

Abstract
We report that interferon γ (IFN-γ) inhibits transcription of the macrophage scavenger receptor gene by antagonizing the Ras-dependent activities of AP-1 and cooperating ets domain transcription factors, apparently as a result of competition between AP-1/ets factors and activated STAT1 for limiting amounts of CBP and p300. Consistent with this model, STAT1α interacts directly with CBP in cells, and microinjection of anti-CBP and anti-p300 antibodies blocks transcriptional responses to IFN-γ. Cells lacking STAT1 fail to inhibit AP-1/ets activity, and overexpression of CBP both potentiates IFN-γ-dependent transcription and relieves AP-1/ets repression. Thus, CBP and p300 integrate both positive and negative effects of IFN-γ on gene expression by serving as essential coactivators of STAT1α, modulating gene-specific responses to simultaneous activation of two or more signal transduction pathways.
Interferon γ (IFN-γ) exerts antiviral and antiproliferative effects in many cell types and cooperates with other cytokines and regulatory molecules to coordinate diverse aspects of macrophage activation (1). Transcriptional responses to IFN-γ require STAT1α, which becomes tyrosine-phosphorylated, dimerizes, and binds to specific gamma activated sites (GASs) in target genes (2–5). To investigate the mechanisms by which IFN-γ also exerts antagonistic effects on gene expression, we have used the scavenger receptor A (SR-A) gene as a model. The SR-A gene encodes a macrophage-specific integral membrane protein that has been proposed to play roles in cell adhesion (6) and the clearance of oxidatively modified proteins and other polyanionic molecules (7). Expression of the SR-A gene is positively regulated by macrophage colony-stimulating factor (M-CSF; refs. 8–10) and is inhibited by IFN-γ (11).
Transcriptional activation of the scavenger receptor gene in response to M-CSF has been proposed to be mediated by members of the AP-1 and ets domain families of transcription factors that bind as ternary complexes to regulatory elements within the SR-A promoter and enhancer (12). The binding of M-CSF to its receptor initiates, among other events, a Ras-dependent protein kinase cascade that results in the activation of a family of mitogen-activated protein kinases (13). These kinases in turn phosphorylate serine and threonine residues within the transcriptional activation domains of AP-1 and ets factors (14), resulting in increased transcriptional activity. Recent studies suggest that transcriptional activation by AP-1 and ets domain proteins requires interactions with the coactivator proteins CREB binding protein (CBP) and the adenovirus E1A-associated protein, p300 (15). CBP and p300 are large, structurally and functionally conserved proteins (16) that have been demonstrated to play essential coactivator roles for several classes of regulated transcription factors, including CREB (17), nuclear receptors (18) and myogenic basic helix–loop–helix transcription factors (19). In this manuscript, we present evidence indicating that CBP and p300 serve to mediate both positive and negative transcriptional responses to the JAK/STAT signal transduction pathway by serving as essential coactivators of STAT1α, suggesting a mechanism for the integration of transcriptional responses to the simultaneous activation of the JAK/STAT and Ras/AP-1 signal transduction pathways during macrophage development.
MATERIALS AND METHODS
Analysis of SR–Human Growth Hormone (hGH) Expression in Transgenic Mice and Solid Phase Kinase Assays.
Bone marrow cells enriched for progenitor cells (20) were obtained from transgenic mice expressing the SR6.5–hGH and SR-enhancer/promoter (EP) transgenes and treated with human M-CSF (20 ng/ml) for 3 days before determination of hGH content as described (10, 21). At least three independently derived transgenic lines were studied for each transgene to exclude integration site artifacts. In solid phase kinase assays, equivalent amounts of glutathione S-transferase (GST)–c-Jun1–79 and GST–ets2 fusion proteins bound to glutathione-agarose beads were incubated with cell lysates prepared from serum-starved HeLa cells or serum-starved cells treated with serum, 12-O-tetradecanoylphorbol 13-acetate (TPA), and/or IFN-γ for 15 min and washed. Kinase assays were performed as described (22), and the GST fusion proteins were resolved by SDS/PAGE and subjected to autoradiography.
Transient Transfection Analysis and Antibody Microinjection.
THP-1, U937, and HeLa cells were transfected (23) with the described promoter constructs (12, 23) and treated with 1000 units/ml IFN-γ and/or 0.1 μM TPA unless otherwise indicated. Eight copies of the consensus GAS sequence TTCTCGGAA (24) were placed upstream of the minimal prolactin promoter to generate 8 × GAS–luciferase. Insulin-responsive Rat-1 fibroblasts were injected with plasmids and CBP-specific or p300-specific antibodies as previously described (25). Approximately 1 hr after injection, the cells were stimulated as indicated with 1000 units/ml IFN-γ and analyzed the next day as described (25). In peptide neutralization experiments, the CBP 2–22 peptide was preincubated with the antibody in the same buffer at approximately a 2:1 molar ratio of peptide to antibody.
Coimmunoprecipitation and in Vitro Protein–Protein Interaction Assays.
The 293 fibroblast cell line was transiently transfected with the full-length CBP–flag construct described by Kamei et al. (18) or with the indicated GAL4–CBP fusion proteins. Immunoprecipitations, Western blotting, and detection were carried out using a monoclonal anti-FLAG antibody or an anti-GAL4 DNA binding domain antibody exactly as previously described (18). For in vitro assays, equal amounts of GST fusion proteins bound to glutathione-agarose beads (Sigma) were incubated with 7 × 105 cpm of 35S-STAT1α or 35S-STAT1ΔC705 produced by translation in vitro for 2 hr at 4°C. After washing, bound STAT proteins were visualized by SDS/PAGE and autoradiography.
RESULTS
To identify genomic regulatory elements responsible for inhibitory effects of IFN-γ, experiments were performed in transgenic mice containing scavenger receptor regulatory elements linked to an hGH reporter gene (Fig. 1A). These regulatory elements contain binding sites for AP-1, AP-1/ets2 ternary complexes and PU.1 (Fig. 1A), that have been shown to be required for macrophage-specific expression in vivo and in vitro (10, 12, 23). Culture of bone marrow progenitor cells in the presence of M-CSF stimulated reporter gene expression in transgenic lines containing regulatory information extending to −6.5 kb from the transcriptional start site (SR6.5–hGH) or minimal enhancer-promoter information (SR-EP–hGH). This response depended on the AP-1 and ets2 binding sites, as specific mutations of these sites abolished M-CSF-dependent expression (SRmAE–hGH; Fig. 1B). IFN-γ inhibited M-CSF-dependent activation of both the SR6.5–hGH and the SR-EP–hGH transgenes, indicating that cis-active elements necessary for negative regulation were contained within the minimal enhancer-promoter regions (Fig. 1C).
Figure 1.

IFN-γ antagonizes M-CSF-dependent expression of scavenger receptor hGH transgenes. (A) SR–hGH fusion genes used to generate transgenic mice are illustrated, indicating the approximate positions of binding sites for AP-1, ets2, and PU.1 proteins. SRmAE–hGH contains point mutations in the AP-1/ets binding motifs that abolish AP-1 and ets2 interaction. (B) Regulation of the SR6.5-hGH, SR-EP–hGH, and SRmAE–hGH transgenes in bone marrow progenitor cells and progenitor cells treated for 3 days with M-CSF (20 ng/ml). (C) IFN-γ inhibition of M-CSF-dependent expression of SR6.5-hGH and SRmEP–hGH transgenes in bone-marrow-derived macrophages. (D) Inhibition of TPA-dependent expression of SR6.5–luciferase and SR-EP–luciferase fusion genes in THP-1 cells. The SR6.5 and SR-EP regulatory elements illustrated in A were fused to a luciferase reporter gene and transfected into THP-1 cells. Cells were treated with TPA and/or IFN-γ, and luciferase activity was quantitated 12 hr later. (E) IFN-γ inhibits TPA and Val-12 Ras-dependent activation of an AP-1/ets-dependent promoter. A luciferase reporter gene containing three copies of the composite AP-1/ets element from the SR-A promoter fused to the minimal (−36 to +33) rat prolactin promoter was transfected into HeLa cells. To stimulate AP-1 and ets activity, cells were either cotransfected with a vector directing the expression of a constitutively active form of Ras (Val-12 Ras) or treated with TPA.
Consistent with these data, TPA treatment of THP-1 monocytic leukemia cells, which undergo macrophage differentiation and up-regulate scavenger receptor gene expression following treatment with phorbol ester (26), markedly stimulated the activities of the SR6.5 and SR-EP regulatory elements, and this expression was strongly inhibited by IFN-γ (Fig. 1D). Based on the observation that M-CSF and TPA-dependent activation of the scavenger receptor gene is primarily mediated by AP-1 and cooperating ets domain transcription factors, the possibility that these transcription factors represented the targets of the inhibitory effects of IFN-γ was considered. To directly test this possibility, three copies of the AP-1/ets site from the scavenger receptor promoter were linked to a minimal TATA box-containing promoter. TPA treatment of transfected cells resulted in a 20-fold increase in promoter activity, and this increase was inhibited by approximately 75% by treatment with IFN-γ (Fig. 1E). Similar results were observed using analogous promoters that lacked either the AP-1 or the ets binding sites (data not shown). IFN-γ also inhibited activation of the AP-1/ets promoter in response to a constitutively active form of Ras (Fig. 1E). Electrophoretic mobility-shift assays failed to demonstrate effects of IFN-γ on the levels or composition of proteins binding to the AP-1/ets element (data not shown), suggesting that IFN-γ inhibited their transcriptional activities. Together, these results indicated that IFN-γ effectively inhibits transcription mediated by AP-1 and ets transcription factors.
To assess the transactivation functions of c-Jun and ets2, which are the major components of the AP-1/ets ternary complex that binds to the SR AP-1/ets motif in macrophage cell lines (12), the N-terminal 79 aa of c-Jun and the N-terminal 340 aa of ets2 were fused to the DNA binding domain of GAL4. These fusion proteins were tested for their ability to activate a a promoter containing four GAL4 binding sites. The GAL4–ets2 chimera was not sufficiently active to measure transcriptional inhibition (data not shown). As illustrated in Fig. 2A, however, the activity of the c-Jun transactivation domain was markedly potentiated by cotransfection of a constitutively active form of Ras, and this activity was in turn strongly inhibited by IFN-γ .
Figure 2.

IFN-γ inhibits the transcriptional activities of AP-1 and cooperating ets factors. (A) IFN-γ inhibits the activities of the c-jun transactivation domain. The N-terminal transactivation domain of c-jun was fused in frame to the GAL4 DNA binding domain and used to direct expression of a Gal4-dependent promoter. Transactivation domain function was stimulated by cotransfection of a Val-12 Ras expression plasmid. (B) Effects of IFN-γ on c-Jun kinase and ets2 kinase activities. HeLa cells were rendered quiescent by culture in serum-free media for 16 hr and then treated with IFN-γ, TPA, and/or serum as indicated. Cell lysates were then assayed for Jun N-terminal kinase and ets2 kinase activities (24). (C) Overexpression of CBP relieves IFN-γ antagonism of AP-1/ets activities. The 3× AP-1/ets-promoter was cotransfected into THP-1 cells with 1 μg of a CBP expression plasmid or an equivalent amount of empty expression plasmid and treated with IFN-γ and/or TPA. (D) Overexpression of CBP potentiates transcriptional responses to IFN-γ. A luciferase reporter gene containing eight GASs linked to a minimal prolactin promoter was cotransfected into HeLa cells with increasing amounts of a vector directing expression of CBP (24). Error bars for A, C, and D are standard deviations.
Ras-dependent activation of AP-1 and cooperating ets domain transcription factors is thought to result, in part, from phosphorylation of their respective transactivation domains by members of the mitogen-activated protein kinase family (14). IFN-γ has been reported to stimulate or inhibit mitogen-activated protein kinase activities, depending on cell type and culture conditions (27–30). Thus, antagonistic effects of IFN-γ on AP-1 and ets activities could potentially result from inhibition of activating kinases. However, using extracts prepared from cells in which inhibitory effects of IFN-γ on AP-1 activity were observed, IFN-γ treatment had little or no effect on Jun N-terminal kinase activity or ets2 kinase activity in TPA-treated or serum-stimulated cells (Fig. 2B), suggesting that alternative mechanisms account for the antagonistic effects of IFN-γ.
Transcriptional activation by AP-1 and ets proteins has been demonstrated to require CBP (15), which serves an essential coactivator role for several classes of transcription factors (17, 19, 31) and is structurally and functionally related to p300 (16, 32). Because CBP and p300 appear to be expressed at rate-limiting concentrations in most cells, the antagonistic effects of IFN-γ on AP-1/ets activity could potentially result from competition with activated STAT1α for coactivator interactions. Consistent with this possibility, overexpression of STAT1α potentiated AP-1/ets antagonism by IFN-γ (data not shown) and co-transfection of a CBP expression plasmid abolished the inhibitory effect of IFN-γ on AP-1/ets activity (Fig. 2C). Furthermore, cotransfection of increasing amounts of the CBP expression plasmid led to increasing levels of IFN-γ-dependent transcriptional activation from a minimal promoter containing multiple GAS elements (Fig. 2D), but had little or no effect on the activity of the minimal promoter alone (data not shown).
To directly determine whether CBP is functionally required for IFN-γ signaling, antibody microinjection experiments were performed (Fig. 3). IFN-γ-dependent transcription was assessed by co-injection of a lacZ reporter gene driven by a minimal TATA-containing promoter fused to multiple GASs. IFN-γ treatment of cells co-injected with control antibody resulted in detection of β-galactosidase activity in 60% of the injected cells. In contrast, co-injection of a specific anti-CBP antibody reduced the percentage of cells exhibiting detectable β-galactosidase activity to 29% (Fig. 3B). Preincubation of the CBP antibody with the immunizing peptide abolished its ability to inhibit IFN-γ-dependent transcription (Fig. 3B). Because the anti-CBP antibody did not completely block IFN-γ signaling, we considered the possibility that p300 might also contribute to STAT1α-dependent transcription. Consistent with this possibility, co-injection of a p300-specific antibody with the GAS-lacZ reporter gene reduced the percentage of cells exhibiting detectable β-galactosidase activity in response to IFN-γ from 60% to 25% (Fig. 2B). When the anti-CBP and anti-p300 antibodies were co-injected together with the GAS–lacZ reporter gene, IFN-γ-dependent expression was reduced to near basal levels (Fig. 2B). In contrast, co-injection of anti-CBP and/or anti-p300 antibodies had no effect on a comparable promoter in which transcription depends on multiple SP-1 binding sites (SV40; Fig. 3C) or on a dioxin-responsive promoter (data not shown).
Figure 3.

CBP and p300 are required for IFN-γ-dependent transcription. (A) Microinjection of a CBP-specific antiserum inhibits IFN-γ-dependent transcription. Rat-1 fibroblasts were injected with a lacZ reporter gene under the transcriptional control of a minimal promoter containing eight copies of a consensus GAS. Cells were co-injected with control rabbit IgG (1, 2, 4, 5) or rabbit anti-CBP IgG (3, 6), treated with vehicle (1, 4) or IFN-γ (2, 3, 5, 6), and analyzed 12 hr later. Expression of the lacZ reporter gene was determined by 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-Gal) staining (4–6). Injected cells were visualized by nuclear staining for IgG using a rhodamine anti-rabbit IgG (1, 2, 3) and by the presence of X-Gal staining, which partially quenches the rhodamine signal. Photomicrographs of representative injected cells are shown. (B) Co-injection of anti-CBP and anti-p300 IgG strongly inhibits IFN-γ-dependent transcription. Cells were injected with the GAS–lacZ reporter gene and anti-CBP IgG, anti-CBP preincubated with the CBP peptide antigen, anti-p300 IgG, or a combination of anti-CBP IgG and anti-p300 IgG as shown. The bar graph indicates the percentage of injected cells that exhibited detectable X-Gal staining. (C) Injection of anti-p300 IgG has no effect on basal levels of expression of a SP1-dependent (SV40)–lacZ reporter gene. For B and C, error bars represent the mean ± SEM of a minimum of three experiments in which at least 200 cells were injected.
To determine whether STAT1α interacts with a CBP-containing complex, cells were transfected with an epitope-tagged version of CBP and treated with IFN-γ for 15 min, and whole-cell extracts were immunoprecipitated with an antiserum to STAT1 or preimmune antiserum and assessed following Western blot analysis. CBP was selectively detected in lysates immunoprecipitated with anti-STAT1 antiserum (Fig. 4A, lanes 2 and 3). To identify regions of CBP that are required for STAT1 interaction, cells were transfected with plasmids directing expression of GAL4–CBP fusion proteins and treated with IFN-γ. Western blotting of STAT1α immunoprecipitates using an anti-GAL4 antibody indicated an IFN-γ-dependent interaction of STAT1α with an N-terminal 451-aa region (GAL–CBP-NR) as well as a constitutive interaction with an 830-aa central region of CBP that encompasses the bromo domain and E1A interaction domains of CBP (GAL–CBP-NS; Fig. 4B). In vitro translated STAT1α was also found to interact with a GST–CBP fusion protein containing the N-terminal 451 aa of CBP. This interaction was significantly reduced by truncation of 34 C-terminal aa that are required for transcriptional activity of STAT1α (ref. 33; STAT1ΔC705; Fig. 4C).
Figure 4.

Interactions of STAT1α with CBP (A) STAT1α interacts with CBP in cells. 293 cells were transfected with a vector directing expression of an epitope (FLAG)-tagged form of CBP and then treated with IFN-γ or vehicle for 15 min, and whole-cell lysates were prepared. Lysates were immunoprecipitated with an anti-STAT1 antiserum (lanes 2 and 3) or preimmune serum (lane 4). Immunoprecipitates or unprecipitated lysate (lane 1) were resolved by SDS/PAGE, transferred to nitrocellulose membranes, and subjected to Western blotting using an anti-CBP antiserum. (B) IFN-γ-dependent interactions of STAT1α with the N terminus of CBP. The indicated regions of CBP were linked in frame to the DNA binding domain of GAL4 and transfected into 293 cells. Cells were subsequently treated with IFN-γ or vehicle for 15 min, and whole-cell lysates were prepared. Lysates were immunoprecipitated with an anti-STAT1 antiserum and subjected to Western blotting using an anti-GAL4 DNA binding domain antibody. (C) Direct interactions of STAT1α with the N terminus of CBP. 35S-labeled STAT1α or STAT1ΔC truncated at amino acid 705 were produced by translation in vitro and incubated with GST–CBP1–451, GST–CBP-bromo domain, or GST alone. After washing and SDS/PAGE, specifically associated proteins were detected by autoradiography.
If competition between STAT1α and AP-1/ets factors for limiting amounts of CBP and p300 accounts for IFN-γ-dependent inhibition of AP-1/ets-dependent promoters, such antagonism should not occur in cells that lack STAT1. To test this prediction, U3A cells that lack STAT1 (4) were transfected with an AP-1/ets-dependent promoter and treated with combinations of TPA and IFN-γ. As illustrated in Fig. 5, treatment of these cells with IFN-γ had no effect on AP-1/ets-dependent transcription. In contrast, 2fTGH parental cells, which contain STAT1, were similar to the other cells tested in these studies in that IFN-γ treatment resulted in inhibition of AP-1/ets activity. Thus, AP-1/ets antagonism requires activated STAT1.
Figure 5.
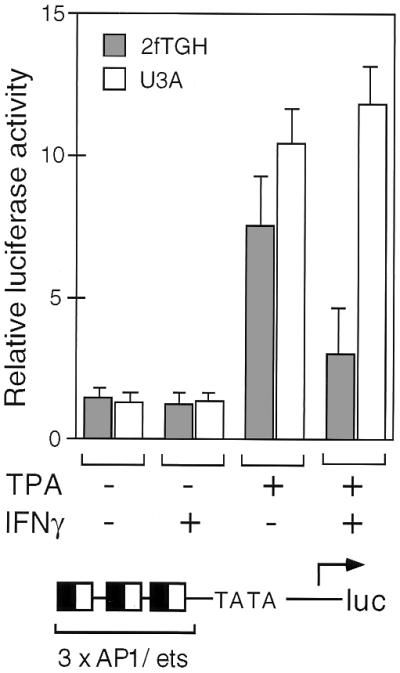
AP-1/ets inhibition requires STAT1. The luciferase reporter gene containing three copies of the composite AP-1/ets element described in Fig. 1E was transfected into U3A cells that lack STAT1 or parental 2fTGH cells that contain functional STAT1. Cells were treated with TPA and/or IFN-γ, as indicated, before assay for relative luciferase activity. Error bars are standard deviations
DISCUSSION
Macrophages develop specialized immunologic and homeostatic functions in response to specific combinations of cytokines, colony-stimulating factors, and other regulatory molecules. Thus, information provided by these molecules must be integrated at the level of transcription to achieve appropriate physiologic responses. Recent studies have suggested several potential mechanisms of cross-talk between these pathways that could mediate integrative functions proximal to the level of transcriptional activation (34–36). The studies presented in this manuscript support the existence of an additional nuclear mechanism for integration of the JAK/STAT and Ras/AP-1 signaling pathways, based on differential recruitment of CBP-containing and p300-containing coactivator complexes. Thus, the transcriptional outcomes to simultaneous stimulation of two or more signal transduction pathways will depend on the abundance of these factors and their relative affinities for CBP and p300. Indeed, some of the antiviral and growth inhibitory properties of IFNs result from competition between STAT factors and other classes of transcription factors for interaction with CBP and p300. Because AP-1 and ets factors contribute to transcriptional activation of some viral immediate/early genes, inhibition of AP-1/ets activities by IFN-γ could thus potentially account for some of its antiviral properties. The importance of this level of regulation between pathways is confirmed by failure of inhibition of AP-1/ets activities in cells lacking STAT1. The recent observation that haplo insufficiency of CBP is the apparent cause of the Rubenstein–Taybi Syndrome (37), characterized by mental retardation, dysmorphic facial features, and broad thumbs, also supports the idea that CBP levels are both limiting and critical for normal development. In concert, the present studies indicate that competition between various classes of regulated transcription factors for interaction with p300 and CBP, initially suggested to provide the basis for AP-1 antagonism by nuclear receptors (18), surprisingly represents a general mechanism for integration of additional signal transduction pathways required for establishing specific cellular phenotypes.
Acknowledgments
We thank Dr. Chris Schindler for a STAT1α cDNA expression vector, Dr. George Stark for U3A and 2fTGH cells, and Tanya Schneiderman for help in preparation of the manuscript. A.E.H. is supported by the National Institute of General Medical Sciences Training Program. G.B. is supported by a National Institute of Health Physician Scientist Award to the University of California at San Diego. D.W.R. is supported by an American Diabetes Association Career Development Award. M.G.R. is an Investigator with the Howard Hughes Medical Institute. C.K.G. is an Established Investigator of the American Heart Association. These studies were supported by grants from the National Institutes of Health to M.G.R. and C.K.G.
Footnotes
Abbreviations: IFN-γ, interferon γ; M-CSF, macrophage colony-stimulating factor; GAS, gamma activated site; hGH, human growth hormone; GST, glutathione S-transferase; TPA, 12-O-tetradecanoylphorbol 13-acetate.
References
- 1.Young H A, Hardy K J. J Leukocyte Biol. 1995;58:373–381. [PubMed] [Google Scholar]
- 2.Schindler C, Darnell J E J. Annu Rev Biochem. 1995;64:621–651. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 3.Pearse R N, Feinman R, Shuai K, Darnell J E, Jr, Ravetch J V. Proc Natl Acad Sci USA. 1993;90:4314–4318. doi: 10.1073/pnas.90.9.4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Müller M, Laxton C, Briscoe J, Schindler C, Improta T, Darnell J E J, Stark G R, Kerr I M. EMBO J. 1993;12:4221–4228. doi: 10.1002/j.1460-2075.1993.tb06106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shuai K, Horvath C M, Huang L H, Qureshi S A, Cowburn D, Darnell J E., Jr Cell. 1994;76:821–828. doi: 10.1016/0092-8674(94)90357-3. [DOI] [PubMed] [Google Scholar]
- 6.Fraser I, Hughes D, Gordon S. Nature (London) 1993;364:343–346. doi: 10.1038/364343a0. [DOI] [PubMed] [Google Scholar]
- 7.Davis W J, Sousa P A, Schultz R M. Dev Biol. 1996;174:190–201. doi: 10.1006/dbio.1996.0065. [DOI] [PubMed] [Google Scholar]
- 8.Fogelman A M, Haberland M E, Seager J, Hokom M, Edwards P A. J Lipid Res. 1981;22:1131–1141. [PubMed] [Google Scholar]
- 9.Ishibashi S, Inaba T, Shimano H, Harada K, Inoue I, Mokuno H, Mori N, Gotoda T, Takaku F, Yamada N. J Biol Chem. 1990;265:14109–14117. [PubMed] [Google Scholar]
- 10.Horvai A, Palinski W, Wu H, Moulton K S, Kalla K, Glass C K. Proc Natl Acad Sci USA. 1995;92:5391–5395. doi: 10.1073/pnas.92.12.5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geng Y J, Hansson G K. J Clin Invest. 1992;89:1322–1330. doi: 10.1172/JCI115718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu H, Moulton K, Horvai A, Parik S, Glass C K. Mol Cell Biol. 1994;14:2129–2139. doi: 10.1128/mcb.14.3.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roussel M. J Cell Sci. 1994;18:105–108. doi: 10.1242/jcs.1994.supplement_18.15. [DOI] [PubMed] [Google Scholar]
- 14.Karin M. Curr Opin Cell Biol. 1994;6:415–424. doi: 10.1016/0955-0674(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 15.Arias J, Alberts A S, Brindle P, Claret F X, Smeal T, Karin M, Feramisco J, Montminy M. Nature (London) 1994;370:226–229. doi: 10.1038/370226a0. [DOI] [PubMed] [Google Scholar]
- 16.Arany Z, Sellers W R, Livingston D M, Eckner R. Cell. 1994;77:799–800. doi: 10.1016/0092-8674(94)90127-9. [DOI] [PubMed] [Google Scholar]
- 17.Kwok R P, Lundblad J R, Chrivia J C, Richards J P, Bächinger H P, Brennan R G, Roberts S G, Green M R, Goodman R H. Nature (London) 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 18.Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin S-C, Heyman R, Rose D, Glass C, Rosenfeld M. Cell. 1996;85:1–12. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 19.Eckner R, Yao T-P, Oldread E, Livingston D M. Genes Dev. 1996;10:2478–2490. doi: 10.1101/gad.10.19.2478. [DOI] [PubMed] [Google Scholar]
- 20.Daley G Q, Van Etten R A, Baltimore D. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 21.Lira S A, Crenshaw E B, Glass C K, Swanson L W, Rosenfeld M G. Proc Natl Acad Sci USA. 1988;85:4755–4759. doi: 10.1073/pnas.85.13.4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hibi M, Lin A, Smeal T, Minden A, Karin M. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 23.Moulton K S, Semple K, Wu H, Glass C K. Mol Cell Biol. 1994;14:4408–4418. doi: 10.1128/mcb.14.7.4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanno Y, Kozak C A, Schindler C, Driggers P H, Ennist D L, Gleason S L, Darnell J E, Ozato K. Mol Cell Biol. 1993;13:3951–3963. doi: 10.1128/mcb.13.7.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rose D, McCabe G, Feramisco J, Adler M. J Cell Biol. 1992;119:1405–1411. doi: 10.1083/jcb.119.6.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moulton K S, Wu H, Barnett J, Parthasarathy S, Glass C K. Proc Natl Acad Sci USA. 1992;89:8102–8106. doi: 10.1073/pnas.89.17.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu J, Rockow S, Kim S, Xiong W, Li W. Mol Cell Biol. 1994;14:8018–8027. doi: 10.1128/mcb.14.12.8018. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Liu M K, Brownsey R W, Reiner N E. Infect Immun. 1994;62:2722–2731. doi: 10.1128/iai.62.7.2722-2731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J, Kim S, Chen M, Rockow S, Yi S E, Wagner A M, Hay N, Weichselbaum R R, Li W. Blood. 1995;86:2774–2788. [PubMed] [Google Scholar]
- 30.Singh K, Balligand J-L, Fischer T A, Smith T W, Kelly R A. J Biol Chem. 1996;271:1111–1117. doi: 10.1074/jbc.271.2.1111. [DOI] [PubMed] [Google Scholar]
- 31.Dai P, Akimaru H, Tanaka Y, Hou D-X, Yasukawa T, Kanei-Ishii C, Takahashi T, Ishii S. Genes Dev. 1996;10:528–540. doi: 10.1101/gad.10.5.528. [DOI] [PubMed] [Google Scholar]
- 32.Lundblad J R, Kwok P S, Laurance M E, Harter M L, Goodman R H. Nature (London) 1995;374:85–88. doi: 10.1038/374085a0. [DOI] [PubMed] [Google Scholar]
- 33.Wen Z, Zhong Z, Darnell J E J. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 34.Wu H, Klingmüller U, Besmer P, Lodish H F. Nature (London) 1995;377:242–246. doi: 10.1038/377242a0. [DOI] [PubMed] [Google Scholar]
- 35.David M, Petricoin E I, Benjamin C, Pine R, Weber M J, Larner A C. Science. 1995;269:1721–1723. doi: 10.1126/science.7569900. [DOI] [PubMed] [Google Scholar]
- 36.Darnell J E, Jr, Kerr I M, Stark G R. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 37.Petrij F, Giles R H, Dauwerse H G, Saris J J, Hennekam R C M, Masuno M, Tommerup N, van Ommen G-J, Goodman R H, Peters D J M, Breuning M H. Nature (London) 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]