB) were used at concentrations of 1 mM and 0.2 mM, respectively.
Recombinant DNA Techniques.
DNA manipulations were carried out by standard procedures (13). P. putida was transformed by a RbCl method (14) or by electroporation (Gene Pulser apparatus; Bio-Rad).
Construction of Each Element of the System.
The rationale of the genetic design is given in Results. The stv gene, the Ptac promoter, and the bacteriophage T7 transcription system (i.e., the φ10 promoter, and the RNA polymerase and lysozyme genes), were placed on two compatible plasmids, pCC102 (an RSF1010 derivative bearing the xylS2 gene and the Pm::lacI fusion) and pRO1614 (pMB1 and pRO1600 replicon), as shown in Fig. 2A. Fragments of pUC18 and pGEM-luc plasmids, bearing convenient cloning sites, were inserted into pCC102, downstream of the lacI gene, to facilitate subcloning of the φ10::stv fusion and subsequent DNA manipulations. The stv gene used here encodes a core streptavidin consisting of amino acid residues 16–133 of the mature streptavidin (11). This protein has a higher binding stoichiometry to biotinylated macromolecules than natural core streptavidin. A 1.3-kb fragment of the luc gene, derived from pGEM-luc, was inserted into pRO1614 as a spacer to reduce expression of the T7 lysozyme gene. This also makes the construct comparable to the corresponding region of pRO-llp, in which the lysozyme gene is separated from the Ptet promoter by the 1-kb lacI gene (lacI inserted between the EcoRI and BamHI sites of the Tetr locus).
Figure 2.

Construction of plasmids pCC-s05 and pRO-ilp (A), and physical maps of their killing and regulatory elements (B). stv or s, Streptavidin gene; T7 RNA pol or pol, T7 RNA polymerase gene (T7 gene 1); lys, T7 lysozyme gene (T7 gene 3.5); kan, kanamycin-resistance gene; lucΔC, a truncated luc gene encoding an N-terminal fragment of luciferase; MCS, a fragment of pUC18 containing part of the multiple cloning site. B, BamHI; Bg, BglI; D, DraI; Ea, EagI; EI, EcoRI; EV, EcoRV; H, HindIII; Hp, HpaI; K, KpnI; N, NheI; Nr, NruI; P, PstI; Sa, ScaI; Sc, SacI; Sl, SalI; Sm, SmaI. Unique cleavage sites are marked with asterisks.
For construction of pCC-s04 carrying the lac operator (Olac) immediately upstream of φ10::stv, a BglII–HindIII fragment of pTSA-13 carrying φ10::stv was first inserted into the SmaI–HindIII site of pKK223-3. Then, a BamHI fragment carrying Olacφ10::stv was cloned into the BamHI site of pVLT33. Finally, a KpnI–SalI fragment of the resulting plasmid was placed in the KpnI–SalI site of pCC-s05.
Fluctuation Tests.
Frequency of the appearance of clones resistant to the induction of cell death was estimated following the Luria–Delbrück approach as described (15).
RESULTS
Design of a Conditionally Lethal Construct.
The killing function in our system is based on the almost irreversible binding (Kd ≈ 10−15 M) of d-biotin by streptavidin (16), a tetrameric protein produced by S. avidinii. Cell death results from depletion of free biotin and direct inhibition of biotin-dependent carboxylases, decarboxylases, and transcarboxylases (17). Inactivation of these enzymes blocks the first committed step of fatty acid biosynthesis and affects gluconeogenesis, amino acid metabolism, replenishment of the Krebs cycle, and substrate uptake by some anaerobes.
A simple strategy for using streptavidin as a suicide factor would be to place its gene directly under the control of a promoter negatively regulated by a repressor protein synthesized in response to an environmental signal. The most serious drawback of such a design is incomplete repression (leakiness) of regulatory promoters. To achieve tighter control of the induction of a lethal phenotype, an additional regulatory circuits, involving a heterologous RNA polymerase and its inhibitor, were generated by coupling expression of the stv gene to the bacteriophage T7 transcription system (7) (Fig. 1). In this case, the stv gene was transcribed from the T7 gene 10 promoter (φ10) by T7 RNA polymerase. The T7 gene 1, encoding RNA polymerase, was fused to the E. coli hybrid trp-lac (tac) promoter, negatively regulated by the LacI repressor. The E. coli LacI-Olac system has been shown to be active in a broad range of microorganisms, including P. putida (18) and yeasts (19). The leakiness of the Ptac promoter was compensated by an inhibitor of T7 RNA polymerase, T7 lysozyme (20). The lysozyme, in addition to its muramidase activity, binds to T7 RNA polymerase and blocks its transcription activity. The T7 gene 3.5, encoding lysozyme, was constitutively transcribed from the Ptet promoter. The lacI gene was transcribed from the Pm promoter induced by XylS protein-aromatic carboxylic acid complexes. Both the Pm promoter and xylS regulatory gene (used xylS2 allele encodes XylSthr45 with altered effector specificity and increased affinity for benzoates) are derived from the P. putida TOL plasmid where they control expression of genes clustered in the meta-cleavage pathway operon (5). Streptavidin should be synthesized upon inactivation of LacI with IPTG or by interception of LacI synthesis in response to depletion of a benzoic acid effector of the XylS protein, such as 3MB. Positive effectors of the XylS protein can be taken up by P. putida or produced within the cell by oxidation of toluene, m- and p-xylenes, and their derivatives, through the TOL-encoded upper catabolic pathway (3MB from m-xylene) (5). Note that the bacteriophage T7 system, in the configuration described above, by itself causes stress to the cell when induced. Transcription from the strong φ10 and Ptac promoters, and overexpression of T7 RNA polymerase, for example, engage a large pool of ribonucleotides, and subsequently amino acyl tRNAs, and ribosomes.
Figure 1.

Scheme of a tightly regulated biological containment system to control survival of bacteria by availability of 3MB or other hydrocarbon effectors of the XylS protein. (A) Survival. (B) Induction of a lethal phenotype.
An additional level of regulation of stv gene expression was achieved by placing the φ10::stv fusion immediately downstream of the lacI gene, but in the opposite orientation (Figs. 1 and 2B). This should further decrease uninduced expression of the stv gene by generating an antisense RNA (21) complementary to the stv transcript. On the other hand, RNA synthesized from φ10 promoter upon induction of suicide should similarly block the remaining lacI mRNA and improve the kinetics of bacterial culture decay.
The T7 lysozyme and T7 RNA polymerase genes are also oppositely oriented. However, in this case, the level of accumulating countertranscript is likely to be lower because of the distance between Ptet and Ptac promoters (≈6 kb).
Induction of the Lethal Phenotype.
The entire suicide construct was prepared as a pCC-s05 (xylS2, lacI, and stv genes)/pRO-ilp (T7 RNA polymerase and lysozyme genes) broad host-range two-plasmid system (Fig. 2). Copy numbers of pCC-s05 and pRO-ilp in P. putida were estimated to be 1–3 and 10–20, respectively. Transfer of P. putida (pCC-s05, pRO-ilp) from a 3MB-containing medium to a 3MB-free medium resulted in inhibition of the culture growth within 3 h at 30°C (Fig. 3A). Replacement of the stv gene with a truncated gene encoding only the N-terminal half of the protein or the use of pCC102 plasmid lacking the φ10::stv fusion, instead of pCC-s05, caused almost no response of P. putida to the absence of 3MB inducer (data not shown).
Figure 3.

Kinetics of the induced suicide of P. putida cultures. Bacteria were grown to a mid-exponential growth phase in LB medium containing 3MB, ampicillin, and kanamycin. After washing with LB, cells were diluted 100-fold with LB supplemented with antibiotics with or without 3MB (A), or resuspended in the same medium without 3MB (B), and further incubated. Broken lines with open symbols refer to the additional presence of IPTG in LB. Plasmid combinations: s04, pCC-s04/pRO-ilp; s05, pCC-s05/pRO-ilp. CFU, colony forming unit. Each data point is the mean of two or three independent experiments.
The efficiency of host cell killing by intracellularly synthesized streptavidin was quantitated by counting viable cells before and after the removal of 3MB (Fig. 3B). Bacterial samples were periodically collected and incubated for a week on agar plates supplemented with 3MB, 50 μg/ml biotin, and appropriate antibiotics. At 4–8 h after induction of the stv gene expression by the removal of 3MB, up to 99.9% of the P. putida cells did not renew growth even after prolonged incubation in the presence of biotin. Longer incubation times were needed to contain the culture at lower temperatures. For example, 8–9 h were required to reduce the bacterial population by 90% at 20°C. As expected, removal of 3MB, together with the addition of IPTG, induced faster and more efficient cell death.
Replacement of pCC-s05 with pCC-s04 containing the Olac (with Ptac) 41 bp upstream of φ10, at the edge of the DNA sequence covered by a promoter-bound T7 RNA polymerase, resulted in remarkably slower killing upon removal of 3MB. However, in the additional presence of IPTG it resulted in killing of as much as 6 orders of magnitude of the initial bacterial population in 4 h. This represents one of the most rapid and efficient eliminations of a bacterial culture reported. Derepression of Ptac preceding φ10 on pCC-s04 by removal of 3MB did not induce a lethal level of the stv gene expression by bacterial RNA polymerase, presumably because of the presence of a putative transcription terminator/attenuator immediately downstream of φ10 and residual antisense expression in the absence of 3MB. The need for the T7 lysozyme also in P. putida with the Olacφ10 fusion on pCC-s04 was verified by removing the lysozyme gene from pRO-ilp (cutting the plasmid with SphI and religating). Although the resulting P. putida (pCC-s04, pRO-ip) was virtually contained upon removal of 3MB, the cell population grew very slowly, and the accumulation of mutants was significantly higher.
Rates of escape of P. putida from killing were estimated by fluctuation tests. Table 1 compares bacteria carrying plasmid constructs (i) producing different uninduced basal levels of active T7 RNA polymerase—i.e., with and without T7 lysozyme (pCC-s04/pRO-ilp or pCC-s04/pRO-ip), (ii) with or without direct LacI-Olac-dependent accessibility of the φ10 promoter for RNA polymerase (pCC-s05/pRO-ilp or pCC-s04/pRO-ilp), and (iii) with or without countertranscript protection against basal expression of the stv gene (pCC-s05/pRO-ilp and pVLTΔ-s02/pRO-llp). The construction of pVLTΔ-s02 is shown in Fig. 2A. pRO-llp differs from pRO-ilp by having the pCC102-derived lacI gene instead of a 1.3-kb luc DNA spacer. The level of accumulation of killing-resistant clones in P. putida with the pCC-s05/pRO-ilp plasmid combination was about two orders of magnitude lower than in the construct without antisense expression, although pCC- and pVLT-based hybrid plasmids (both RSF1010 derivatives) are not completely isogenic. For the pCC-s04/pRO-ilp combination, which has the Olac also next to φ10, killing-resistant mutants appeared with the lowest frequency. The absence of T7 lysozyme within a cell increased the level of mutation by two orders of magnitude.
Table 1.
Survival of P. putida, carrying different constructs for protection against uninduced expression of the stv gene, upon induction of suicide
| Plasmid combination | Surroundings of the stv gene | Survival per cell per generation | |
|---|---|---|---|
| pVLTΔ-s02, pRO-llp |
|
10−4–10−5 | |
| pCC-s05, pRO-ilp |
|
10−6–10−7 | |
| pCC-s04, pRO-ilp |
|
10−7–10−8 | |
| pCC-s04, pRO-ip |
|
10−5–10−6 |
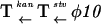
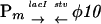
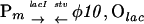
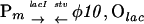
Each arrow indicates the direction of transcription. T, bacterial transcription terminator; Olac, lac operator.
Restriction nuclease cleavage analysis of plasmids from 20 killing-resistant clones showed, in most cases, changes in DNA digestion patterns involving the stv and T7 RNA polymerase gene regions (data not shown).
DISCUSSION
The conditional lethal system presented here consists of the streptavidin gene as a key suicide element. The biotin-binding ability of streptavidin in P. putida is notable because it requires not only proper peptide folding but also formation of the correct tetrameric structure. So far, there are only several genes that have been proven to be bactericidal upon expression in the members of genus Pseudomonas. These are genes encoding cell membrane destabilizing peptides Hok (E. coli plasmid R1) (22) and Gef (E. coli) (23), lysis genes of bacteriophages λ and φX174 (24), and the colE3 gene encoding an RNase (E. coli) (25). It is also worth mentioning that the T7 lysozyme maintains its T7 RNA polymerase inhibition ability in a heterologous P. putida system.
The rate of inactivation, a basic determinant of the efficiency of suicide designs, in our stv-based system is 10−7–10−8 per cell per generation. This value is lower than those already reported for constructs based on single toxic functions (10−2–10−6), even in E. coli, which is easier to contain (9, 23, 26, 27). It apparently reflects the very low basal level of streptavidin in cells. Despite the fact that the coupled T7 transcription/LacI-Olac system is less leaky than the lac system alone (Table 1), an additional explanation of the better protection of cells against uninduced expression of the stv gene is synthesis of the antisense RNA originating from the Pm promoter. The lack of such protection, especially without a transcription terminator (e.g., LacI-Olac complex) upstream of φ10 as in pVLTΔ-s02, resulted in a remarkably higher mutation frequency of 10−4–10−5 per cell per generation. The LacI-Olac complex formed upstream of the φ10 in pCC-s04 apparently interferes also with binding of bacteriophage RNA polymerase to the φ10 promoter (slower induction of the stv gene expression upon removal of 3MB) and further decreases the frequency of appearance of killing-resistant clones by reducing uninduced transcription of the stv gene. Preliminary data on the killing of E. coli by IPTG-induced expression of the stv gene suggest that, as expected, this system should be effective also in enteric bacteria.
A temperature shift from 30°C to 20°C alters the kinetics of the culture decay. This effect was more pronounced in a bacterial population depleted of 3MB in the absence of IPTG, indicating a contribution of both weaker interaction of T7 RNA polymerase with φ10 and the longer lifetime of the LacI repressor and/or its mRNA within a cell. Continuing decline of the culture after the addition of IPTG indicates a lower rate of mutational inactivation of the construct at 20°C than at 30°C.
The suicide design was tested under rich nutrient conditions—i.e., in LB medium containing some biotin. The growth conditions were favorable for cell survival, since killing occurs by depletion of biotin. In fact, slightly higher levels of IPTG-induced cell death were observed in minimal M9 medium (not shown). Under mostly starving conditions in, for instance, soil or seawater, this system may perform even more effectively. A SmaI fragment of pCC-s05 containing xylS2, lacI, and stv genes (Fig. 2B) has been already stably integrated into P. putida chromosome via mini-Tn5-mediated transposition (data not shown). Other known killing genes will be placed under control of φ10 promoter and integrated into the chromosome of P. putida {hsdR1 stv lacI xylS2 Kanr} to create multiple back up systems following incorporation of the T7 RNA polymerase-lysozyme regulatory module. Alternatively, stability of pRO-ilp upon induction of suicide in the absence of selection for plasmid maintenance can be achieved, for instance, by inserting into it the parB (hok/sok) locus of R1.
To summarize, additional regulatory circuits arranged to reduce the basal level of the expression of killing gene can remarkably increase the effectiveness of cell suicide machinery. Because of the general demand of biotin as a carboxyl carrier in the living world, the stv gene can serve as a universal cassette for programmed cell death. The stv-based system in combination with, for example, biotinylated solid supports and biotinylated fluorescent probes could also allow very sensitive monitoring of the presence of recombinant microorganisms.
Acknowledgments
We are indebted to R. H. Olsen for providing pRO1614, F. W. Studier for pLysE, S. Tabor for mGP1-2, and K. N. Timmis for pCC102, pVLT33, and P. putida mt-2. This work was supported by U.S. Department of Defense Grant DAAH04-94-2004.
Footnotes
Abbreviations: GEM, genetically engineered microorganism; IPTG, isopropyl β-d-thiogalactopyranoside; 3MB, m-methylbenzoate; LB, Luria–Bertani.
References
- 1.Molin S. Curr Opin Biotechnol. 1993;4:299–305. doi: 10.1016/0958-1669(93)90099-i. [DOI] [PubMed] [Google Scholar]
- 2.Molin S, Boe L, Jensen L B, Kristensen C S, Givskov M, Ramos J L, Bej A K. Annu Rev Microbiol. 1993;47:139–166. doi: 10.1146/annurev.mi.47.100193.001035. [DOI] [PubMed] [Google Scholar]
- 3.Ramos J L, Andersson P, Jensen L B, Ramos C, Ronchel M C, Díaz E, Timmis K N, Molin S. Bio/Technology. 1995;13:35–37. doi: 10.1038/nbt0195-35. [DOI] [PubMed] [Google Scholar]
- 4.Argaraña C E, Kuntz I D, Birken S, Axel R, Cantor C R. Nucleic Acids Res. 1986;14:1871–1882. doi: 10.1093/nar/14.4.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marqués S, Ramos J L. Mol Microbiol. 1993;9:923–929. doi: 10.1111/j.1365-2958.1993.tb01222.x. [DOI] [PubMed] [Google Scholar]
- 6.Franklin F C H, Bagdasarian M, Bagdasarian M M, Timmis K N. Proc Natl Acad Sci USA. 1981;78:7458–7462. doi: 10.1073/pnas.78.12.7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Studier F W, Rosenberg A H, Dunn J J, Dubendorff J W. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 8.Brosius J, Holy A. Proc Natl Acad Sci USA. 1984;81:6929–6933. doi: 10.1073/pnas.81.22.6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Contreras A, Molin S, Ramos J-L. Appl Environ Microbiol. 1991;57:1504–1508. doi: 10.1128/aem.57.5.1504-1508.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olsen R H, DeBusscher G, McCombie W R. J Bacteriol. 1982;150:60–69. doi: 10.1128/jb.150.1.60-69.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sano T, Pandori M W, Chen X, Smith C L, Cantor C R. J Biol Chem. 1995;270:28204–28209. doi: 10.1074/jbc.270.47.28204. [DOI] [PubMed] [Google Scholar]
- 12.de Lorenzo V, Eltis L, Kessler B, Timmis K N. Gene. 1993;123:17–24. doi: 10.1016/0378-1119(93)90533-9. [DOI] [PubMed] [Google Scholar]
- 13.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 14.Bagdasarian M, Timmis K N. Curr Top Microbiol Immunol. 1982;96:47–67. doi: 10.1007/978-3-642-68315-2_4. [DOI] [PubMed] [Google Scholar]
- 15.Knudsen S M, Karlstrom O H. Appl Environ Microbiol. 1991;57:85–92. doi: 10.1128/aem.57.1.85-92.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weber P C, Ohlendorf D H, Wendolski J J, Salemme F R. Science. 1989;243:85–88. doi: 10.1126/science.2911722. [DOI] [PubMed] [Google Scholar]
- 17.Fall R R. Methods Enzymol. 1979;62:390–398. doi: 10.1016/0076-6879(79)62246-2. [DOI] [PubMed] [Google Scholar]
- 18.Bagdasarian M M, Amann E, Lurz R, Ruckert B, Bagdasarian M. Gene. 1983;26:273–282. doi: 10.1016/0378-1119(83)90197-x. [DOI] [PubMed] [Google Scholar]
- 19.Amore R, Kotter P, Kuster C, Ciriacy M, Hollenberg C P. Gene. 1991;109:89–97. doi: 10.1016/0378-1119(91)90592-y. [DOI] [PubMed] [Google Scholar]
- 20.Moffat B A, Studier F W. Cell. 1987;49:221–227. doi: 10.1016/0092-8674(87)90563-0. [DOI] [PubMed] [Google Scholar]
- 21.Eguchi Y, Itoh T, Tomizawa J. Annu Rev Biochem. 1991;60:631–652. doi: 10.1146/annurev.bi.60.070191.003215. [DOI] [PubMed] [Google Scholar]
- 22.Molin S, Klemm P, Poulsen L K, Biehl H, Gerdes K, Andersson P. Bio/Technology. 1987;5:1315–1318. [Google Scholar]
- 23.Jensen L B, Ramos J L, Kaneva Z, Molin S. Appl Environ Microbiol. 1993;59:3713–3717. doi: 10.1128/aem.59.11.3713-3717.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kloos D-U, Strätz M, Güttler A, Steffan R J, Timmis K N. J Bacteriol. 1994;176:7352–7361. doi: 10.1128/jb.176.23.7352-7361.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Díaz E, Munthali M, de Lorenzo V, Timmis K N. Mol Microbiol. 1994;13:855–861. doi: 10.1111/j.1365-2958.1994.tb00477.x. [DOI] [PubMed] [Google Scholar]
- 26.Ahrenholtz I, Lorenz M G, Wackernagel W. Appl Environ Microbiol. 1994;60:3746–3751. doi: 10.1128/aem.60.10.3746-3751.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Recorbet G, Robert C, Givaudan A, Kudla B, Normand P, Faurie G. Appl Environ Microbiol. 1993;59:1361–1366. doi: 10.1128/aem.59.5.1361-1366.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]