

Abstract
Delta-like protein 1 (Dlk1) is a transmembrane protein characterized by epidermal growth factor (EGF)-like repeats. Dlk1, which is also known as preadipocyte factor 1 (pref-1) because of its ability to inhibit preadipocyte differentiation, regulates the differentiation of several other cell types through unknown mechanisms. To elucidate Dlk1 functions, identification of Dlk1-regulated target genes is critical. The observation that Dlk1 is expressed in many endocrine tissues suggests that Dlk1 may have endocrine-related functions. Because Dlk1 is expressed in GH producing cells, we hypothesize that one function of Dlk1 is to regulate GH expression. We found that GH mRNA, protein, and secretion were significantly decreased in GH3 pituitary cell clones that stably express Dlk1. In contrast, Dlk1 expression was unable to alter prolactin expression. Co-transfection of GH3 cells with a GH promoter-regulated reporter gene showed that Dlk1 repressed GH promoter activity. Deletion and mutation analysis of the GH promoter indicated that Pit-1 binding sites in the GH promoter are required for Dlk1-mediated repression. Furthermore, Dlk1 expression represses Pit-1-mediated transcription when both proteins are co-expressed in MCF-7 cells. Deletion analysis of Dlk1 revealed that the ability of Dlk1 to regulate GH promoter activity is independent of both its EGF-like repeats and its ability to modulate MAP kinase activity. The observation that Dlk1 regulates GH expression identifies the first endocrine function of Dlk1, establishes GH as a Dlk1-regulated target gene, and provides a model system to facilitate studies of Dlk1-mediated signaling.
Keywords: Dlk1, growth hormone, promoter
Introduction
Growth hormone (GH), which is mainly produced in the somatotroph cells of the anterior pituitary, is involved in the regulation of fat mass. When administered to GH-deficient adults, GH increases lean body mass and decreases body fat [1]. Treatment of the 3T3-L1 preadipocyte cell line with GH prevents these cells from differentiating to adipocytes, suggesting that blocking preadipocyte differentiation is one mechanism whereby GH can regulate body fat homeostasis. The ability of GH to block preadipocyte differentiation has been attributed to its ability to increase expression of Delta-like protein 1 (Dlk1) [2, 3], a known inhibitor of adipogenesis [4].
Dlk1, also known as pref-1 [4], pG2 [5] and zona glomerulosa specific mRNA [6], contains 6 EGF-like repeat domains in the extracellular domain, a transmembrane domain and a short cytoplasmic tail. It is cleaved by a yet unidentified protease to release a soluble circulating peptide containing the EGF-like repeats [7-10]; but its function remains elusive. This circulating peptide was originally identified as fetal antigen 1 [7]. By virtue of its EGF-like motifs, Dlk1 is a member of the EGF-like repeat containing protein family that includes other members such as notch, delta, and serrate [6]. Like other EGF-like repeat protein family members, Dlk1 is involved in regulating differentiation and cell fate determination. Dlk1 regulates the differentiation of several different cell lineages including skeletal stem cells [11], thymocytes [12] and adrenal gland cells [6], but the most studied function of Dlk1 is the regulation of preadipocyte differentiation [13].
Although several biological functions of Dlk1 have been described, how Dlk1 elicits its biological responses remains unknown because Dlk1 acts through signaling systems that differ from other EGF-like family members. Several other EGF-like protein family members exert their effects by binding to notch receptors via the DSL motif [14, 15]. Because Dlk1 lacks the DSL motif, it is unlikely that Dlk1 works through this pathway. In addition, a Dlk1 receptor has not been found to date.
A key step in the investigation of Dlk1 function is to identify a Dlk1-regulated target gene, thus establishing a model system that can be used to investigate Dlk1-mediated signaling. In addition to being expressed in preadipocytes, Dlk1 is also expressed in many endocrine tissues, including the growth hormone-producing somatotroph cells of the pituitary gland [16], insulin-producing β cells [17], sex hormone-producing leydig cells of the testis, and the theca interna and the Hilus cells of the ovary [18]. Dlk1 expression in such a large number of endocrine cells suggests that Dlk1 has endocrine functions. Because Dlk1 is expressed in somatotroph cells, we hypothesized that one function of Dlk1 is to regulate GH expression.
We found that GH mRNA, protein and secretion were significantly repressed in GH3 rat pituitary cell clones that stably express Dlk1. Our data showed that Dlk1 could repress Pit-1-medated transcription and that the ability of Dlk1 to repress GH promoter activity is independent of its EGF-like repeats. Therefore we have for the first time identified an endocrine function of Dlk1 and established GH as a Dlk1-regulated target gene. This study also provides a model system that can facilitate studies of Dlk1-mediated signaling.
Materials and Methods
Plasmids
All PCR primer sequences are written 5’ to 3’. All constructs were verified by DNA sequence analysis.
Full-length mouse Dlk1 (accession # BC052159) cDNA in the vector sport6 (sp-Dlk1) was purchased from Open Biosystems (Huntsville, AL). The hemaglutinin (HA) epitope was added to the 3’ end of Dlk1 cDNA and the resulting DNA was cloned into the expression vector pCI-neo (Promega, Madison, WI) expression vector that confers puromycin resistance to produce HA-Dlk1. This construct was used to establish Dlk1-expressing GH3 cell clones. The pCI-Dlk1 and FLAG-Dlk1 vectors were constructed by cloning the Dlk1 cDNA into pCI-neo with and without a C-terminal FLAG epitope respectively. These vectors were used to express Dlk1 in reporter assays. A construct which expresses amino acids 1-272 of Dlk1 (272Dlk1) has previously been shown to be sufficient to regulate adipogenesis [8, 19]. This 272Dlk1 construct was prepared utilizing the SmaI restriction site located in the extracellular domain and cloning the extracellular domain into pCI-neo. The dEGFDlk1 vector was prepared by PCR amplifying the nucleotides that code for amino acids 247-284 of FLAG-DLK1 using sense primer TGTCTGCAGGAAGAAGCGCGGGGCTAGCCCCG and antisense primer TAATACGACTCACTATAGGG and cloning into pCI-neo. The resultant Dlk1 mutant expresses a protein that contains amino acids 1-43 fused in frame to amino acids 247-385.
The C-terminal truncation of Dlk1 (340Dlk1), which codes for amino acids 1-340, was prepared by PCR amplification of pCI-Dlk1 with the sense primer CCCGTGCAGGTCACCCACCTGC and the antisense primer TTTGCGGCCGCTTAGCGCAGGTTGGACACCCAGG.
Single amino acids mutants of Dlk1 were prepared using FLAG-Dlk1 as template and PCR-based site-directed mutagenesis as previously described [20]. The sense and antisense primers used to make each construct are listed below:
T334A: GCCTGGGTGTCCAACCTGCGCTACAAC and
TTCGCACTTGTTGAGAAAGAC;
S337A: GCCAACCTGCGCTACAACCACATG and
CACCCAGGTTTCGCACTTGTTG;
Y341A: GCAAACCACATGCTTCGCAAGAAGAAG and
GCGCAGGTTGGACACCCAGGTTTC;
Y355A: GCTAACAGCGGCGAGGAGCTGGCGGTCAATATCATC and
CTGCAACAGGAGGTTCTTCTTCTTGCGAAGC;
S357A: GCCGGCGAGGAGCTGGCGGTCAATATC and
GTTATACTGCAACAGGAGGTTCTTC;
TT374/5AA: GCCGCTTTCAACAAGGAGGCTGGTG and
CATGTCAATCTTCTCGGGGAAG.
For reporter vectors, nucleotide numbering is relative to the transcriptional start site. The -500 GHp-luciferase (-500 GHp-Luc) reporter vector, kindly provided by Dr. Norman Eberhardt (Mayo Clinic, Rochester, MN), has been described previously [21]. The reporter constructs containing GH promoter deletions were prepared using PCR. The sense primers were GGGAGGAGCTTCTAGATTATCC for -130GHp-Luc, ATTAGCACAAGCCCGGGAGTGGCCCCATG for -100 GHp-Luc, AAGCTAGCCCCATGCATAAATGTACAC for -90 GHp-Luc and AAGCTAGCGAAACAGGTGGGGTCAACAG for -70 GHp-Luc. For all constructs the antisense primer was CTTTATGTTTTTGGCGTCTTCCA. After amplification, the promoter fragments were cloned into PGL3 basic luciferase reporter vector (Promega, Madison, WI).
The GH promoter reporter with a deletion of nucleotides from -130 to -65 (d130-65GHp-Luc) was prepared using PCR-based mutagenesis as previously described [20] with a sense primer AGGTGGGGTCAACAGTGGGAGAGAAGG and an antisense primer CCTCCCACACATGCTGGGATCATGCC.
The mutant construct -130PmtGHp-Luc was generated by destroying the two Pit-1 binding sites (-88/-75 and-124/-111) on 130GHp-Luc, using PCR-based mutagenesis as previously described [20]. First, a mutant ProxmtGHp-Luc that disrupted the proximal Pit-1 site (-88/-75) was constructed using sense primer GGGTTGAAATGTACACAGAAACAGGTGG and antisense primer GGGGCCACTGACGGGCTTGTGCTAATG. Next, it was used as the template to introduce the second mutation which destroys the distal Pit-1 site (-124/-111), using sense primer CCCGGGTAGCACAAGCCCGTCAGTGGCCC and antisense primer AATTTAGAAGCTCCTCCCACACATGC, thus generating -130PmtGHp-Luc, in which both Pit-1 binding sites are mutated. These mutations change the proximal Pit-1 binding site from ATGCATAAATG to GGGTTGAAATG and the distal Pit-1 binding from TATCCAT to TCCCGGG, with the mutated based underlined.
The Nrf2 expression vector and the 4X antioxidant response element luciferase reporter have been previously described [22].
Cell Culture and Transfections
Rat pituitary-derived GH3 cells, human breast cancer MCF-7 cells, and the retroviral packaging line 90.74 were obtained from American Tissue Type Collections (Manassas, VA). GH3 and MCF-7 cells were cultured in DMEM, 10% fetal bovine serum, penicillin, streptomycin, glutamine and incubated at 37 °C, 95% relative humidity and 5% CO2. 90.74 cells were cultured as above with the exception that calf serum was used instead of fetal bovine serum.
To generate stable Dlk1-expressing cell clones, 100 mm dishes of GH3 cells were transfected with 6 μg of the HA-Dlk1 expression construct using TransIT-LT1 transfection reagent (Mirus, Madison, WI) following the manufacturer’s instructions. Forty-eight hours post transfection, cells were cultured in regular media containing 2.5 ug/mL puromycin (Sigma, St. Louis, MO), individual clones were expanded and western blotting performed to determine Dlk1 expression. Clones that failed to express Dlk1 were used as Dlk1 negative controls.
To make GH3 stable cell clones using a retrovirus, 12 μg of empty retroviral expression vector or the same vector containing HA-tagged Dlk1 under the control of the CMV promoter were transfected into 100 mm dishes of the 90.74 retroviral packing line. Forty-eight hours post transfection, media from the packing line was filtered onto GH3 cells and 8μg/mL polybrene added to allow the viral infection. Forty-eight hours post infection the cells were treated with 2 μg/mL puromycin. Cell lysate from the surviving pool of cells was used for western blot analysis or for ELISA (see below).
For reporter assays, GH3 and MCF-7 cells were plated in 24 well plates and transiently transfected using TransIT-LT1. For GH3 cells, each well was transfected with 200 ng of expression vector (200 ng of the empty pCI vector for control; 100 ng Dlk1-expressing vector plus 100 ng of the empty pCI vector for Dlk1; 100 ng Pit-1-expressing vector plus 100 ng of the empty pCI vector for Pit-1; 100 ng Dlk1-expressing vector plus 100 ng of Pit-1-expressing vector for Dlk1 + Pit-1) and 100 ng of reporter vector, plus 200 ng of Lac Z expressing vector. For MCF-7 cells, each well was transfected with 100 ng of expression vector (100 ng of the empty pCI vector for control; 50 ng Dlk1-expressing vector plus 50 ng of the empty pCI vector for Dlk1; 50 ng Pit-1-expressing vector plus 50 ng of the empty pCI vector for Pit-1; 50 ng Dlk1-expressing vector plus 50 ng of Pit-1-expressing vector for Dlk1 + Pit-1) and 100 ng of reporter vector, plus 200 ng of Lac Z expressing vector. Two days post transfection, cells were lysed and luciferase activity, normalized with β-galactosidase activity, was measured as previously described [23]. For studies involving the inhibition of the MAP kinase pathway, the cells were treated with 40 μM PD98059 (New England Biolabs, Beverly, MA) for 4 hours prior to lysis. Statistical significance was calculated using a T-test.
Western Blotting
Cells were lysed with triton lysis buffer as previously described [24]. Total protein concentration was determined using the BCA Protein Assay Kit (Pierce, Rockford, IL). Thirty micrograms of total protein was separated on a 12.5 % acrylamide gel and transferred to PVDF. Membranes were probed with antibodies that recognize Dlk1 (Santa Cruz Biotechnology, Santa Cruz, CA), GH, prolactin (Prl) (National Hormone and Pituitary Program, Torrance, CA), or β-actin (Sigma). After incubation with appropriate secondary antibodies, specific proteins were detected using ECL Plus chemiluminescent reagent (Amersham Pharmacia, Piscataway, NJ).
Northern Blotting
Total RNA from GH3 cell clones was isolated using Trizol reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. Ten micrograms of total RNA was separated on a 1 % formaldehyde-agarose gel and transferred to a nylon membrane. Prehybridization, hybridization and washing was performed using the NorthernMax protocol (Ambion, Austin, TX) following manufacturers instructions. To generate a Rat GH probe, GH3 cDNA was PCR amplified using GCACCCTCGAGCCCAGATTCCAAACTGCTC as the sense primer and ATGATGCATCTTAATTTTATTAGGACAAAGTGTAGGGG as the antisense primer. The amplified product was then cloned into TOPO-TA vector (Invitrogen) and excised before labeling. The probe was labeled using the RadPrime DNA labeling System (Invitrogen) and purified using a Bio-spin 30 (BioRad, Hercules, CA).
ELISA
Pooled GH3 cell populations (105) infected with empty viral expression vector as a control or viral vector with Dlk1 expression were plated in triplicate into a 24-well plate. The following day, the cells were fed with fresh media. After overnight incubation, conditioned media were collected and the GH concentration was measured in duplicate using a GH ELISA kit (Amersham Biosciences). After conditioned media collection, the cells were lysed in triton lysis buffer as previously described [24] and the total protein concentration determined using the BCA Protein Assay Kit (Pierce, Rockford, IL). The secreted GH concentration measured in the conditioned media was then normalized to total cellular protein.
Results
Dlk1 regulates GH expression in GH3 Cells
To determine if Dlk1 can regulate GH expression, we generated independent GH3 cell clones that stably express mouse Dlk1 by stable transfection. For these experiments, rat-derived GH3 cells were used because they are a widely studied model of GH regulation. Mouse Dlk1 was used because it is the best-studied form of Dlk1. The expression of endogenously produced GH in Dlk1 expressing cell clones was determined by western blot analysis and compared to GH expression in Dlk1 negative clones. Two independent GH3 cell clones that stably express Dlk1 both demonstrated a marked reduction of endogenous GH protein levels when compared to the Dlk1 negative clones. GH expression from one Dlk1 expressing cell clone and two independent Dlk1 negative cell clones are shown in Figure 1A. GH3 cells also produce the pituitary hormone prolactin (Prl). To determine if the Dlk1-mediated repression is specific for GH, endogenous Prl expression was also tested in these cell clones. In contrast to GH, Dlk1 expression was unable to repress Prl expression (Figure 1A).
Figure 1. Dlk1 Represses GH Expression in GH3 Cells.
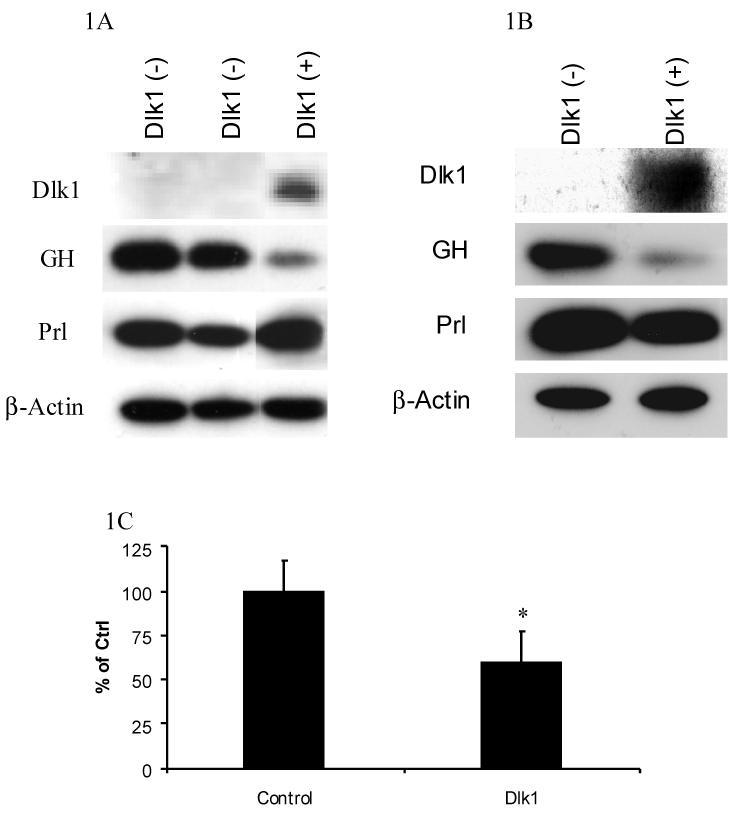
A. Western blot showing the expression of Dlk1, GH, Prl and the β-actin control in two Dlk1 negative (-) cell clones and in a Dlk1 expressing (+) cell clone. B. Western blot showing the expression of Dlk1, GH, Prl and the β-actin control in pooled GH3 cell populations that were produced by transducing with either a retrovirus containing empty vector or a Dlk1 expression vector. C. ELISA was used to measure the concentration of GH secreted into the media from pooled GH3 cell populations that were produced by transducing with a retrovirus containing empty vector or Dlk1. The secreted GH values were normalized to total cellular protein.
To confirm that the GH repression was due to the expression of Dlk1 and not because of an unrelated event such as clonality, we used an alternative method to express Dlk1 in GH3 cells. Using a retroviral-mediated approach, Dlk1 was expressed in GH3 cells and, using a pooled cell population, we compared endogenous GH expression in empty vector transduced cells to that in Dlk1 expressing cells. Western blot analysis showed that the cells transduced with a Dlk1 expression vector express less GH than cells transduced with empty vector alone (Figure 1B). Again, Dlk1 expression was unable to repress endogenous Prl expression, demonstrating that Dlk1-mediated gene regulation is specific for GH expression (Figure 1B).
We next determined if the Dlk1-mediated repression of GH expression resulted in a corresponding decrease in secreted GH. Using a retroviral-mediated approach, we expressed Dlk1 in GH3 cells and compared the GH concentrations in the media to that from empty vector transduced cells. Dlk1 expression resulted in a 40 % reduction in the concentrations of GH in the media, indicating that Dlk1 expression results in reduced GH secretion (Figure 1C).
Dlk1 Represses GH mRNA Expression and GH Promoter Activity
To determine if Dlk1 represses GH expression transcriptionally, northern blot analysis was performed to measure endogenous GH mRNA from one Dlk1 expressing GH3 clone and two Dlk1 negative GH3 clones. The Dlk1-expressing clone showed a reduction in endogenous GH mRNA levels, indicating that Dlk1 expression can repress GH gene transcription (Figure 2A).
Figure 2. Dlk1 Represses GH mRNA Expression and GH promoter Activity.
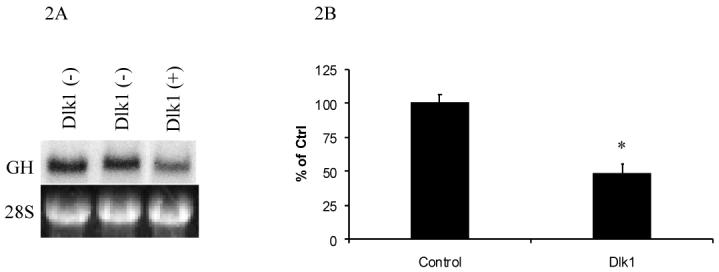
GH mRNA expression was examined by northern blotting in independent GH3 stably transfected cell clones that are Dlk1 negative or in a Dlk1 positive GH3 cell clone (A), showing the repression of GH mRNA in a Dlk1 expressing clone as compared to cells that do not express Dlk1. The 28S RNA loading control is shown immediately below. B. GH3 cells were transiently co-transfected with a luciferase reporter whose expression is regulated by the -500 to +6 region of the human growth hormone promoter (-500GHp-Luc) along with empty expression vector control or Dlk1 expressing vector, and luciferase activities were measured. Values are the mean ± standard deviation of three 3 separate experiments with duplicate samples per experiment. *: P<0.05.
Having determined that Dlk1 expression regulates GH mRNA expression, we tested the ability of Dlk1 to regulate GH promoter activity. A human GH promoter was used for this study because the human GH promoter has been well characterized in GH3 cells. A construct in which expression of the luciferase reporter gene is controlled by the -500 to +6 bp region of the human GH promoter (-500GHp-Luc) was transfected into GH3 cells. Luciferase activity was then monitored in the absence or presence of co-transfected Dlk1. As shown in Figure 2B, Dlk1 expression resulted in a 50% suppression of the activity of this GH promoter in GH3 cells.
A Pit-1 Response Element is Required for Dlk1-Mediated Regulation of the GH Promoter
To determine what cis-acting DNA element is required for Dlk1-mediated suppression of the GH promoter, reporter constructs containing successive deletions of the GH promoter were transiently transfected into GH3 cells in the absence or presence of co-transfected Dlk1. The constructs tested contained the -130 to +6 (-130GHp-Luc), the -90 to +6 (-90GHp-Luc), or the -70 to +6 (-70GHp-Luc) regions of the GH promoter controlling luciferase gene expression. Using this method, we have found that the sequence between -90 and -70 of the GH promoter is required for Dlk1-mediated regulation of the GH promoter (Figure 3).
Figure 3. Dlk1-Mediated Repression of GH Promoter Activity Requires Pit-1 Binding Sites.

The left panel depicts a schematic representation of the reporters being tested. Wild type proximal and distal Pit-1 binding sites are depicted as filled squares; mutated sites are depicted as unfilled squares. The names of the reporter vectors tested in order from top to bottom are -500GHp-Luc, -130GHp-Luc, -90GHp-Luc, -70GHp-Luc, d130-65GHp-Luc and -130PmtGHp-Luc. The right panel shows luciferase activities in the absence or presence of co-transfected Dlk1. Values are the mean ± standard deviation of three separate experiments with duplicate samples per experiment. *: P<0.05.
Previous reports have shown that the -90 to -70 region of the GH promoter contains a binding site for the transcription factor Pit-1/GHF-1 [25]. The overlap of the Pit-1 response element with the Dlk1 responsive region strongly implies that Dlk1-mediated GH repression occurs through a Pit-1-dependent mechanism. The GH promoter contains two separate Pit-1 response elements, a proximal site that maps to -88/-75 and a distal site that maps to -124/-111 [25]. A reporter construct that had the -130 to -65 region of the GH promoter deleted (d130-65GHp-Luc) was generated to determine if Dlk1 could regulate the activity of the GH promoter in the absence of the Pit-1 sites. Using this mutant, we found that Dlk1 failed to repress GH promoter activity when the -130 to -65 region was deleted, suggesting that Dlk1 represses GH promoter activity in a Pit-1-dependent manner (Figure 3).
To further confirm that Dlk1 regulates GH promoter activity through a Pit-1-mediated mechanism, the Pit-1 binding sites in the -130GHp-Luc were abolished by site-directed mutagenesis to generate -130PmtGHp-Luc. Dlk1 was unable to repress expression of this reporter construct, demonstrating that Pit-1 binding sites are required for Dlk1-mediated repression of GH promoter activity (Figure 3).
Dlk1 Regulates Pit-1-Mediated Transcriptional Activation
As expected, when the Pit-1 binding sites are mutated, GH promoter activity is relatively low. Therefore, the inability of Dlk1 to regulate the GH promoter in the absence of Pit-1 binding sites could be due to the fact that the promoter activity is already so low that Dlk1 cannot further repress activity. To confirm the involvement of Pit-1, we tested the ability of Dlk1 to repress Pit-1 mediated transcription in a different cell type. MCF-7 cells were chosen to co-express Dlk1 and Pit-1 because this cell line contains a very low concentration of endogenous Pit-1 [26]. Furthermore, when Pit-1 was transfected into MCF-7 cells, it can activate transcription [27].
MCF-7 cells were co-transfected with the -130GHp-Luc reporter along with either Pit-1 or Dlk1 alone, or Pit-1 and Dlk1 together, to examine if co-expressed Dlk1 would suppress Pit-1-dependent transcription. We found that transfection of Dlk1 alone has no effect on the reporter activity in MCF-7 cells (Figure 4). Transfection with Pit-1 alone resulted in an approximately 2-fold induction of reporter activity compared to the transfection with an empty expression vector, indicating that Pit-1 is capable of activating the -130GHp-Luc reporter in MCF-7 cells. When Dlk1 and Pit-1 were co-expressed, the ability of Pit-1 to activate expression of the reporter was greatly diminished (Figure 4), indicating that Dlk1 is capable of repressing Pit-1-dependent transcription in MCF-7 cells.
Figure 4. Dlk1 Regulates Pit-1 Transcriptional Activation in MCF7 cells.
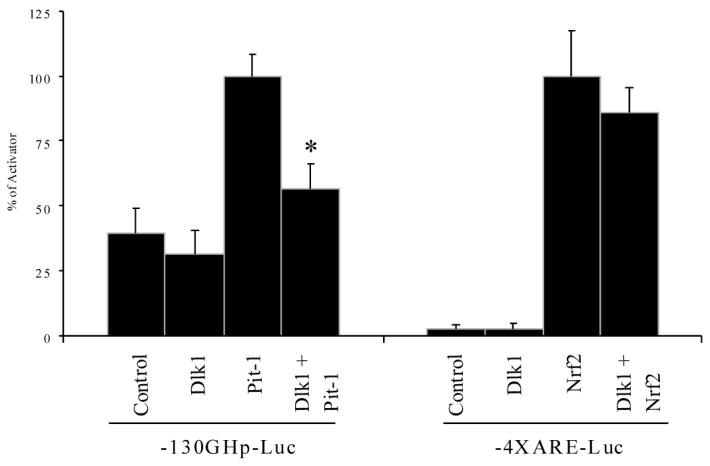
-130GHp-Luc was transfected into MCF-7 cells along with Dlk1 or Pit-1 alone, or with Dlk1 and Pit-1 together to determine if Dlk1 can repress Pit-1 activated transcription. As a control, the 4X antioxidant response element reporter (4XARE-Luc) was transfected into MCF-7 cells along with Dlk1 or Nrf2 alone, or with Dlk1 and Nrf2 together to determine if Dlk1 can repress transcription mediated by another unrelated transcription factor. Empty PCI expression vector was used to ensure that each well was transfected with equal amounts of DNA. Values are the mean ± standard deviation of three 3 separate experiments with duplicate samples per experiment. *: P<0.05.
To ensure that the ability of Dlk1 to repress transcriptional activation is specific for Pit-1 and not a nonspecific phenomenon, we tested whether Dlk1 could repress the function of an unrelated transcription factor Nrf2, which binds and activates transcription of promoters containing an antioxidant response element (ARE). This transcription factor gives robust transcriptional activation so any alteration in its activity will readily be detected [22, 28]. As shown in Figure 4, Dlk1 was unable to significantly repress transcription mediated by Nrf2, indicating that the ability of Dlk1 to repress Pit-1-induced transcription is specific.
Dlk1-Mediated GH Promoter Repression is Independent of its EGF repeats
We next examined which portion of Dlk1 is sufficient to repress GH promoter activity. Mouse Dlk1 is a protein of 385 amino acids that consists a signal peptide (amino acids 8-17), 6 EGF-like repeats (amino acids 26-246), a juxtamembrane domain which contains a protease cleavage site (amino acids 282-303), a transmembrane domain (amino acids 300-330), and a cytoplasmic tail (amino acids 331-385) [4, 8] (Figure 5). Expression vectors containing either wild type Dlk1 or different truncated Dlk1 mutants were transfected into GH3 cells to determine which portion of Dlk1 is required to regulate the activity of -500GHp-luc.
Figure 5. Repression of GH Promoter Activity by Dlk1 Deletion Mutants.

The left panel depicts a schematic representation of wild type Dlk1 and Dlk1 deletion constructs. Grey boxes represent the 6 EGF-like domains, the black box represents the juxtamembrane region and the dotted box represents the transmembrane region. The right panel shows luciferase activities in GH3 cells in the absence or presence of cotransfected Dlk1 mutants. The empty vector control is the top bar and set to 1, and the abilities of various Dlk1 mutants to regulate promoter activity are compared to the control. Values are the mean ± standard deviation of three 3 separate experiments with duplicate samples per experiment. *: P<0.05.
A Dlk1 mutant (272Dlk1) that expresses amino acids 1-272 of Dlk1, which includes all 6 EGF-like repeats but lacks the juxtamembrane, transmembrane and intracellular domains, was unable to repress GH promoter activity (Figure 5). Western blotting confirmed proper expression of this truncated protein (data not shown), indicating that this truncated protein lacks a functional domain required to regulate GH promoter activity.
A Dlk1 mutant, 340mDlk1, that expresses amino acids 1-340 of Dlk1 with the intracellular domain deleted, and another Dlk1 mutant, dEGFDlk1, with the EGF-like repeats deleted, were both able to repress GH promoter activity (Figure 5). Collectively these results show that the ability of Dlk1 to regulate GH promoter activity is independent of the EGF-like repeats, and the functional domain is located between amino acids 272-340.
Dlk1-Mediated Repression of the GH Promoter is Independent of its Phosphorylation
We next investigated whether potential phosphorylation of the intracellular domain of Dlk1 plays a role in its function to suppress GH promoter activity. The intracellular domain of Dlk1 contains a total of 7 serine, threonine and tyrosine residues. Site-directed mutagenesis was used to mutate each residue to alanine. Each site-directed mutant repressed activity of the GH promoter; indicating that abolishing potential phosphorylation sites individually does not affect the ability of Dlk1 to regulate GH expression (Figure 6).
Figure 6. Dlk1-Mediated GH promoter Repression is Independent of Dlk1 Phosphorylation.
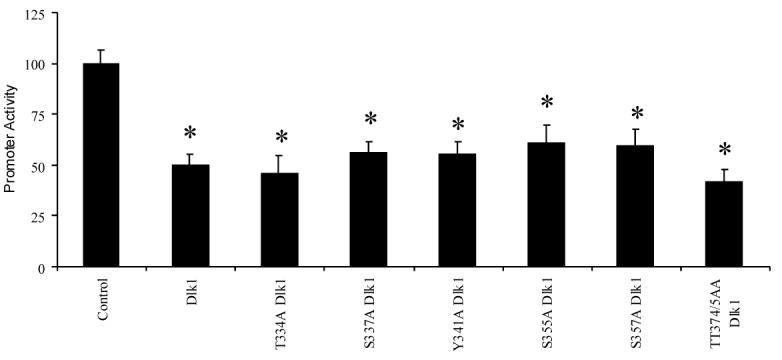
GH3 cells were transfected with -500GHp-Luc along with empty vector control or various phosphorylation-deficient Dlk1 mutants. The empty vector control (PCI) is set to 1, and the abilities of various Dlk1 mutants to regulate promoter activity are compared to the control. Values are the mean ± standard deviation of three separate experiments with duplicate samples per experiment. *: P<0.05.
Dlk1-Mediated GH Regulation is Independent of MAP Kinase
It has previously been shown that in cells with Dlk-1 expression knocked-down by antisense technology, IGF-I/insulin treatment resulted in a faster and higher-level activation of ERK/MAPK compared to cells with normal Dlk-1 expression [29]. Furthermore, inhibition of MAP kinase activity has been shown to repress GH promoter activity [30]. To determine if Dlk1 represses GH promoter activity through modulation of the MAP kinase pathway, we inhibited the MAP kinase pathway in GH3 cells with the specific blocker of this pathway, PD98059, and examined the effect of Dlk1 on GH promoter activity. PD98059 treatment alone of GH3 cells transfected with -500GHp-luc resulted in approximately a 50 % repression of luciferase activity (Figure 7A), similar to previously reported data [30]. In the presence of PD98059, Dlk1 represses further the GH promoter activity by approximately an additional 50%. The repression by PD98059 and by Dlk1 was additive (Figure 7A). As shown in Figure 7B, when the Dlk1-mediated GH promoter repression in the absence of PD98059 was compared with that in the presence of PD98059, it is clear that Dlk1-mediated repression of the GH promoter activity in the presence of PD98059 is very similar to its repression in the absence of PD98059. Therefore, Dlk1 can repress GH promoter activity when the MAP kinase pathway is blocked. This finding indicates that Dlk1 regulates the GH promoter through a MAP kinase-independent mechanism.
Figure 7. Dlk1-Mediated Regulation of GH promoter is MAP Kinase Independent.
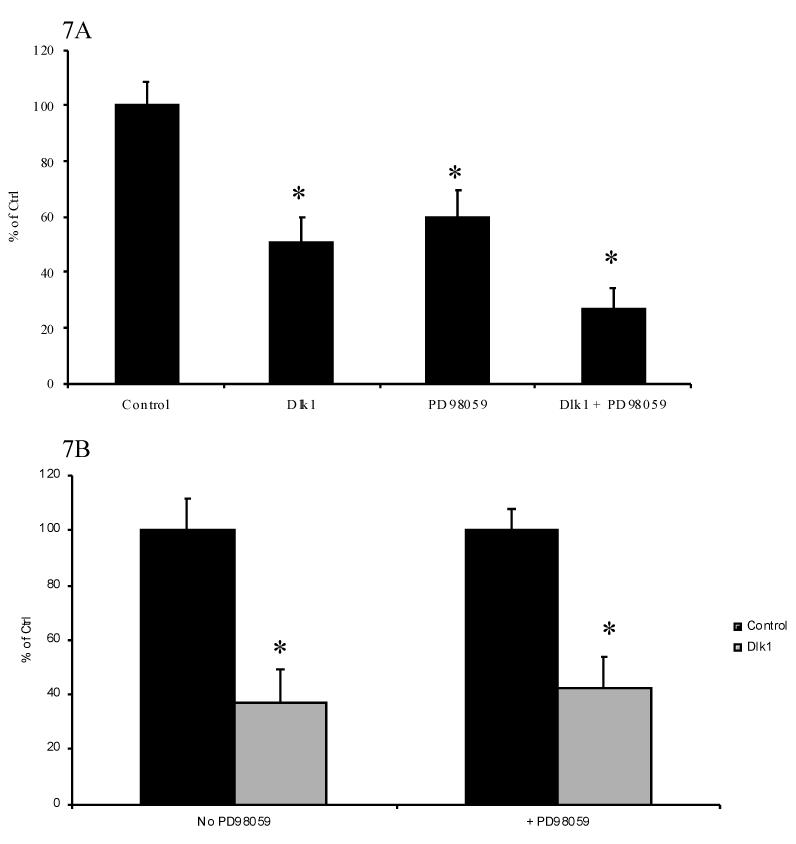
A. The ability of Dlk1 to regulate -500GHp-Luc was tested in GH3 Cells in the absence or presence of 40 μM PD98059. The empty vector control is set to 1. B. Comparison of Dlk1-mediated repression of GH promoter in the absence and presence of PD98059. Black bars: control, empty expression vector co-transfected. Grey bars: Dlk1-expressing vector co-transfected. Values are the mean ± standard deviation of three separate experiments with duplicate samples per experiment. *: P<0.05.
Discussion
Dlk1 is expressed in the normal pituitary gland [31, 32], specifically in the GH - producing somatotroph cells [16, 33]. Dlk1 is also expressed in pancreatic β cells [17], leydig cells and the theca interna and Hilus cells of the ovary [18], suggesting that Dlk1 has endocrine-related functions. Previous studies investigating potential endocrine links to Dlk1 have focused primarily on its regulation by a number of hormones. Dexamethasone increases Dlk1 expression in neuroblastoma cells [34] and downregulates Dlk1 expression in preadipocytes [35]. In pancreatic islets, both GH and prolactin induce Dlk1 expression [9, 36], and GH induces Dlk1 expression in preadipocytes [3]. Additionally, Andersen et al. have determined that either a pharmacological increase or decrease in GH levels results in a corresponding change in the concentration of circulating Dlk1 [37]. Our results show that GH mRNA, protein, and secretion are significantly decreased in pituitary-derived somatotroph GH3 cells that express Dlk1. Studies with the GH promoter indicate that Dlk1 represses GH promoter activity. Taken together, these results imply that Dlk1 represses GH transcription and therefore reveal a feedback regulatory loop to keep GH function precisely modulated.
Deletion analysis of the GH promoter revealed that Dlk1 repressed GH promoter activity through Pit-1 binding sites. Furthermore, Dlk1 is able to repress Pit-1-mediated transcription in MCF-7 cells. Collectively, these data suggest that Dlk1 inhibits GH expression by inhibiting the ability of Pit-1 to activate transcription of the GH promoter. Pit-1 is a pituitary-specific transcription factor that belongs to the POU domain class of proteins [38]. Members of the POU family are characterized by a DNA binding domain referred to as the POU domain [39]. Pit-1 is required for the terminal differentiation of somatotroph cells [40] and is responsible for the somatotroph-specific expression of GH [25]. It is interesting to note that Dlk1, a gene proposed to keep cells in an undifferentiated state [41] can repress the activity of a transcription factor known to promote terminal differentiation. In the developing mouse pituitary gland, Dlk1 is highly expressed while GH levels remain low [33]. As the pituitary develops, Dlk1 expression levels decrease. At the same developmental time when Dlk1 levels are decreasing, GH levels rise in a Pit-1-dependent manner [42]. The down-regulation of Dlk1 and the corresponding up-regulation of Pit-1-regulated GH at the same developmental timeframe is not only consistent with a GH regulatory role of Dlk1 in the normal pituitary gland, but also suggests a role for Dlk1 in the regulation of normal pituitary gland development.
It has been reported that Dlk1-overexpressing mice are smaller and weigh less than wild type littermates [10]. It has been suggested that this weight reduction is attributed to less fat (including brown adipose tissue, renal fat pad, inguinal fat pad, parametrial fat pad and epididymal fat pad). However, the overall decrease in body weight is much more substantial than the reported decrease in fat pad weight, suggesting that other factors are also involved in weight reduction. One of such factor could be the suppression of GH expression by over-expressed Dlk1. Interestingly, Dlk1 knockout animals are also smaller that the wild-type littermates [43]. This paradox could be explained in part by the ability of Dlk1 to control differentiation. In Dlk1-deficient animals, Dlk1 expressing tissues such as the pituitary gland may not differentiate and form properly. As a result, normal physiological processes such as growth would be affected.
The molecular mechanism underlying Dlk1 function remains unknown. Although Dlk1 belongs to the EGF-like family, it lacks the DSL motif by which other EGF-like protein family members exert their effects by binding to notch receptors through this motif [14, 15]. Therefore, Dlk1 must act through different signaling system(s). In addition, no receptor for the circulating form of Dlk1 has been found. We have generated constructs that express truncated Dlk1 to identify which region of Dlk1 is required to regulate GH promoter activity. We found that the functional motif required to regulate GH promoter activity was independent of the EGF-like repeats and its intracellular domain, and mapped to the juxtamembrane or transmembrane region of Dlk1. Consistent with our findings, previous experiments have shown that a construct encoding all EGF-like repeats, the transmembrane domain and the intracellular domain but lacking the juxtamembrane domain is unable to regulate adipocyte differentiation [44]. These data indicate that the juxtamembrane region of Dlk1 contains the functional motif required to regulate adipogenesis. If the same functional domains of Dlk1 are required to regulate GH expression as well as adipogenesis, it likely indicates that Dlk1 uses similar signaling pathways to regulate both processes. Therefore, understanding how Dlk1 regulates GH expression could provide insight as to how Dlk1 regulates adipocyte differentiation and possibly the differentiation of other Dlk1-regulated cell types, thus providing the molecular basis for Dlk1 function.
The juxtamembrane sequence of Dlk1 contains a series of lysine repeats characteristic of a leucine zipper protein dimerization domain [8]. Previous studies have shown that Dlk1 functions as a dimer, although the dimerization domain has yet to be mapped [12, 45]. It is possible that the juxtamembrane region functions as a protein dimerization domain whereby Dlk1 is able to homodimerize with itself or form a heterodimer with a yet to be identified signaling partner, and the resulting dimer transduces Dlk1-mediated signaling. Using site-directed mutagenesis, we determined if phosphorylation is responsible for mediating Dlk1 signaling; our data suggests that it is not. We also investigated if Dlk1 regulates GH expression via a MAP kinase-dependent mechanism. It has been shown that Dlk1 can modulate the activity of the MAP kinase pathway in the presence of growth factors such as insulin and IGF-I [29]. In 3T3 cells, when Dlk-1 expression was knocked down by antisense technology, IGF-I/insulin treatment resulted in a faster and higher-level activation of ERK/MAPK comparing to that in cells with normal Dlk-1 expression. In addition, an inhibition of the MAP kinase pathway has been shown to result in repression of the GH promoter by approximately 40% in GH3 cells [30]. However, using a chemical inhibitor PD98059, we have found that Dlk1-mediated GH repression is independent of the MAP kinase pathway. It has been shown that the MAPK response sequence in the GH promoter is located between -196 and -132 bp, and that Pit-1 is not involved in MAPK-mediated regulation of GH expression [30]. We have found that Dlk1-mediated GH repression requires Pit-1 and the response region is located between -90 and -70 bp. Therefore all these data are consistent with our conclusion that Dlk1-mediated GH repression is independent of the MAPK pathway.
In conclusion, we have shown that Dlk1 is able to repress GH expression in GH3 cells, likely through a Pit-1-dependent mechanism. The observation that Dlk1 regulates GH expression identifies the first endocrine function of Dlk1, establishes GH as a Dlk1-regulated target gene, and provides a model system that should facilitate studies of Dlk1-mediated signaling.
Acknowledgements
The authors would like to thank Mark Hannink (University of Missouri-Columbia) for providing the 4X-ARE and HA-NRF2 vectors and Norman Eberhardt (Mayo Clinic) for providing the -500GHp-Luc reporter vector.
Grant Support: National Institute of Health Grant R01-DK-40947 and the Jarislowsky Foundation.
Abbreviations
- Dlk1
Delta-like protein 1
- GH
growth hormone
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baum HB, et al. Effects of physiologic growth hormone therapy on bone density and body composition in patients with adult-onset growth hormone deficiency. A randomized, placebo-controlled trial. Ann Intern Med. 1996;125(11):883–90. doi: 10.7326/0003-4819-125-11-199612010-00003. [DOI] [PubMed] [Google Scholar]
- 2.Hansen LH, et al. Characterization of the inhibitory effect of growth hormone on primary preadipocyte differentiation. Mol Endocrinol. 1998;12(8):1140–9. doi: 10.1210/mend.12.8.0154. [DOI] [PubMed] [Google Scholar]
- 3.Wolfrum C, et al. Role of Foxa-2 in adipocyte metabolism and differentiation. J Clin Invest. 2003;112(3):345–56. doi: 10.1172/JCI18698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smas CM, Sul HS. Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell. 1993;73(4):725–34. doi: 10.1016/0092-8674(93)90252-l. [DOI] [PubMed] [Google Scholar]
- 5.Helman LJ, et al. The sequence of an adrenal specific human cDNA, pG2. Nucleic Acids Res. 1990;18(3):685. doi: 10.1093/nar/18.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okamoto M, et al. Zona glomerulosa-specific factor: cloning and function. Steroids. 1997;62(1):73–6. doi: 10.1016/s0039-128x(96)00162-6. [DOI] [PubMed] [Google Scholar]
- 7.Jensen CH, et al. Protein structure of fetal antigen 1 (FA1). A novel circulating human epidermal-growth-factor-like protein expressed in neuroendocrine tumors and its relation to the gene products of dlk and pG2. Eur J Biochem. 1994;225(1):83–92. doi: 10.1111/j.1432-1033.1994.00083.x. [DOI] [PubMed] [Google Scholar]
- 8.Smas CM, Chen L, Sul HS. Cleavage of membrane-associated pref-1 generates a soluble inhibitor of adipocyte differentiation. Mol Cell Biol. 1997;17(2):977–88. doi: 10.1128/mcb.17.2.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedrichsen BN, et al. Expression, biosynthesis and release of preadipocyte factor-1/ delta-like protein/fetal antigen-1 in pancreatic beta-cells: possible physiological implications. J Endocrinol. 2003;176(2):257–66. doi: 10.1677/joe.0.1760257. [DOI] [PubMed] [Google Scholar]
- 10.Lee K, et al. Inhibition of adipogenesis and development of glucose intolerance by soluble preadipocyte factor-1 (Pref-1) J Clin Invest. 2003;111(4):453–61. doi: 10.1172/JCI15924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdallah BM, et al. Regulation of human skeletal stem cells differentiation by Dlk1/Pref-1. J Bone Miner Res. 2004;19(5):841–52. doi: 10.1359/JBMR.040118. [DOI] [PubMed] [Google Scholar]
- 12.Kaneta M, et al. A role for pref-1 and HES-1 in thymocyte development. J Immunol. 2000;164(1):256–64. doi: 10.4049/jimmunol.164.1.256. [DOI] [PubMed] [Google Scholar]
- 13.Laborda J. The role of the epidermal growth factor-like protein dlk in cell differentiation. Histol Histopathol. 2000;15(1):119–29. doi: 10.14670/HH-15.119. [DOI] [PubMed] [Google Scholar]
- 14.Henderson ST, et al. Functional domains of LAG-2, a putative signaling ligand for LIN-12 and GLP-1 receptors in Caenorhabditis elegans. Mol Biol Cell. 1997;8(9):1751–62. doi: 10.1091/mbc.8.9.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simpson P. Developmental genetics. The Notch connection. Nature. 1995;375(6534):736–7. doi: 10.1038/375736a0. [DOI] [PubMed] [Google Scholar]
- 16.Larsen JB, et al. Fetal antigen 1 and growth hormone in pituitary somatotroph cells. Lancet. 1996;347(8995):191. doi: 10.1016/s0140-6736(96)90374-8. [DOI] [PubMed] [Google Scholar]
- 17.Tornehave D, et al. Fetal antigen 1 (FA1) in the human pancreas: cell type expression, topological and quantitative variations during development. Anat Embryol (Berl) 1993;187(4):335–41. doi: 10.1007/BF00185891. [DOI] [PubMed] [Google Scholar]
- 18.Jensen CH, et al. Fetal antigen 1, an EGF multidomain protein in the sex hormone-producing cells of the gonads and the microenvironment of germ cells. Mol Hum Reprod. 1999;5(10):908–13. doi: 10.1093/molehr/5.10.908. [DOI] [PubMed] [Google Scholar]
- 19.Garces C, et al. Adipocyte differentiation is modulated by secreted delta-like (dlk) variants and requires the expression of membrane-associated dlk. Differentiation. 1999;64(2):103–14. doi: 10.1046/j.1432-0436.1999.6420103.x. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Y, et al. Receptor internalization-independent activation of Smad2 in activin signaling. Mol Endocrinol. 2004;18(7):1818–26. doi: 10.1210/me.2004-0079. [DOI] [PubMed] [Google Scholar]
- 21.Shepard AR, Zhang W, Eberhardt NL. Two CGTCA motifs and a GHF1/Pit1 binding site mediate cAMP-dependent protein kinase A regulation of human growth hormone gene expression in rat anterior pituitary GC cells. J Biol Chem. 1994;269(3):1804–14. [PubMed] [Google Scholar]
- 22.Ansell PJ, et al. Repression of cancer protective genes by 17beta-estradiol: ligand-dependent interaction between human Nrf2 and estrogen receptor alpha. Mol Cell Endocrinol. 2005;243(12):27–34. doi: 10.1016/j.mce.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Willis SA, et al. Formation and activation by phosphorylation of activin receptor complexes. Mol Endocrinol. 1996;10(4):367–79. doi: 10.1210/mend.10.4.8721982. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Y, et al. Truncated activin type I receptor Alk4 isoforms are dominant negative receptors inhibiting activin signaling. Mol Endocrinol. 2000;14(12):2066–75. doi: 10.1210/mend.14.12.0570. [DOI] [PubMed] [Google Scholar]
- 25.Lefevre C, et al. Tissue-specific expression of the human growth hormone gene is conferred in part by the binding of a specific trans-acting factor. Embo J. 1987;6(4):971–81. doi: 10.1002/j.1460-2075.1987.tb04847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gil-Puig C, et al. Pit-1/GHF-1 and GH expression in the MCF-7 human breast adenocarcinoma cell line. J Endocrinol. 2002;173(1):161–7. doi: 10.1677/joe.0.1730161. [DOI] [PubMed] [Google Scholar]
- 27.Gonzalez MM, Carlberg C. Cross-repression, a functional consequence of the physical interaction of non-liganded nuclear receptors and POU domain transcription factors. J Biol Chem. 2002;277(21):18501–9. doi: 10.1074/jbc.M200205200. [DOI] [PubMed] [Google Scholar]
- 28.Ansell PJ, et al. In vitro and in vivo regulation of antioxidant response element-dependent gene expression by estrogens. Endocrinology. 2004;145(1):311–7. doi: 10.1210/en.2003-0817. [DOI] [PubMed] [Google Scholar]
- 29.Ruiz-Hidalgo MJ, et al. dlk modulates mitogen-activated protein kinase signaling to allow or prevent differentiation. Exp Cell Res. 2002;274(2):178–88. doi: 10.1006/excr.2001.5464. [DOI] [PubMed] [Google Scholar]
- 30.Gong FY, Deng JY, Shi YF. Stimulatory effect of interleukin-1beta on growth hormone gene expression and growth hormone release from rat GH3 cells. Neuroendocrinology. 2005;81(4):217–28. doi: 10.1159/000087160. [DOI] [PubMed] [Google Scholar]
- 31.Moreno CS, et al. Novel molecular signaling and classification of human clinically nonfunctional pituitary adenomas identified by gene expression profiling and proteomic analyses. Cancer Res. 2005;65(22):10214–22. doi: 10.1158/0008-5472.CAN-05-0884. [DOI] [PubMed] [Google Scholar]
- 32.Altenberger T, et al. Identification of DLK1 variants in pituitary- and neuroendocrine tumors. Biochem Biophys Res Commun. 2006;340(3):995–1005. doi: 10.1016/j.bbrc.2005.12.094. [DOI] [PubMed] [Google Scholar]
- 33.Yevtodiyenko A, Schmidt JV. Dlk1 expression marks developing endothelium and sites of branching morphogenesis in the mouse embryo and placenta. Dev Dyn. 2006 doi: 10.1002/dvdy.20705. [DOI] [PubMed] [Google Scholar]
- 34.Helman LJ, et al. Molecular markers of neuroendocrine development and evidence of environmental regulation. Proc Natl Acad Sci U S A. 1987;84(8):2336–9. doi: 10.1073/pnas.84.8.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smas CM, et al. Transcriptional repression of pref-1 by glucocorticoids promotes 3T3-L1 adipocyte differentiation. J Biol Chem. 1999;274(18):12632–41. doi: 10.1074/jbc.274.18.12632. [DOI] [PubMed] [Google Scholar]
- 36.Carlsson C, et al. Growth hormone and prolactin stimulate the expression of rat preadipocyte factor-1/delta-like protein in pancreatic islets: molecular cloning and expression pattern during development and growth of the endocrine pancreas. Endocrinology. 1997;138(9):3940–8. doi: 10.1210/endo.138.9.5408. [DOI] [PubMed] [Google Scholar]
- 37.Andersen M, et al. Fetal antigen 1 in healthy adults and patients with pituitary disease: relation to physiological, pathological, and pharmacological GH levels. J Clin Endocrinol Metab. 2001;86(11):5465–70. doi: 10.1210/jcem.86.11.7990. [DOI] [PubMed] [Google Scholar]
- 38.Andersen B, Rosenfeld MG. POU domain factors in the neuroendocrine system: lessons from developmental biology provide insights into human disease. Endocr Rev. 2001;22(1):2–35. doi: 10.1210/edrv.22.1.0421. [DOI] [PubMed] [Google Scholar]
- 39.Herr W, et al. The POU domain: a large conserved region in the mammalian pit-1, oct-1, oct-2, and Caenorhabditis elegans unc-86 gene products. Genes Dev. 1988;2(12A):1513–6. doi: 10.1101/gad.2.12a.1513. [DOI] [PubMed] [Google Scholar]
- 40.Li S, et al. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nature. 1990;347(6293):528–33. doi: 10.1038/347528a0. [DOI] [PubMed] [Google Scholar]
- 41.Floridon C, et al. Does fetal antigen 1 (FA1) identify cells with regenerative, endocrine and neuroendocrine potentials? A study of FA1 in embryonic, fetal, and placental tissue and in maternal circulation. Differentiation. 2000;66(1):49–59. doi: 10.1046/j.1432-0436.2000.066001049.x. [DOI] [PubMed] [Google Scholar]
- 42.Simmons DM, et al. Pituitary cell phenotypes involve cell-specific Pit-1 mRNA translation and synergistic interactions with other classes of transcription factors. Genes Dev. 1990;4(5):695–711. doi: 10.1101/gad.4.5.695. [DOI] [PubMed] [Google Scholar]
- 43.Moon YS, et al. Mice lacking paternally expressed Pref-1/Dlk1 display growth retardation and accelerated adiposity. Mol Cell Biol. 2002;22(15):5585–92. doi: 10.1128/MCB.22.15.5585-5592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mei B, et al. Only the large soluble form of preadipocyte factor-1 (Pref-1), but not the small soluble and membrane forms, inhibits adipocyte differentiation: role of alternative splicing. Biochem J. 2002;364(Pt 1):137–44. doi: 10.1042/bj3640137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohno N, et al. dlk inhibits stem cell factor-induced colony formation of murine hematopoietic progenitors: Hes-1-independent effect. Stem Cells. 2001;19(1):71–9. doi: 10.1634/stemcells.19-1-71. [DOI] [PubMed] [Google Scholar]