

Abstract
Filamentous bacteriophage are widely used as immunogenic carriers for “phage-displayed” recombinant peptides. Here we report that they are an effective immunogenic carrier for synthetic peptides. The f1.K phage was engineered to have an additional Lys residue near the N-terminus of the major coat protein, pVIII, so as to enhance access to chemical cross-linking agents. The dimeric synthetic peptide, B2.1, was conjugated to f1.K (f1.K/B2.1) in high copy number and compared as an immunogen to B2.1 conjugated to ovalbumin (OVA/B2.1) and to phage-displayed, recombinant B2.1 peptide. All immunogens were administered without adjuvant. The serum antibody titers were measured against: the peptide, the carrier, and, if appropriate, the cross-linker. All immunogens elicited anti-peptide antibody titers, with those elicited by OVA/B2.1 exceeding those by f1.K/B2.1; both titers were greater than that elicited by recombinant B2.1 phage. Comparison of the anti-peptide and anti-carrier antibody responses showed that f1.K/B2.1 elicited a more focused anti-peptide antibody response than OVA/B2.1. The anti-peptide antibody response against f1.K/B2.1 was optimized for the injection route, dose and adjuvant. Dose and adjuvant did not have a significant effect on anti-peptide antibody titers, but a change in injection route from intraperitoneal (IP) to subcutaneous (SC) enhanced anti-peptide antibody titers after seven immunizations. The optimized anti-peptide antibody response exceeded the anti-carrier one by 21-fold, compared to 0.07-fold elicited by OVA/B2.1. This indicates that phage as a carrier can focus the antibody response against the peptide. The results are discussed with respect to the advantages of phage as an alternative to traditional carrier proteins for synthetic peptides, carbohydrates and haptens, and to further improvements in phage as immunogenic carriers.
Keywords: Filamentous phage, Synthetic peptide, Immunization
1. Introduction
Synthetic peptides have been used in developing immunogens that target the humoral immune response; typically, to elicit antibodies (Abs) that cross-react with a discrete epitope on a protein (and sometimes on carbohydrate (CHO) [1-6] or DNA [7-9]). Currently, there are no FDA-approved conjugate vaccines like this; however, there is noteworthy research focused on developing peptides as immunogenic B-cell epitopes (BCEs). The goal of these studies is to use a peptide immunogen, instead of whole antigen (Agn), to produce Abs that cross-react with a single restricted epitope on a protein. For example, peptides are being used to produce Abs against specific epitopes on tumor Agns that decrease cell proliferation [10-13]. Peptides are also used as alternatives to CHO Agns because they can elicit a T-cell dependent immune response along with CHO-cross-reactive Abs [1-6]. Others have proposed using peptide immunogens to produce HIV-1 neutralizing Abs [14-16]. These few examples of epitope targeting illustrate the breadth of research that is focused on using BCE peptides to develop vaccines.
To effectively elicit an Ab response and develop immunological memory, most BCE peptides should be connected to other peptides or proteins that stimulate CD4 T-cell help. To do this, vaccine-lead peptides are chemically-conjugated to strongly-immunogenic “carrier” proteins that provide T-cell epitopes (TCEs). These typically include: ovalbumin (OVA) [17-19], tetanus toxoid (TT, which is FDA-approved) [2,12,20], BSA [3,18,21], and keyhole limpet hemocyanin (KLH) [5,6,18]. However, these types of carrier protein themselves elicit strong Ab responses such that immundominant BCEs on the carrier may undermine the Ab response against the synthetic peptide, especially if that peptide is less immunogenic than carrier BCEs. Furthermore, Abs directed against a carrier protein may bind non-specifically to the cognate Agn and give false positive results in tests for cross-reactivity [22]. These problems can be partially controlled by: (i) the choice of carrier protein; (ii) creating a particulate antigen, to enhance immunogenicity; and (iii) maximizing copy number of the peptide on the carrier. Other measures include optimization of dose, injection route, and adjuvant. Despite these measures, the Ab response against a peptide/carrier conjugate may be dominated by Abs against the carrier. However, this problem could be avoided by using a large, particulate carrier that elicits a relatively restricted B-cell response while providing CD4 T-cell help.
The immunogenic characteristics of filamentous bacteriophage make them good candidates for targeting a focused Ab response against synthetic peptides. Phage comprise 2000–4000 overlapping copies of a single, 50 amino acid protein (pVIII), which dominates the Ab response over three of the four other minor coat proteins present at the tips of the virion in low copy number (three to five per virion). The exception to this is the minor coat protein, pIII, whose ~200-residue outer domains are immunogenic. Unlike complex carrier proteins that contain many BCEs, the Ab response against phage is restricted to the twelve N-terminal residues of pVIII [23] and the outer domains of pIII. Phage have also been shown to elicit CD4 T-cell help, form immunological memory [24] and are well-documented as immunogenic carriers for recombinant peptides [25-35] (for reviews see [36-38]). Furthermore, phage can be engineered to enhance the immune response, for example, by the addition of foreign CD4 T-cell epitopes to pIII or pVIII [39,40].
In this study, we assessed whether the filamentous phage is an effective carrier for the dimeric synthetic peptide, B2.1 [14], and how phage compared as an immunogen to the traditional carrier protein, OVA. The f1.K phage were engineered to have a well-exposed Lys residue near the N-terminus of all copies of pVIII, to allow efficient conjugation to peptide. Mice were immunized with (i) synthetic B2.1 peptide conjugated to f1.K phage (f1.K/B2.1); (ii) recombinant phage bearing the B2.1 peptide fused to the N-terminus of pVIII (B2.1 phage); or (iii) synthetic B2.1 peptide conjugated to OVA (OVA/B2.1). The analysis of the Ab response comprised measuring the serum Abs against the B2.1 peptide and against the appropriate carrier (OVA or phage); for conjugates, the anti-cross-linker Ab response was also measured. The extent to which the Ab response was focused against the B2.1 peptide was calculated as the ratio of the anti-peptide Ab response to the anti-carrier Ab response, and is meant to reflect the relative strength of Ab response against the peptide. We found that the OVA/B2.1 conjugate was the best immunogen, since it elicited stronger anti-peptide Ab responses compared to the two-phage immunogens. However, higher peptide-to-carrier Ab ratios were elicited by the f1.K/B2.1 conjugate, indicating that the phage conjugate was best at focusing the Ab response against the peptide. Thus, a more focused Ab response against the B2.1 peptide was produced, perhaps by virtue of decreased BCE diversity on the carrier. The results suggest that phage are useful carriers for synthetic vaccine-lead peptides, and perhaps, by extension, for molecules such as CHOs, DNA, and haptens. Further modifications to phage for enhanced immunogenicity are discussed.
2. Materials and methods
2.1. Materials and animals
The oligonucleotide Primer 1 has the sequence: 5′-GCCGCTTTTGCGGGATCGTCCGAAGCTTTNGMACC-CTCAGCAGCGAAAGAC-3′ (where N = A, C, G, T, and M = C or A; GibcoBRL Life Technologies, Inc., Gaithersburg, MD). Escherichia coli strain, K91, the filamentous phage f1 [41], and f88-4 [42] were kindly supplied by G. P. Smith (University of Missouri-Columbia). Recombinant phage, B2.1, from which the B2.1 peptide was derived, is described in Zwick et al. [14]. Reagents include: the homobifunctional cross-linker, bis[sulfosuccinimidyl] suberate (BS3, contains an 11.4 Å spacer arm), (Pierce Chemical Co., Rockford, IL) and the Adj emulsion Monophosphoryl-lipid A + Trehalose dicorynomycolate (MPL® + TDM, Adj) (Sigma–Aldrich, Oakville, Ont., Canada). The monoclonal (M)Ab b12 was kindly supplied by Burton et al. [43,44] (The Scripps Research Institute, La Jolla, CA). Five-week old, female BALB/c mice were purchased from Charles River Laboratories (St. Constant, Que.) and allowed to mature at the SFU Animal Care Facility. All animal studies were performed in accordance with institutional guidelines.
Synthetic B2.1 peptide (H-HERSYMFSDLENRCIA-AEGK-NH2) was purchased as purified homodimer from Neo-MPS (San Diego, CA). Purity was ≥95% as assessed by analytical HPLC, and the molecular weight (MW) was confirmed to be 4709.2 g/mol by electrospray mass spectrometry.
2.2. Construction of f1.K phage
The f1.K phage was derived from phage f1 [41] by site-directed mutagenesis [45] using Primer 1. Following mutagenesis, the double-stranded product was used to transform the E. coli K91 cells following the heat-shock method described by Sambrook et al. [46]. The transformed cells were mixed with melted top agar, and poured onto agar plates containing NZY medium [46]. Single plaques were picked and amplified in culture, and their replicative-form DNA was isolated and tested for the presence of the desired mutation by digestion with the restriction endonuclease HindIII (New England Bio-labs, Beverly, MA) following the manufacturer’s instructions. A single clone was selected that bore a HindIII site, and the expected sequence was confirmed by DNA sequence analysis [47].
2.3. Large-scale preparation of f1.K phage and recombinant B2.1
Methods for large-scale preparation of recombinant B2.1 phage are described by Bonnycastle et al. [48]. Briefly, a single colony of K91 cells infected with B2.1 phage was used to inoculate 500 ml volumes of NZY medium containing 15 μg/ml tetracycline and 1 mM IPTG; the cultures were shaken at 250 rpm for 21 h at 37 °C. Phage were concentrated from the cultures by precipitation with polyethylene glycol (PEG) [48] followed by purification by CsCl gradient purification [48]. The phage concentration was determined by absorbance measurement as described in Smith and Scott [49] and re-confirmed by electrophoresis on a 0.8% agarose gel in 4×-GBB buffer.
Large-scale preparation of f1.K phage followed the same protocol with the exception that plaques were used to inoculate the culture medium and no selection or inducing agents were used.
2.4. Coupling of synthetic B2.1 peptide to f1.K and OVA using BS3 cross-linking agent
2.4.1. Coupling of synthetic B2.1 peptide to f1.K
In the preparation of the f1.K/B2.1 conjugate (see Table 1), an 11-fold molar excess of peptide (MW 4709.2) was supplied for each molecule of pVIII (there are 2700 copies of pVIII per phage particle); this is equivalent to ~3.5 molecules of peptide per amine (each pVIII molecule on f1.K phage contains 2 Lys and 1 N-terminal amine). Specifically, 1.2 × 1013 particles of f1.K phage were mixed with 2.9 mg synthetic B2.1 peptide, and 4.2 mg BS3 cross-linker in a total volume of 1.4 ml PBS, pH 7.4. This brought the concentrations of phage protein, peptide and BS3 cross-linker to 260 μg/ml (8.6 × 1012 particles/ml), 2.1 mg/ml (0.5 mM) and 3.0 mg/ml (5.2 mM), respectively. The reactions containing phage, peptide and cross-linker were rotated slowly at room temperature for 1 h, and quenched by the addition of 145 μl 1 M Tris–HCl, pH 7.4. The phage were precipitated from the reaction by mixing with 0.15 vol. PEG/NaCl (16.7%/3.3 M) followed by an overnight incubation at 4°C and centrifugation at 13,000 × g for 40 min at 4 °C; the supernatants were removed. To remove PEG/NaCl, the conjugate was washed by resuspending the pellet in 12 ml PBS followed by transfer to a 12 ml polyallomer quick-seal tube (Beckman Coulter Inc., Fullerton, CA). The f1.K/B2.1 conjugate was pelleted by ultracentrifugation in a 70 Ti.1 rotor at 50,000 rpm for 4 h at 4 °C in a L8-80 ultracentrifuge (Beckman Coulter), and resuspended in 1 ml PBS. Aliquots of conjugate were removed and analyzed by electrophoresis on a 4× GBB, 0.8% agarose gel [48]. Phage conjugates and recombinant B2.1 phage were analyzed using a modified SDS–PAGE system [53].
Table 1.
The copy number of B2.1 peptide molecules displayed for the synthetic-conjugate and recombinant phage immunogens
| Name | Description of peptide-carrier | Lys and N-terminal amino group (N-term.) | Number of peptides displayed or coupleda | Molecules of peptide per dosee | Coupling efficiency (peptide per kDa carrier/Lys per kDa carrier) | |||
|---|---|---|---|---|---|---|---|---|
| Per pVIII or OVA molecule | Per kDa of carrier | Per pVIII or OVA molecule | Per phageb | Per kDa of carrierc | ||||
| Study 1 | ||||||||
| B2.1 Phage | Recombinant B2.1 peptide displayed on pVIII | N/Af | N/A | 0.05 ± 0.004d (amino acid analysis) 0.04 (SDS–PAGE) | 195; 156 | 0.01; 0.01 | 5.0 × 1014 (100); 4.0 × 1014 (100) | N/A |
| f1.K/B2.1 (Batch A) | Synthetic B2.1 peptide conjugated to f1.K phage | 2 Lys, 1 N-term. | 0.51 | 0.44 ± 0.04 | 1188 | 0.07 | 4.5 × 1014 (10) | 0.13 |
| OVA/B2.1 | Synthetic B2.1 peptide conjugated to OVA | 20 Lys, 1 N-term. | 0.47 | 10 ± 1.83 | N/A | 0.23 | 1.4 × 1015 (10) | 0.49 |
| Study 2 | ||||||||
| f1.K/B2.1 (Batch B) | Synthetic B2.1 peptide conjugated to f1.K phage | 2 Lys, 1 N-term. | 0.47 | 0.46 ± 0.04 | 1242 | 0.08 | 4.7 × 1014 (10); 1.2 × 1015 (25) | 0.16 |
For B2.1 the number of peptides refers to copies of dimer present.
The number of copies of peptide per carrier was calculated assuming that f1.K has 2700 pVIII molecules (6.4 kb) and f88-4 based recombinant phage: 3900 pVIII molecules (9.2 kb).
The calculated molecular weights for B2.1 peptide, f1.K, f88-4 and OVA are 4709, 16,000,000, 23,000,000 and 42,881 Da, respectively.
Whereas 5% of the total pVIII of the recombinant B2.1 phage carry the dimer, 10% of the pVIII molecules of these phage carry a single chain of the B2.1 dimer; thus, 10% of the total pVIII is recombinant.
Dose, in μg, is shown in brackets.
Not applicable.
2.4.2. Coupling of synthetic B2.1 peptide to OVA
The OVA/B2.1 conjugate was prepared by mixing a 60-fold molar excess of B2.1 peptide with OVA, allowing ~3 molecules of peptide per Lys residue (OVA contains 20 Lys and 1 N-terminal amine). Specifically, 298 μg OVA were mixed with 2.0 mg peptide and 2.8 mg BS3 cross-linker in 1 ml PBS, and the conjugation reaction was treated as described above. Instead of purification by PEG precipitation and ultracentrifugation, the conjugates were washed three times with 3 ml PBS using a Centricon-30 ultrafiltration device (Amicon, Inc., Beverly, MA) according to the manufacturer’s instructions, and stored at 4 °C. The OVA/B2.1 conjugate was analyzed using conventional SDS–PAGE [46].
2.5. Immunization of mice
2.5.1. Study 1
As shown in Table 2, groups of 5 eight-week-old BALB/c mice were given intraperitoneal (IP) immunizations with: (i) 100 μg recombinant B2.1 phage for five immunizations; (ii) 10 μg f1.K/B2.1 conjugate for seven immunizations; or (iii) 10 μg OVA/B2.1 conjugate for seven immunizations. All immunogens were administered in 100 μl PBS without Adj. For all groups, the mice were immunized on Days 0, 14, 28, 42, 63, 98, and 119; every two weeks for the first four immunizations and every three weeks for immunizations 5, 6, and 7. The exception was the B2.1 phage group; these mice received five immunizations. Blood was collected from the tail vein, two-weeks after each inoculation, and sera were collected after centrifugation. Samples were stored at −20°. Final bleeds and euthanasia were performed on Day 140 (Day 77 for B2.1 phage group) by cardiac puncture under CO2 anaesthesia.
Table 2.
Description of immunization Study 1 and 2
| Immunogen | Dose (μg) | Adjuvant | Number of immunizations |
|---|---|---|---|
| Study 1 | |||
| f1.K/B2.1 conjugate | 10 | 7 | |
| OVA/B2.1 conjugate | 10 | 7 | |
| Recombinant B2.1 phage | 100 | 5 | |
| Study 2 | |||
| f1.K/B2.1 conjugate | 10 | No | 7 |
| f1.K/B2.1 conjugate | 25 | No | 7 |
| f1.K/B2.1 conjugate | 10 | Yes | 7 |
| f1.K/B2.1 conjugate | 25 | Yes | 7 |
For Study 1, groups of five BALB/c mice were given IP immunizations with either 100 μg B2.1 phage or 10 μg f1.K/B2.1 or 10 μg OVA/B2.1 conjugate. All immunogens were administered without Adj. In Study 2, groups of five BALB/c mice were given SC immunizations with either 10 μg or 25 μg f1.K/B2.1 conjugate with or without Adj. The timing for both studies is described in Section 2.
2.5.2. Study 2
For Study 2 (see Table 2), groups of 5 eight-week-old BALB/c mice were given a total of seven subcutaneous (SC) immunizations with: (i) 10 μg f1.K/B2.1, no-Adj; (ii) 25 μg f1.K/B2.1, no-Adj; (iii) 10 μg f1.K/B2.1, with Adj; or (iv) 25 μg f1.K/B2.1, with Adj. The adjuvant/immunogen emulsions were prepared according to the manufacturer’s instructions. The first four immunizations were given every three weeks, on Days 0, 21, 42, 63, followed by a four-week interval before the fifth immunization on Day 91. The sixth immunization was given on Day 147, 8 weeks after the fifth immunization, and the seventh was given 7 weeks after the sixth on Day 210. Tail bleeds were conducted two weeks after each immunization, on Days 0, 14, 35, 56, and 105. Final bleeds and euthanasia were performed on Day 244 as described above. Blood was treated as described above.
2.6. Western blot analysis
The antigenicity of the conjugates and the recombinant phage was confirmed by Western blot. Following SDS–PAGE, proteins were transferred onto PVDF Immobilon-PSQ transfer membranes (Millipore Corporation, Bedford, MA) using a Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad Laboratories, Richmond, CA) [50]. The membranes were blocked overnight at 4 °C with 5% (w/v) skim milk in TBS (50 mM Tris–HCl, pH 7.4, 150 mM NaCl), then incubated with polyclonal anti-phage IgG (5 nM) or MAb b12 (2.5 nM) in TBS containing 2.5% skim milk and 0.05% (v/v) Tween 20 (Sigma) for 2 h at room temperature. Membranes were washed with TBS containing 5% skim milk for 20 min, then four times in TBS containing 0.1% Tween 20. The primary Abs were detected with protein A/G-horseradish peroxidase (HRP) conjugate from Pierce at a dilution of 1/1500. After washing, the bound HRP conjugate was detected with an ECL Western blot Detection Reagents kit (Amersham International plc, Buckinghamshire, England), and exposed to Safelight Medical X-ray film (Fuji photofilm Co., Ltd., Tokyo) for visualization.
2.7. Amino acid analysis
Amino acid analyses of B2.1 peptide dimer conjugated to f1.K phage and to OVA, and recombinant B2.1 phage, f1.K and f88-4 phage and OVA were performed at the Alberta Peptide Institute (University of Alberta, Edmonton) using standard hydrolysis. Amino acid analysis data were used to determine the B2.1 peptide copy number for the peptide-bearing immunogens, and to provide baseline data for the carriers f1.K and f88-4 phage and OVA.
2.8. ELISA and determination of serum titers
Titration serum ELISAs were performed as follows: 1010 f88-4 or f1.K particles, 1 μg OVA, 200 ng B2.1 peptide or 1 μg BS3-treated BSA in 35 μl TBS were adsorbed overnight at 4 °C, to wells of microtiter/96-well plates (Costar Highbinding, Easy Wash plates; Corning Inc., Corning, NY). Each well was blocked with 200 μl TBS containing 2% BSA for 1 h at 37 °C, and then washed three times with TBS containing 0.1% Tween 20. Sera were diluted in TBS containing 0.1% Tween 20 and 1% BSA, starting at a 1/100 dilution, and titrated in 1/4 dilutions. The titrated sera, in a volume of 35 μl, were added to each well and incubated 2 h at room temperature. The wells were washed six times and bound Ab was detected by a 45 min incubation at room temperature with goat anti-mouse IgG (Fc specific):HRP conjugate (Pierce), diluted 1/1500 in TBS containing 0.1% Tween 20. HRP was detected with 35 μl 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid (ABTS; Sigma) solution containing H2O2[51]. The OD405–490 of each reaction was measured by an EL 312e ELISA plate reader (Bio-Tek). The data from each titration, after subtraction of background signals from sera plated on BSA-coated wells, were analyzed to give the dilution factor (the inverse of the titer) corresponding to the half-maximum OD405–490 reading; this was about 0.55 OD405–490 for the majority of serum samples. Serum samples that showed weak binding, but did reach the half-maximum OD405–490, were assigned a titer of 50. The arithmetic mean and standard error of the mean are reported for each treatment group.
2.9. Statistical analysis
The data were further analyzed by ANOVA [52] using MINITAB™ statistical software to determine the variance within and between groups. Differences between groups were considered significant if the ANOVA p-value was below 0.05. While ANOVA can indicate significant differences between groups, it does not describe the hierarchy of those differences. The Tukey post hoc analysis [52] was used to establish the groups that showed the largest statistical differences.
3. Results
3.1. Preparation and analysis of phage and OVA conjugates
The phage f1.K was developed to enhance chemical cross-linking to the phage surface via a well-exposed Lys residue. The partial amino acid sequence of the N-terminal region of mature pVIII of wild-type f1 (AEGDDPAKAA) was modified to include a four-residue insert that places a Lys residue near the N-terminus of pVIII. The resulting f1.K N-terminal sequence (new residues in bold) is: AEGAKASDDPAKAA. The insertion of four residues into the N-terminal segment of all copies of pVIII was well tolerated by f1.K phage, as indicated by a high phage yield (~2 × 1015 phage particles/L) and its stability over multiple passages (data not shown).
Six cross-linking agents were tested for their ability to conjugate the B2.1 dimeric peptide to the f1.K phage and to OVA. The cross-linker BS3 produced the largest amount of conjugated B2.1 peptide and the strongest peptide antigenicity after conjugation as detected by ELISA with MAb b12 (data not shown). Fig. 1A shows the results of SDS–PAGE of f1.K/B2.1 conjugate followed by Western blot with MAb b12 (Fig. 1A). Also shown is f1.K/B2.1 conjugate detected by polyclonal anti-phage Ab (Fig. 1B). Fig. 1A confirms the presence of B2.1 on the phage and revealed that the mobility of pVIII from f1.K phage was affected by the BS3 cross-linker, resulting in a ladder of cross-linked pVIII molecules having slower mobility than monomeric pVIII. Coupling of synthetic B2.1 peptide to OVA (OVA/B2.1) with BS3 also resulted in conjugates that showed significant cross-linking of B2.1 peptide to OVA. However, the OVA/B2.1 conjugate did not show the same ladder effect that was observed for the f1.K/B2.1 conjugate; this is probably due to the size of the OVA multimers and the resolution capabilities of the acrylamide gel (not shown). Antigenicity of recombinant B2.1 phage was confirmed by Western blot with MAb b12 [14] (not shown).
Fig. 1.
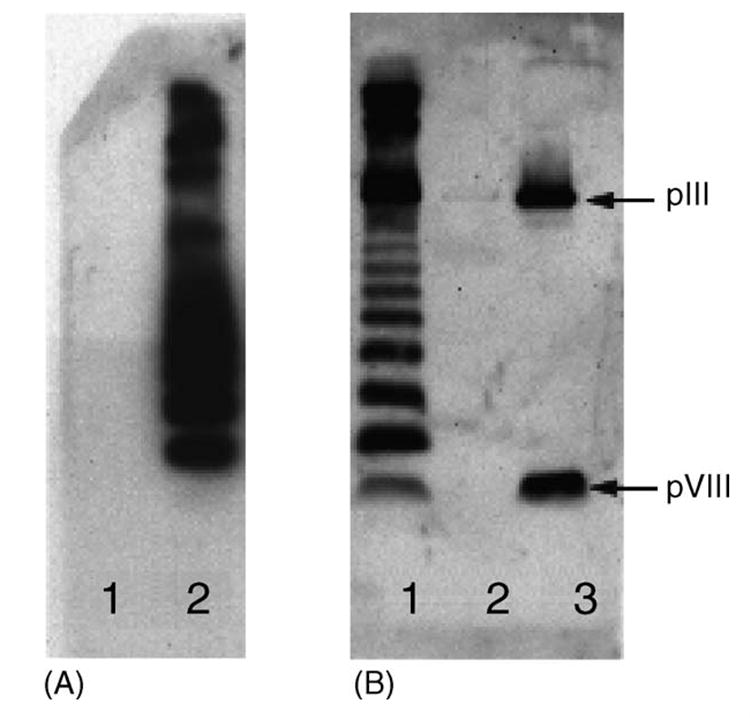
Western blots of f1.K/B2.1 peptide conjugate. (A) Detected with MAb b12. Lanes: (1) f1.K phage, negative control; (2) f1.K/B2.1 peptide conjugate, 5 × 1010 phage particles. (B) Detected with rabbit anti-phage polyclonal Ab. Lanes: (1) f1.K/B2.1 peptide conjugate, 5 × 1010 phage particles; (2) OVA/B2.1 conjugate-negative control; (3) f1.K phage-positive control, 2 × 1010 phage particles. The positions of the pIII (MW 42.5 kDa) and pVIII (MW 5.5 kDa) bands are indicated by arrows.
3.2. Peptide copy number of phage and OVA immunogens
Table 1 shows the peptide copy number of the immunogens. The number of B2.1 peptides displayed per carrier molecule for the f1.K/B2.1 and OVA/B2.1 conjugates was determined by amino acid analysis, whereas the copy number for recombinant B2.1 phage was estimated from analysis by SDS–PAGE of titration of recombinant and wild-type pVIII proteins [53], and confirmed by amino acid analysis. The recombinant peptide covered 4%–5% of the phage surface whereas synthetic B2.1 peptide coupled to f1.K phage resulted in 44%–46% coverage, depending on the batch. Given that there are two exposed Lys residues per pVIII molecule, and one N-terminal amine, the peptide conjugated to 20% or greater of the available sites. Importantly, the f1.K/B2.1 conjugates had a >6-fold higher copy number of peptide per phage than recombinant display.
There is no way of knowing whether there was preference for the well-exposed Lys; however, a comparison of conjugations to f1.K and f88.4 (which lacks the exposed Lys) showed significant differences. A 20-mer monomeric synthetic peptide, representing the major antigenic repeat of the malarial circumsporozoite protein (NANP), was conjugated to the phage using the cross-linker sulfosuccinimidyl 4-N-maleimidomethyl cyclohexane-1-carboxylate (sulfo-SMCC). Amino acid analysis for copy number indicated that almost twice as much peptide coupled to f1.K than to f88.4 (0.48 copies of peptide for f88.4 and 0.90 copies of peptide per pVIII for f1.K). Furthermore, SDS–PAGE of the cross-linked phage without peptide showed pVIII multimers (up to six) forming in a ladder for the f1.K treated with sulfo-SMCC, whereas the f88.4 treated with sulfo-SMCC conjugated pVIII only to a dimer (our unpublished data).
Conversion of the data to peptide copy number per kDa-equivalent of protein (Table 1) allowed comparison between the three protein carriers. OVA by far coupled the largest number of peptides per mass-equivalent of protein. As OVA contains 20 Lys residues (the majority are surface exposed as seen in the crystal structure [PDB file 1OVA]) the coupling of ~10 peptides per molecule was probably near the limit. Furthermore, the coupling efficiency of peptide to the OVA carrier (0.49, see Table 1) was 3.5-fold greater than that of peptide coupled to f1.K carrier (0.13 and 0.16).
3.3. Analysis of Study 1: comparison between phage and OVA as synthetic peptide-carriers
As shown in Table 2 and Section 2, mice were immunized with the f1.K/B2.1 and OVA/B2.1 conjugates, and B2.1 phage. The dosage of recombinant phage used was high, reflective of those used in published studies (e.g., see Felici et al. [31]). The conjugates were used in much lower doses to produce a slower rise in anti-peptide Ab titers. Adj was purposely not used, so as to compare the intrinsic immunogenicity of each immunogen, and so that the baseline immune responses against the immunogens could be tested over time. This also allowed Ab titers to reach a plateau so that immunogens could be compared through multiple rounds of immunization. Sera were analyzed by titration ELISA for the presence of Abs against: the B2.1 peptide, the appropriate carrier (f1.K phage, OVA, or f88.4 phage) and the BS3 cross-linker (for conjugates only). Fig. 2 summarizes the half-maximal titers from the third, fifth, and seventh immunizations. The data were analyzed by ANOVA (Fig. 2), which identifies significant differences between immunization groups. To clarify the hierarchical relationship among groups that significantly differed from one another, Tukey post hoc analysis was used (discussed below).
Fig. 2.
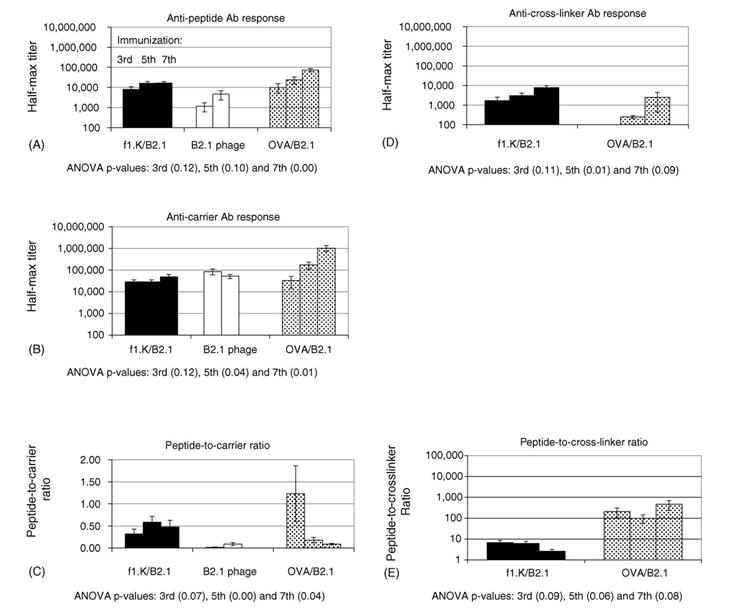
Study 1 Ab titers: shown are the mean antibody titer±standard error after third, fifth and seventh immunizations against: (A) B2.1 peptide; (B) f1.K, f88-4; and OVA carriers (C) peptide-to-carrier ratios; (D) BS3 cross-linker for f1.K/B2.1 and OVA/B2.1; and (E) peptide-to-cross-linker ratio. Also shown are ANOVA p-values that indicate the existence of significant differences between groups that received different antigens at the third, fifth and seventh immunizations.
Statistical analysis of anti-peptide Ab titers (Fig. 2A), elicited by f1.K/B2.1, recombinant B2.1 phage and OVA/B2.1 immunogens, revealed no significant differences between groups after the third and fifth immunizations. This was due to large within group variation in the OVA/B2.1 group; however, when this group’s data were omitted from the analysis, it was revealed that the f1.K/B2.1 conjugate elicited significantly stronger anti-peptide Ab responses than the recombinant B2.1 phage, even though the dose of phage conjugate was one-tenth that of the recombinant phage. This result was surprising, since the f1.K/B2.1 conjugate and recombinant B2.1 delivered comparable amounts of total peptide molecules (~4.5 × 1014 molecules, see Table 1) even though the doses were 10 and 100 μg phage, respectively. After the seventh immunization, OVA/B2.1 elicited anti-peptide Ab titers that were >4-fold higher than those elicited by f1.K/B2.1. It was expected that the anti-peptide Ab response produced by the OVA/B2.1 conjugate would be stronger than that produced by the f1.K/B2.1 conjugate, since the OVA/B2.1 conjugate delivered three-times more peptide molecules per dose. Unexpectedly, the anti-peptide and anti-carrier Ab responses for the f1.K/B2.1 group plateaued after the fifth immunization (Fig. 2B). These Ab responses did not plateau for the OVA/B2.1 conjugate, indicating that higher Ab titers could have been achieved with more rounds of immunization.
Ab responses to the carrier proteins produced a different trend (Fig. 2B). Within group variation was too large to reveal significant trends after the third immunization. However, the fifth immunization with OVA/B2.1 produced an anti-carrier response that was 5-fold stronger than the one elicited by f1.K/B2.1, but it was not significantly stronger than that elicited by B2.1 phage. This trend continued after the seventh immunization, when the anti-OVA Ab response was 20-fold greater than the anti-phage one. Anti-carrier Ab response from the recombinant B2.1 phage group peaked at the third immunization, whereas that of the phage conjugate did not do so until after the fifth immunization; this probably reflects the difference in phage dosage used. The f1.K/B2.1 and OVA/B2.1 conjugates were administered at the same dosage (10 μg), thus, it was unexpected that the anti-OVA Ab response exceeded the anti-phage Ab response. Furthermore, the anti-OVA Ab response did not reach a peak after seven immunizations whereas the anti-phage Ab response plateaued after the fifth immunization. Perhaps this is due to increased variation of BCEs on OVA, whereas the BCEs on phage are restricted and contribute to a preset plateau in the anti-phage Ab response.
Besides identifying the immunogen that produced the strongest anti-peptide Ab titers, we also wanted to identify the immunogens that produced a relatively strong response against the peptide compared to the response against the carrier and the cross-linker, reasoning that an important parameter of any anti-peptide Ab response is the titer produced against unimportant and/or unwanted parts of the immunogen. Thus, the peptide-to-carrier ratios were calculated for each individual serum (Fig. 2C). Statistical analysis of the third immunization peptide-to-carrier ratios did not reveal significant differences due to high variation within the OVA/B2.1 group. However, after the fifth immunization, the peptide-to-carrier ratio elicited by f1.K/B2.1 was 0.58; thus, the anti-peptide Ab titer was 58% of the anti-phage Ab titer. In contrast, the peptide-to-OVA ratio was 0.2, indicating a less focused response against the peptide, even though the OVA/B2.1 conjugate had a higher copy number of peptide per amount of protein (Table 1). After the seventh immunization, this trend increased with both groups dropping to 0.48 and 0.09, respectively. Even though the same total amount of peptide molecules were delivered by the recombinant B2.1 phage and f1.K/B2.1 conjugate, the peptide-to-carrier ratio was lowest for the recombinant B2.1 phage. This is probably due to low peptide copy number on the recombinant phage relative to the f1.K/B2.1 conjugate. Surprising, OVA/B2.1 conjugate delivered more peptide per dose, but produced lower peptide-to-carrier ratios than f1.K/B2.1. This difference in ratios may be due to the relatively large number of BCEs on OVA versus phage.
As reported by Briand et al. [19], cross-linking agents can elicit an Ab response; thus, we evaluated the Ab response against the BS3 cross-linker (Fig. 2D). Data from the third immunization showed that the phage conjugate elicited a response against BS3 that was 33-times stronger than that recalled by OVA conjugate. After the seventh immunization, the anti-BS3 response elicited by f1.K/B2.1 was only three-times greater than those elicited by OVA/B2.1, and was not considered significantly different. The strong anti-cross-linker responses produced by the f1.K/B2.1 may be due to coverage by excess cross-linker that did not link peptide, but bound exclusively to the pVIII major coat protein. This is supported by cross-linked pVIII multimers, as shown by SDS–PAGE (Fig. 1), and low coupling efficiency of peptide to phage compared to OVA (Table 1). Furthermore, the peptide-to-cross-linker ratio (Fig. 2E) for the f1.K/B2.1 group was much lower than that produced by the OVA/B2.1 conjugate. This effect was most pronounced after the seventh immunization where the f1.K/B2.1 conjugate produced anti-peptide Ab titers that were only three-times greater than the anti-cross-linker Abs. Those for the OVA/B2.1 group were almost 500-times stronger than the anti-cross-linker Ab response.
In conclusion, Study 1 revealed that the strength of the anti-peptide Ab response depends on immunogen peptide copy number and the strength of the Ab response against the whole immunogen, rather than on the total dose of peptide. In contrast, the ability to focus the Ab response against a peptide (as revealed by the peptide-to-carrier ratios) appears related to the immunological complexity of the carrier, as well as the peptide copy number. For example, the restricted BCEs on the f1.K/B2.1 conjugate appeared to enhance peptide-to-carrier ratios. Since our goal is to develop an immunogen that focuses a strong Ab response against a restricted BCE, such as a peptide, we conducted a second study to optimize the anti-peptide Ab responses produced by the f1.K/B2.1 conjugate.
3.4. Analysis of Study 2: effect of route, dose, and Adj on Ab response against f1.K/B2.1
The first immunization study compared three carriers for synthetic and recombinant peptides (f1.K phage, OVA, and recombinant phage) and showed that immunization with f1.K/B2.1 conjugate elicited a more focused anti-peptide Ab response than OVA/B2.1, but at comparatively low anti-peptide Ab titers. To further optimize the anti-peptide Ab response against f1.K/B2.1, we compared the effects of dose and the Adj, MPL® + TDM, on the Ab response against: the peptide, the carrier, and the cross-linker; peptide-to-carrier ratios were also assessed. Since we were concerned about Adj causing IP lesions [54] that could be further aggravated by frequent boosting immunizations, we changed the injection route to SC and increased the time between boosts to reduce potential discomfort in the mice due to adjuvant toxicity.
The effects of dose and Adj on anti-peptide Ab titers elicited by f1.K/B2.1 are summarized in Fig. 3A. An increase in dose of f1.K/B2.1 conjugate did not significantly affect anti-peptide Ab titers. In contrast, Adj enhanced anti-peptide Ab titers after the third immunization, but did not enhance the magnitude of the anti-peptide Ab response at plateau. At the fifth and seventh immunizations, comparison of all of the anti-peptide Ab titers showed no statistically significant differences between immunization groups. Thus, Adj appears to increase the strength of the early Ab responses, but not the final titer.
Fig. 3.
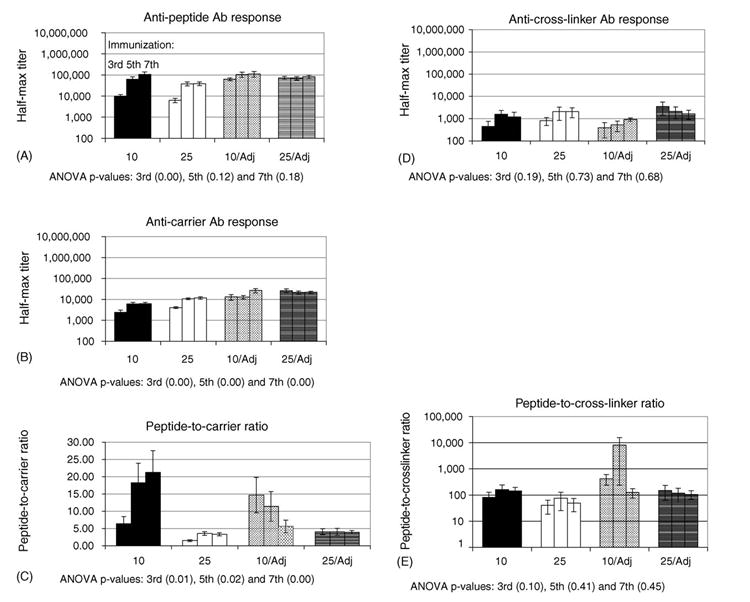
Study 2 Ab titers: shown are the mean antibody titer±standard error after third, fifth and seventh immunizations against: (A) B2.1 peptide; (B) f1.K carrier; (C) peptide-to-carrier ratios; (D) BS3 cross-linker; and (E) peptide-to-cross-linker ratio. Also shown are ANOVA p-values that indicate the existence of significant differences between groups that received different immunization conditions at the third, fifth and seventh immunizations.
Anti-carrier Ab responses (Fig. 3B) were influenced by a combination of dose and Adj. After the third and fifth immunizations, the 25 μg dose with Adj elicited anti-carrier Ab titers that were significantly stronger than those elicited by the 10 and 25 μg doses without Adj. By the seventh immunization, the anti-carrier responses for the 10 μg with Adj group had reached the same level as the high dose with Adj. It can be inferred from these trends that the addition of Adj increases the total anti-carrier Ab response compared to groups that did not receive Adj; this is in contradiction to the observations that Adj and dose did not affect the magnitude of anti-peptide Ab titers.
The effects of dose and Adj on the anti-carrier Ab responses are reflected by the peptide-to-carrier ratios shown in Fig. 3C. Peptide-to carrier-ratios in Study 2 dramatically exceeded those reached in the Study 1 (Fig. 2C). The highest peptide-to-carrier ratio in Study 1 was 0.58 for the 10 μg dose of f1.K/B2.1 with no-Adj compared to 21-times the anti-carrier response in the second study with the same immunogen and dose. In fact, the anti-peptide Ab titers reached levels similar to those elicited by OVA/B2.1 in Study 1, although, the titers that resulted from these different studies cannot be compared directly. The highest peptide-to-carrier ratios in Study 2 were produced by the 10 μg dose in which the addition or absence of Adj produced opposite effects. The no-Adj group produced the highest peptide-to-carrier ratios after the seventh immunization, whereas the Adj group produced the highest peptide-to-carrier ratios after the third immunization. This trend suggests that the 10 μg dose is optimal for high peptide-to-carrier ratios, and that that Adj enhances the rate of Ab response, but not its plateau, thus, requiring fewer rounds of immunization.
The anti-cross-linker Ab responses did not yield significant trends due to large within group variation (Fig. 3D) and was consistent with anti-cross-linker Ab titers reported in Study 1 (compare Fig. 2D and Fig. 3D). However, the peptide-to-cross-linker ratios were much higher than those in the first study (compare Fig. 2E and Fig. 3E); this reflects the increase in anti-peptide Ab responses produced by the immunizations given in Study 2.
In conclusion, it appears that the change in injection routes from IP to SC dramatically enhanced the anti-peptide Ab titers in Study 2 compared to Study 1. Also unexpected, was that Adj and increased dose did not enhance anti-peptide Ab titers but did contribute to an increased anti-phage Ab response. Future studies are aimed at further modifying the phage to enhance their use as an immunogen for targeting Ab responses to specific epitopes. These approaches are discussed below.
4. Discussion
4.1. Phage as a carrier for the B2.1 synthetic peptide
We report here the use of filamentous phage as a novel immunogenic carrier for synthetic peptides, and by extension, other non-peptide immunogens such as CHO, DNA and haptens. Based on the hypothesis that the addition of a Lys residue, closer to the N-terminus of pVIII, would enhance access to chemical cross-linking agents, the phage f1.K was constructed. Conjugation of the dimeric synthetic peptide, B2.1, to f1.K (f1.K/B2.1) resulted in a high copy number immunogen that was recognized by MAbb12 (see Table 1 and Fig. 1, respectively). Study 1 compared the immunogenicity of the f1.K carrier to the commonly used “traditional” carrier, OVA [17-19], and to recombinant B2.1 phage. Study 2 further optimized the Ab response by assessing the effects of Adj, dose, and injection route on the anti-peptide Ab titers and peptide-to-carrier ratios. Taken together, the data show that the f1.K/B2.1 conjugate: (i) is immunogenic; (ii) elicits a stronger anti-peptide Ab response than recombinant B2.1 phage; (iii) elicits a more focused anti-peptide Ab response than OVA/B2.1; and (iv) the best results were obtained by using an SC immunization route.
Study 1 was designed to determine if the f1.K/B2.1 conjugate was immunogenic and to compare it to the OVA/B2.1 conjugate. We also sought to compare the Ab responses elicited by f1.K/B2.1 conjugate and recombinant B2.1 phage. Comparison of half-max titers revealed that f1.K/B2.1 conjugate elicited a greater than 3-fold anti-peptide Ab titer compared to recombinant phage after the fifth immunization. However, OVA/B2.1 elicited 4.4-fold greater anti-peptide Ab titers than f1.K/B2.1, after the seventh immunization. Importantly, it is significant that in comparing peptide-to-carrier ratios, the phage conjugate elicited a more focused anti-peptide Ab response than the OVA conjugate. Taken together, these data suggest that OVA is a stronger immunogen (see below), but phage are better at focusing the immune response against the peptide perhaps due to OVA’s more extensive set of BCEs compared to the restricted BCEs on phage. Anti-cross-linker Ab titers were also assessed. The phage carrier elicited anti-cross-linker Ab responses that were not significantly higher than those elicited by OVA after the seventh immunization. However, the peptide-to-cross-linker ratio for the f1.K/B2.1 conjugate was much lower than that for OVA/B2.1. One way to optimize this may be by using shorter cross-linking agents that are less immunogenic (e.g., glutaraldehyde). However, the antigenicity of the peptide must be considered; in our case, we found that BS3 was most effective.
To further optimize the peptide-to-carrier ratios elicited by f1.K/B2.1 in Study 1, Study 2 was designed to test the effects of dosage and Adj on anti-peptide Ab titers. Generally, anti-peptide Ab titers were not significantly enhanced by dose or Adj at the peak of the immune response. In contrast, anti-carrier Ab titers were increased when immunogen was combined with Adj and increased dose; groups that received Adj also reached a plateau in fewer immunizations. Strikingly, both the anti-peptide Ab titers and peptide-to-carrier ratios for Study 2 were much higher than those in Study 1. This observation may be attributed to the change in injection route from IP to SC. Injection route can target different immune cells. For example, intradermal injections target Langerhans dendritic cells [55] that activate helper T-cells and contribute to the humoral response. The SC injection route may also target these specific Agn presenting cells. These results are in agreement with Kenney et al. [56], who observed enhanced Ab titers when using SC injections compared to IP. However, other groups have reported the opposite effect [57,58], suggesting that route requires optimization for each unique Agn and Adj combination.
Assessment of the anti-cross-linker Ab responses for Study 2 showed that the strength the Ab responses were not significantly affected by dose or Adj, and the anti-cross-linker Ab titers were in the same range as Study 1. However, peptide-to-cross-linker ratios were much higher than those elicited by the f1.K/B2.1 conjugate in Study 1, reflecting the strength of the optimized anti-peptide Ab response in Study 2.
In both Studies 1 and 2, the f1.K/B2.1 phage conjugate elicited Ab titers that plateaued after the third or fifth immunizations. Whereas the anti-peptide and anti-carrier Ab responses produced by the OVA conjugate in Study 1 continued to increase after the seventh immunization. This observation may be explained by differences in CD4 TCEs between the two carrier molecules. OVA, a complex protein of 42.9 kDa, probably contains more TCEs per molecular weight protein than pVIII (5.5 kDa). The larger minor coat proteins on phage, such as pIII (42.5 kDa), may contain more TCEs than those found on pVIII, however, these proteins are present in proportionally small amounts. Filamentous phage bear TCEs, since they elicit T-cell dependent class-switching from IgM to IgG [24]. However, the plateau effect may be explained by relatively fewer and/or weaker TCEs on the phage than those on OVA. Ongoing studies that address this hypothesis are discussed below.
Another explanation of the plateau effect is that phage contain fewer BCEs per mg of protein than OVA. As shown in Fig. 1, the Ab response to phage is directed exclusively against pVIII and pIII (the major and minor coat proteins, respectively). The Ab response to pVIII is restricted to 12 residues at the N-terminus [23], thus, there may be a limited number of B-cell clones that could elicit Abs against pVIII. In the case of pIII, there may be more epitope diversity in the exposed N1 and N2 domains; however, this protein is present in limited amounts (3–5 copies/phage), which may reduce its immunogenicity. It appears from the Western blot that the signals on pVIII and pIII are of similar strength, yet there are ~700-fold more copies of pVIII than pIII, reflecting the limitations of the epitopes on pVIII. In contrast to the phage, OVA has a more-varied BCE repertoire, which may enhance the strengthening and broadening of the Ab response over successive immunizations. Taken together, the plateau in the Ab response against phage is likely due to a combination of weak TCEs and restricted BCEs that limit the diversity, expansion and differentiation of phage-specific B-cell clones.
4.2. Modification of phage carriers for optimizing immune responses
The filamentous phage approximate the structure of a viral pathogen as large, particulate molecular scaffolds. We report here that phage appear better at focusing the Ab response against peptide than the traditional carrier, OVA. Furthermore, they elicit relatively weak anti-phage responses due to restricted BCEs on pVIII [23] and low copy number of the outer immunogenic domains of pIII. These advantages suggest that phage are proficient carriers for synthetic and recombinant peptides, CHOs or haptens in situations requiring a focused immune response. A disadvantage of using phage is the relatively weak T-cell help they provide. This issue may be addressed by modifying phage to include immunodominant foreign TCEs with known properties [39,40,59], either by direct fusion to the phage coat proteins or chemical conjugation to pVIII (our unpublished data).
Furthermore, phage could be engineered to reduce existing B-cell epitopes by substituting immunogenic residues on pVIII (identified by epitope mapping with monoclonal antibodies [23]) with amino acids that are typically less immunogenic (e.g., substituting charged residues with polar or apolar ones), or by removing the outer immunogenic domains on pIII. Thus, new epitopes should not be exposed, but the immunogenicity of existing epitopes should be reduced, potentially focusing an Ab response against a poorly-immunogenic target peptide or other small molecule (carbohydrate or hapten). Support for this hypothesis was obtained by Agarwal et al. [60], who showed that substitution of critical binding residues on an immunodominant peptide epitope could redistribute the Ab response to less-dominant epitopes. The combination of the addition of an immunodominant foreign TCE and a further reduction of its BCEs should improve phage as a carrier for eliciting highly targeted Ab responses. This combination could comprise synthetic peptides directly conjugated to the “non-immunogenic” phage resulting in high copy number immunogens that are relatively easy to characterize (see below). As an alternative, hybrid phage could be used to express two recombinant peptides on the same virion [47]; however, this approach may compromise copy number, compared to synthetic peptide conjugation.
The construction and analysis of phage/peptide conjugates is comparable to other peptide/protein conjugates. As with traditional carrier proteins, the chemical conjugation of a synthetic peptide or hapten to phage uses well-established coupling protocols [61]. However, the small size of pVIII allows more extensive characterization of phage conjugates compared to those made with large carrier proteins. Fig. 1A and B show a ladder of extensively cross-linked pVIII molecules and B2.1 peptide conjugated to pVIII multimers, respectively. This ladder represents the biochemical outcome of the coupling reaction and provides visual and immunochemical proof that the cross-linking reaction was successful. This type of ladder is not seen with traditional carrier protein conjugates, since their relatively high molecular weight creates multimers that are not easily resolved by SDS–PAGE.
This paper introduces filamentous bacteriophage as an alternative carrier for synthetic peptides, haptens, and CHO Agns. The data presented indicate that phage/peptide conjugates produce a more focused anti-peptide Ab response than traditional protein carriers. Although phage provide weak T-cell help, further development, such as the addition of foreign TCEs, and the reduction of immunodominant BCEs, should strengthen phage as a useful tool for immunological research and vaccine studies.
Acknowledgments
We thank Loekie van der Wal and the staff at the SFU Animal Care Facility for outstanding care and handling of the mice used in this study. We are grateful to Demetreus Blakemore and Felix Breden for statistical analyses. We also thank Sundeep Chahal for help in completing the serum ELISAs. This work was supported by grants from the Medical Research Council of Canada (MT-14562 to J.K.S.) and from the National Institute of Health (R21 AI44395, and R01 AI4911 to J.K.S).
References
- 1.Grothaus MC, Srivastava N, Smithson SL, Kieber-Emmons T, Williams DB, Carlone GM, et al. Selection of an immunogenic peptide mimic of the capsular polysaccharide of Neisseria meningitidis serogroup A using a peptide display library. Vaccine. 2000;18(13):1253–63. doi: 10.1016/s0264-410x(99)00390-4. [DOI] [PubMed] [Google Scholar]
- 2.Beenhouwer DO, May RJ, Valadon P, Scharff MD. High affinity mimotope of the polysaccharide capsule of Cryptococcus neoformans identified from an evolutionary phage peptide library. J Immunol. 2002;169(12):6992–9. doi: 10.4049/jimmunol.169.12.6992. [DOI] [PubMed] [Google Scholar]
- 3.Pincus SH, Smith MJ, Jennings HJ, Burritt JB, Glee PM. Peptides that mimic the group B streptococcal type III capsular polysaccharide antigen. J Immunol. 1998;160(1):293–8. [PubMed] [Google Scholar]
- 4.Prinz DM, Smithson SL, Westerink MA. Two different methods result in the selection of peptides that induce a protective antibody response to Neisseria meningitidis serogroup C. J Immunol Methods. 2004;285(1):1–14. doi: 10.1016/j.jim.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Hou Y, Gu XX. Development of peptide mimotopes of lipooligosaccharide from nontypeable Haemophilus influenzae as vaccine candidates. J Immunol. 2003;170(8):4373–9. doi: 10.4049/jimmunol.170.8.4373. [DOI] [PubMed] [Google Scholar]
- 6.Melzer H, Baier K, Felici F, von Specht BU, Wiedermann G, Kollaritsch H, et al. Humoral immune response against proteophosphoglycan surface antigens of Entamoeba histolytica elicited by immunization with synthetic mimotope peptides. FEMS Immunol Med Microbiol. 2003;37(2–3):179–83. doi: 10.1016/S0928-8244(03)00074-9. [DOI] [PubMed] [Google Scholar]
- 7.Caton M, Diamond B. Using peptide mimetopes to elucidate anti-polysaccharide and anti-nucleic acid humoral responses. Cell Mol Biol (Noisy-le-grand) 2003;49(2):255–62. [PubMed] [Google Scholar]
- 8.Deocharan B, Qing X, Beger E, Putterman C. Antigenic triggers and molecular targets for anti-double-stranded DNA antibodies. Lupus. 2002;11(12):865–71. doi: 10.1191/0961203302lu308rr. [DOI] [PubMed] [Google Scholar]
- 9.Putterman C, Deocharan B, Diamond B. Molecular analysis of the autoantibody response in peptide-induced autoimmunity. J Immunol. 2000;164(5):2542–9. doi: 10.4049/jimmunol.164.5.2542. [DOI] [PubMed] [Google Scholar]
- 10.Jiang B, Liu W, Qu H, Meng L, Song S, Ouyang T, et al. A novel peptide isolated from a phage display peptide library with trastuzumab can mimic antigen epitope of HER-2. J Biol Chem. 2005;280(6):4656–62. doi: 10.1074/jbc.M411047200. [DOI] [PubMed] [Google Scholar]
- 11.Dakappagari NK, Lute KD, Rawale S, Steele JT, Allen SD, Phillips G, et al. Conformational HER-2/neu b-cell epitope peptide vaccine designed to incorporate two native disulfide bonds enhances tumor cell binding and antitumor activities. J Biol Chem. 2005;280(1):54–63. doi: 10.1074/jbc.M411020200. [DOI] [PubMed] [Google Scholar]
- 12.Riemer AB, Klinger M, Wagner S, Bernhaus A, Mazzucchelli L, Pehamberger H, et al. Generation of peptide mimics of the epitope recognized by trastuzumab on the oncogenic protein Her-2/neu. J Immunol. 2004;173(1):394–401. doi: 10.4049/jimmunol.173.1.394. [DOI] [PubMed] [Google Scholar]
- 13.Yip YL, Smith G, Koch J, Dubel S, Ward RL. Identification of epitope regions recognized by tumor inhibitory and stimulatory anti-ErbB-2 monoclonal antibodies: implications for vaccine design. J Immunol. 2001;166(8):5271–8. doi: 10.4049/jimmunol.166.8.5271. [DOI] [PubMed] [Google Scholar]
- 14.Zwick MB, Bonnycastle LL, Menendez A, Irving MB, Barbas CF, 3rd, Parren PW, et al. Identification and characterization of a peptide that specifically binds the human, broadly neutralizing anti-human immunodeficiency virus type 1 antibody b12. J Virol. 2001;75(14):6692–9. doi: 10.1128/JVI.75.14.6692-6699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menendez A, Chow KC, Pan OC, Scott JK. Human immunodeficiency virus type 1-neutralizing monoclonal antibody 2F5 is multi-specific for sequences flanking the DKW core epitope. J Mol Biol. 2004;338(2):311–27. doi: 10.1016/j.jmb.2004.02.051. [DOI] [PubMed] [Google Scholar]
- 16.Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, Wyatt R, et al. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J Virol. 2004;78(19):10724–37. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geerligs HJ, Weijer WJ, Welling GW, Welling-Wester S. The influence of different adjuvants on the immune response to a synthetic peptide comprising amino acid residues 9-21 of herpes simplex virus type 1 glycoprotein D. J Immunol Methods. 1989;124(1):95–102. doi: 10.1016/0022-1759(89)90190-7. [DOI] [PubMed] [Google Scholar]
- 18.De Silva BS, Egodage KL, Wilson GS. Purified protein derivative (PPD) as an immunogen carrier elicits high antigen specificity to haptens. Bioconjug Chem. 1999;10(3):496–501. doi: 10.1021/bc9800724. [DOI] [PubMed] [Google Scholar]
- 19.Briand JP, Muller S, Van Regenmortel MH. Synthetic peptides as antigens: pitfalls of conjugation methods. J Immunol Methods. 1985;78(1):59–69. doi: 10.1016/0022-1759(85)90329-1. [DOI] [PubMed] [Google Scholar]
- 20.Maitta RW, Datta K, Lees A, Belouski SS, Pirofski LA. Immunogenicity and efficacy of Cryptococcus neoformans capsular polysaccharide glucuronoxylomannan peptide mimotope-protein conjugates in human immunoglobulin transgenic mice. Infect Immun. 2004;72(1):196–208. doi: 10.1128/IAI.72.1.196-208.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rubinchik E, Chow AW. Recombinant expression and neutralizing activity of an MHC class II binding epitope of toxic shock syndrome toxin-1. Vaccine. 2000;18(21):2312–20. doi: 10.1016/s0264-410x(99)00554-x. [DOI] [PubMed] [Google Scholar]
- 22.May RJ, Beenhouwer DO, Scharff MD. Antibodies to keyhole limpet hemocyanin cross-react with an epitope on the polysaccha-ride capsule of Cryptococcus neoformans and other carbohydrates: implications for vaccine development. J Immunol. 2003;171(9):4905–12. doi: 10.4049/jimmunol.171.9.4905. [DOI] [PubMed] [Google Scholar]
- 23.Kneissel S, Queitsch I, Petersen G, Behrsing O, Micheel B, Dubel S. Epitope structures recognised by antibodies against the major coat protein (g8p) of filamentous bacteriophage fd (Inoviridae) J Mol Biol. 1999;288(1):21–8. doi: 10.1006/jmbi.1999.2676. [DOI] [PubMed] [Google Scholar]
- 24.Willis AE, Perham RN, Wraith D. Immunological properties of foreign peptides in multiple display on a filamentous bacteriophage. Gene. 1993;128(1):79–83. doi: 10.1016/0378-1119(93)90156-w. [DOI] [PubMed] [Google Scholar]
- 25.Delmastro P, Meola A, Monaci P, Cortese R, Galfre G. Immunogenicity of filamentous phage displaying peptide mimotopes after oral administration. Vaccine. 1997;15(11):1276–85. doi: 10.1016/s0264-410x(97)00072-8. [DOI] [PubMed] [Google Scholar]
- 26.Bastien N, Trudel M, Simard C. Protective immune responses induced by the immunization of mice with a recombinant bacteriophage displaying an epitope of the human respiratory syncytial virus. Virology. 1997;234(1):118–22. doi: 10.1006/viro.1997.8632. [DOI] [PubMed] [Google Scholar]
- 27.de la Cruz VF, Lal AA, McCutchan TF. Immunogenicity and epitope mapping of foreign sequences via genetically engineered filamentous phage. J Biol Chem. 1988;263(9):4318–22. [PubMed] [Google Scholar]
- 28.Demangel C, Lafaye P, Mazie JC. Reproducing the immune response against the Plasmodium vivax merozoite surface protein 1 with mimotopes selected from a phage-displayed peptide library. Mol Immunol. 1996;33(11–12):909–16. doi: 10.1016/s0161-5890(96)00058-2. [DOI] [PubMed] [Google Scholar]
- 29.di Marzo Veronese F, Willis AE, Boyer-Thompson C, Appella E, Perham RN. Structural mimicry and enhanced immunogenicity of peptide epitopes displayed on filamentous bacteriophage The V3 loop of HIV-1 gp120. J Mol Biol. 1994;243(2):167–72. doi: 10.1006/jmbi.1994.1643. [DOI] [PubMed] [Google Scholar]
- 30.Frenkel D, Katz O, Solomon B. Immunization against Alzheimer’s beta -amyloid plaques via EFRH phage administration. Proc Natl Acad Sci USA. 2000;97(21):11455–9. doi: 10.1073/pnas.97.21.11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Felici F, Luzzago A, Folgori A, Cortese R. Mimicking of discontinuous epitopes by phage-displayed peptides. II. Selection of clones recognized by a protective monoclonal antibody against the Bordetella pertussis toxin from phage peptide libraries. Gene. 1993;128(1):21–7. doi: 10.1016/0378-1119(93)90148-v. [DOI] [PubMed] [Google Scholar]
- 32.Galfre G, Monaci P, Nicosia A, Luzzago A, Felici F, Cortese R. Immunization with phage-displayed mimotopes. Methods Enzymol. 1996;267:109–15. doi: 10.1016/s0076-6879(96)67008-6. [DOI] [PubMed] [Google Scholar]
- 33.Grabowska AM, Jennings R, Laing P, Darsley M, Jameson CL, Swift L, et al. Immunisation with phage displaying peptides representing single epitopes of the glycoprotein G can give rise to partial protective immunity to HSV-2. Virology. 2000;269(1):47–53. doi: 10.1006/viro.2000.0185. [DOI] [PubMed] [Google Scholar]
- 34.Greenwood J, Willis AE, Perham RN. Multiple display of foreign peptides on a filamentous bacteriophage Peptides from Plasmodium falciparum circumsporozoite protein as antigens. J Mol Biol. 1991;220(4):821–7. doi: 10.1016/0022-2836(91)90354-9. [DOI] [PubMed] [Google Scholar]
- 35.Zuercher AW, Miescher SM, Vogel M, Rudolf MP, Stadler MB, Stadler BM. Oral anti-IgE immunization with epitope-displaying phage. Eur J Immunol. 2000;30(1):128–35. doi: 10.1002/1521-4141(200001)30:1<128::AID-IMMU128>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 36.Irving MB, Pan O, Scott JK. Random-peptide libraries and antigen-fragment libraries for epitope mapping and the development of vaccines and diagnostics. Curr Opin Chem Biol. 2001;5(3):314–24. doi: 10.1016/S1367-5931(00)00208-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manoutcharian K, Gevorkian G, Cano A, Almagro JC. Phage displayed biomolecules as preventive and therapeutic agents. Curr Pharm Biotechnol. 2001;2(3):217–23. doi: 10.2174/1389201013378671. [DOI] [PubMed] [Google Scholar]
- 38.van Houten NE, Scott JK. Phage libraries for the development of vaccines and serum based diagnostics. In: Sidhu SS, editor. Phage display in biotechnology and drug discovery. New York: Marcel Dekker; 2004. [Google Scholar]
- 39.Manoutcharian K, Terrazas LI, Gevorkian G, Acero G, Petrossian P, Rodriguez M, et al. Phage-displayed T-cell epitope grafted into immunoglobulin heavy-chain complementarity-determining regions: an effective vaccine design tested in murine cysticercosis. Infect Immun. 1999;67(9):4764–70. doi: 10.1128/iai.67.9.4764-4770.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Berardinis P, D’Apice L, Prisco A, Ombra MN, Barba P, Del Pozzo G, et al. Recognition of HIV-derived B and T cell epitopes displayed on filamentous phages. Vaccine. 1999;17(11–12):1434–41. doi: 10.1016/s0264-410x(98)00377-6. [DOI] [PubMed] [Google Scholar]
- 41.Hill DF, Petersen GB. Nucleotide sequence of bacteriophage f1 DNA. J Virol. 1982;44(1):32–46. doi: 10.1128/jvi.44.1.32-46.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong G, Smith GP, Berry J, Brunham RC. Conformational mimicry of a chlamydial neutralization epitope on filamentous phage. J Biol Chem. 1994;269(39):24183–8. [PubMed] [Google Scholar]
- 43.Burton DR, Pyati J, Koduri R, Sharp SJ, Thornton GB, Parren PW, et al. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science. 1994;266(5187):1024–7. doi: 10.1126/science.7973652. [DOI] [PubMed] [Google Scholar]
- 44.Burton DR, Barbas CF, 3rd, Persson MA, Koenig S, Chanock RM, Lerner RA. A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc Natl Acad Sci USA. 1991;88(22):10134–7. doi: 10.1073/pnas.88.22.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonnycastle LLC, Shen J, Menendez A, Scott JK. Production of peptide libraries. In: Barbas C III, Burton DR, Scott JK, Silverman GJ, editors. Phage display: a laboratory manual Plainview. New York: Cold Spring Harbor Laboratory Press; 2001. pp. 15.3–7. [Google Scholar]
- 46.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual Plainview. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 47.Bonnycastle LLC, Brown KL, Tang J, Scott JK. Assaying phage-borne peptides by phage capture on fibrinogen or streptavidin. Biol Chem. 1997;378(6):509–15. doi: 10.1515/bchm.1997.378.6.509. [DOI] [PubMed] [Google Scholar]
- 48.Bonnycastle LLC, Shen J, Menendez A, Scott JK. General phage methods. In: Barbas C III, Burton DR, Scott JK, Silverman GJ, editors. Phage display: a laboratory manual Plainview. New York: Cold Spring Harbor Laboratory Press; 2001. pp. 15.1–15.30. [Google Scholar]
- 49.Smith GP, Scott JK. Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol. 1993;217:228–57. doi: 10.1016/0076-6879(93)17065-d. [DOI] [PubMed] [Google Scholar]
- 50.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76(9):4350–4. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonnycastle LLC, Mehroke JS, Rashed M, Gong X, Scott JK. Probing the basis of antibody reactivity with a panel of constrained peptide libraries displayed by filamentous phage. J Mol Biol. 1996;258(5):747–62. doi: 10.1006/jmbi.1996.0284. [DOI] [PubMed] [Google Scholar]
- 52.Ashcroft SJH. Practical statistics for the biological sciences: simple pathways to statistical analyses. Basingstoke: Palgrave; 2002. [Google Scholar]
- 53.Zwick MB, Menendez A, Bonnycastle LLC, Scott JK. Analysis of phage-borne peptides. In: Barbas C III, Burton DR, Scott JK, Sil-verman GJ, editors. Phage display: a laboratory manual Plainview. New York: Cold Spring Harbor Laboratory Press; 2001. pp. 18.26–30. [Google Scholar]
- 54.Leenaars M, Koedam MA, Hendriksen CF, Claassen E. Immune responses and side effects of five different oil-based adjuvants in mice. Vet Immunol Immunopathol. 1998;61(2–4):291–304. doi: 10.1016/s0165-2427(97)00133-5. [DOI] [PubMed] [Google Scholar]
- 55.Hanly WC, Artwohl JE, Bennett BT. Review of polyclonal antibody production procedures in mammals and poultry. ILAR J. 1995;37(3):93–118. doi: 10.1093/ilar.37.3.93. [DOI] [PubMed] [Google Scholar]
- 56.Kenney JS, Hughes BW, Masada MP, Allison AC. Influence of adjuvants on the quantity, affinity, isotype and epitope specificity of murine antibodies. J Immunol Methods. 1989;121(2):157–66. doi: 10.1016/0022-1759(89)90156-7. [DOI] [PubMed] [Google Scholar]
- 57.Hu JG, Yokoyama T, Kitagawa T. Studies on the optimal immunization schedule of experimental animals. V. The effects of the route of injection, the content of Mycobacteria in Freund’s adjuvant and the emulsifying antigen. Chem Pharm Bull (Tokyo) 1990;38(7):1961–5. doi: 10.1248/cpb.38.1961. [DOI] [PubMed] [Google Scholar]
- 58.Cooper PD, Steele EJ. Algammulin, a new vaccine adjuvant comprising gamma inulin particles containing alum: preparation and in vitro properties. Vaccine. 1991;9(5):351–7. doi: 10.1016/0264-410x(91)90063-c. [DOI] [PubMed] [Google Scholar]
- 59.Wan Y, Wu Y, Bian J, Wang XZ, Zhou W, Jia ZC, et al. Induction of hepatitis B virus-specific cytotoxic T lymphocytes response in vivo by filamentous phage display vaccine. Vaccine. 2001;19(20–22):2918–23. doi: 10.1016/s0264-410x(00)00561-2. [DOI] [PubMed] [Google Scholar]
- 60.Agarwal A, Sarkar S, Nazabal C, Balasundaram G, Rao KV. B cell responses to a peptide epitope I The cellular basis for restricted recognition. J Immunol. 1996;157(7):2779–88. [PubMed] [Google Scholar]
- 61.Hermanson GT. Bioconjugate techniques. San Diego: Academic Press; 1996. [Google Scholar]