

Abstract
The L1 gene of human papillomavirus-18 (HPV-18) is consistently hypermethylated in cervical carcinomas, but frequently hypo- or unmethylated in exfoliated cells from asymptomatic patients. In precancerous lesions, L1 is sporadically hypermethylated, correlating with the severity of the neoplasia. In order to explore the potential of using L1 methylation as a workable biomarker for carcinogenic progression of HPV-18 infections in routinely taken samples, our aim was to develop methylation-detection techniques that were sensitive and rapid without being overly complex technically. Therein, we developed a methylation-specific PCR (MSP) through the design of primer sets that specifically amplify either methylated or unmethylated HPV-18 L1 DNA within bisulfite-modified sample DNA. Amplification of unmethylated and in vitro methylated HPV-18 DNA by MSP resulted in 2500 copies of either of the two L1 DNA species being detected, a satisfactory sensitivity considering that bisulfite treatment leads to the fragmentation of about 99% of sample DNA. The primers proved specific and did not generate false positive results at concentrations exceeding the lowest limit of detection by a factor of 400. DNA from carcinomas yielded PCR signals only with the methylation specific primers, and not with primers specific for unmethylated L1 genes. The inverse result was obtained with DNA from precursor lesions that contained only hypomethylated DNA. High-grade precursor lesions and carcinomas that contained hyper- as well as hypomethylated L1 DNA yielded PCR signals with both primers. By developing a fluorescence based real-time PCR, we quantitatively analyzed samples with in vitro methylated and unmethylated L1 DNA, and could distinguish clinical samples with hyper- and hypomethylated DNA or mixtures of both DNAs. The methylation-specific and real-time PCR techniques permitted efficient HPV-18 L1 methylation analyses and open the door for larger-scale clinical studies where the utility of methylation status to predict the progression of HPV-18 infection and HPV-18 associated lesions is assessed.
INTRODUCTION
Infection with human papillomaviruses (HPVs) is necessary but not sufficient to cause cervical cancer (International Agency for Research on Cancer, 1995). Many molecular principles of the transformation by HPVs are well researched, notably the multiple functions of the oncoproteins E6 and E7, which include abrogation of the normal functioning of tumor suppressor proteins p53 and pRB (Dyson et al., 1989, Werness et al., 1990). However, the mechanisms that determine whether an HPV infection remains latent, progresses to a dysplasia, or worse, carcinoma, remain largely undefined. They likely include immune surveillance as well as mutations of cellular genes that synergize with the HPV infection. Some of these events occur, however, unquestionably on the level of the virus, notably the recombination between the HPV genomic DNA and chromosomal sequences of the host cell. HPV genomes are very often integrated in the host DNA in cancer but are episomal in precursor lesions (Daniel et al., 1995; Schwarz et al., 1985). This generally accepted observation recently required a slight modification, as stepwise increments of chromosomally integrated HPV genomes rather than singular complete transitions were observed in large-scale studies of disease progression (Arias-Pulido et al., 2006). Integration appears to favor carcinogenesis, as it interferes with negative feedback repression of the transcription of the E6 and E7 oncogenes by the E2 protein (Tan et al., 1994; Demeret et al., 1997) and activates increased transcription of the oncogenes by a nuclear matrix dependent mechanism (Stünkel et al., 2000). Therefore, development of tests that detect HPV genome integration is desirable to improve molecular and etiological diagnosis in both basic research and clinical applications.
In our previous studies of HPV DNA methylation we observed that the L1 gene of HPV-18 is hypermethylated in the carcinomas that were investigated, contrasting with its hypo- or unmethylated state in asymptomatic and a component of precursor lesions. The adjacent long control region (LCR) is hypomethylated in tumors, precursor lesions and in asymptomatic infections (Badal et al., 2004; Turan et al., 2006). Similar studies of HPV-16 suggested a somewhat more complex, two-pronged scenario. In this virus, we observed an intermediate level of methylation of L1 and the LCR in asymptomatic infection, hypomethylation in precursor lesions, and hypermethylation that maximally affected L1, just as in HPV-18, in carcinomas (Badal et al., 2003; Kalantari et al., 2004). These findings suggest that L1 DNA methylation may be a powerful biomarker of the clinical progression of HPV-18 associated disease and possibly also HPV-16 associated lesions, although it is still a matter of inferences whether L1 DNA is methylated as a consequence of recombination between viral and cellular DNA or for yet unknown reasons. We also cannot yet explain the methylation differences between HPV-16 and HPV-18 that we observed during asymptomatic infection. These two HPV types account for more than two thirds of the cervical cancers worldwide (Clifford et al., 2003; Munoz et al., 2003) and are the most frequently studied types among the 18 or so high-risk HPV types. Being the paradigms for high-risk HPV types, they are sometimes considered to be very similar, although a more thorough analysis reveals that they are only remotely related phylogenetically (de Villiers et al., 2004). While they are associated with cervical malignancies, they differ in a wide array of molecular, biological, and pathological properties (for references, see Arias-Pulido, 2005).
The term “DNA-methylation” refers to the transfer of a methyl-group to cytosine residues, which are typically stably maintained in methylated form when they are part of the palindromic cytidine-guanidine (CpG) dinucleotides. DNA methylation is of great general interest, as it affects the conformation of nucleosomes in a complex network of epigenetic interactions with histone methylation, histone acetylation and histone deacetylation (reviewed in Bird, 2002; Fuks, 2005; Goll and Bestor 2005), regulating gene expression on the level of chromatin.
The methylation of HPV-1 genomes and the cottontail rabbit PV was observed more than twenty years ago (Burnett et al., 1984, Sugarawa et al., 1983, Wettstein et al., 1983), but only recently was it shown that methylation of HPV-16 and 18 DNA takes place regularly in vivo in cervical cells, both in clinical samples and in cell cultures (Badal et al., 2003 and 2004; Kim et al., 2003; van Tine et al., 2004; Kalantari et al., 2004; Wiley et al., 2005; Turan et al., 2006; Bhattacharjee and Sengupta, 2006). This phenomenon raises questions of where and how HPV genomes are recognized by the cellular DNA methylation machinery, and about the role of this mechanism during the HPV life cycle and HPV dependent carcinogenesis. During carcinogenesis, HPV DNA may become methylated like any type of foreign DNA in response to the recombination with cellular DNA, pointing toward a correlation of these events, while not yet to the actual cause of methylation (Doerfler et al., 2001). Methylation during the productive HPV-16 life cycle is not yet understood, but may be linked to the binding of sequence specific transcription factors like CDP, that synergize with histone modification mechanisms (O’Connor et al., 2000).
In this publication we report on the development of improved diagnostic tests of HPV-18 L1 methylation that could be used within large scale clinical studies designed to assess the nature and course of HPV-18 mediated cervical disease with respect to methylation. The technical foundation of this work was drawn from our past studies where bisulfite sequencing of HPV genomes was established. This involved the modification of sample DNA with bisulfite, followed by PCR and product cloning into E. coli vectors before sequencing. Application of this approach, referred to as “bisulfite sequencing”, leads to cytosines being detected as thymine residues while methyl cytosines are detected as cytosine residues. Our previous strategies involved the sequencing of multiple E. coli clones from each clinical sample, which was justified in order to evaluate the complex and diverse methylation patterns of HPV genomes both between and even within individual HPV infections. However, this approach would be too labor intensive and complex to apply to larger scale clinical studies designed to assess the impact of methylation using routinely collected patient samples. In order to overcome this, in the present study we explored two techniques designed to detect HPV-18 L1 DNA methylation that could be amenable to high-throughput testing. One of these techniques is based on PCR primers specific for methylated and unmethylated HPV DNA (methylation specific PCR, or “MSP”, Herman et al., 1996). It has the power to detect fairly small amounts of methylated DNA among an excess of unmethylated sequences, and vice versa. Its complexity is reduced compared with the bisulfite sequencing approach described earlier, as it does not require cloning and sequencing. The second technique is based on fluorescence based real-time PCR with TaqMan probes specific for methylated DNA post amplification with primers that do not distinguish between methylated and unmethylated DNA (Eads et al., 2000). This technique is quantitative and even less labor intensive than MSP, as it does not require agarose gel electrophoresis subsequent to the PCR. While neither of these techniques reveals the detailed methylation patterns, they are both useful and suitable tools for the analysis of HPV methylation analyses within the context of large clinical studies.
RESULTS
Selective amplification of unmethylated and in vitro methylated HPV-18 L1 DNA
In order to selectively amplify HPV-18 DNA sequences that were either methylated or unmethylated, we modified the sample DNAs with bisulfite and amplified the modified DNA with either of two primer pairs that were specific for methylated or unmethylated DNA, generating a copy of the HPV-18 genomic segment between the positions 7017 and 7140, respectively. The unmethylated test sample used for the amplificiations was an HPV-18 plasmid clone grown in E. coli. For the methylated test sample we completely modified the same plasmid DNA preparation in vitro with the SssI methyltransferase. Fig. 1 documents the specificity of the amplification reaction, that was targeted at a 12 pg concentration of these DNA preparations. Application of the methylation specific primer pair yielded a PCR product with the methylated but not the unmethylated DNA sample (lanes 2 and 6, respectively). Comparatively, application of the primer pair designed to detect unmethylated DNA yielded a PCR product with the unmethylated (lane 7) but not the methylated (lane 3) DNA. The lack of amplification with the respective negative controls documents a good specificity of the primers, as these reactions targeted approximately 1,000,000 HPV-18 molecules, or about 400 times as many molecules that were minimally necessary to get a detectable signal for the combination of the matching primers and target (see next paragraph).
Fig. 1. Methylation specific PCR (MSP), qualitative analysis: Selective amplification of methylated and unmethylated HPV-18 L1 DNA.
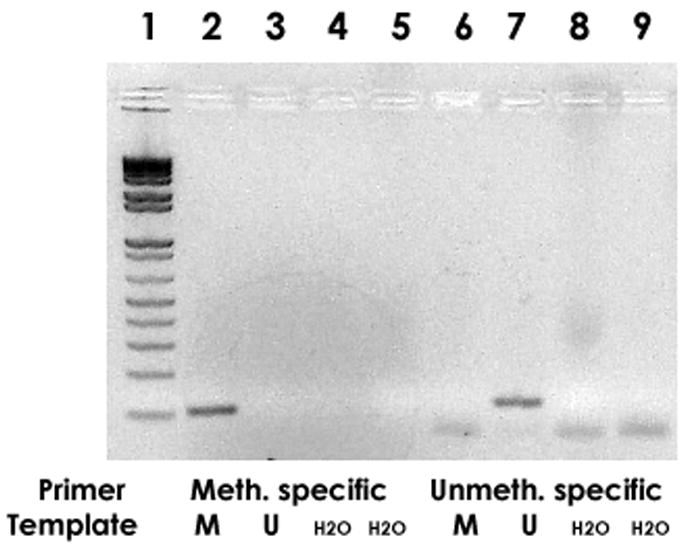
12 pg of HPV-18 plasmid DNA, completely CpG methylated with SssI methyltransferase, were PCR amplified with methylation specific primers (band in lane 2) and primers specific for unmethylated DNA (no band in lane 6). There is no signal with methylated DNA targeted by the primer pair specific for unmethylated DNA (lane 3), while the unmethylated DNA became amplified with these primers (lane 7). Lane 1: size marker, lanes 4,5,8,9: negative control (water).
In order to investigate whether these methods were sensitive enough to analyze clinical samples, we challenged these primers with serially diluted methylated and unmethylated HPV-18 DNA. Fig. 2 shows that the limits of detection were 30 fg of methylated and unmethylated HPV-18 DNA, respectively. This quantity corresponds to approximately 2,500 copies of HPV-18 DNA. This copy number is higher than the typical target sensitivity of a PCR analysis, which is around 1 to 10 copies. However, since about 99 % of bisulfite treated DNA is fragmented to sizes that preclude PCR amplification (Frommer et al., 1992), this detection threshold is satisfactory.
Fig. 2. Methylation specific PCR (MSP), quantitative analysis: Sensitivity of detecting methylated and unmethylated HPV-18 L1 DNA with specific primers.
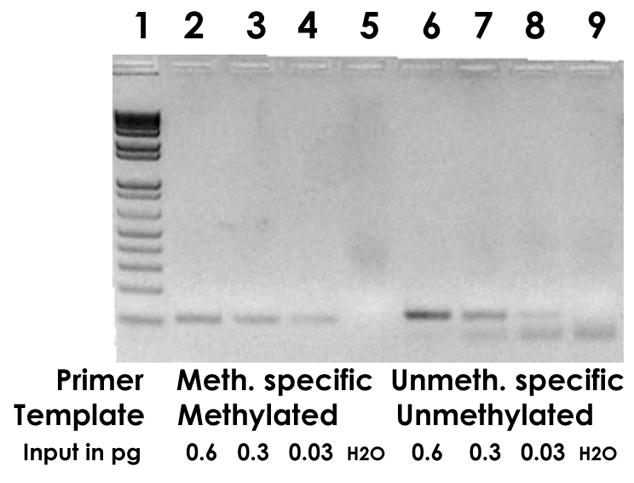
Lanes 2, 3 and 4 show the amplification of 0.6, 0.3, and 0.03 pg of Sss I methylated HPV-18 plasmid with methylation specific primers, lanes 6, 7, and 8 amplification of 0.6, 0.3, and 0.03 pg of unmethylated HPV-18 plasmid with primers specific for unmethylated DNA. The weak bands in slots 4 and 8 were the lowest sensitivity that could be achieved and correspond to about 2,500 molecules, a satisfactory detection limit in consideration of the fact that bisulfite modification fragments about 99% of the target DNA to sizes too small for PCR detection. Slot 1, size marker, slot 5 and 9, negative control (water).
A cervical smear or swab contains roughly 1 million exfoliated cells, and an HPV infected cell has often been estimated to contain approximately 100 HPV genomes, namely as many copies as a cell line with episomal HPV-16 (Stanley et al., 1992). We can therefore extrapolate that DNA preparations from exfoliated cervical cells might maximally contain up to 100 million HPV genomes (although a proportion will contain substantially less). As our test can detect less than 0.01% of the methylated or unmethylated HPV DNA that can be maximally expected in a clinical sample, we anticipate that our method is sufficiently sensitive to be appropriate for the testing of the majority of clinical samples. Within the context of a clinical study, this sensitivity aspect of the technique could be further evaluated by concurrent analysis with a “standard” HPV-18 PCR.
MSP analysis of clinical specimens that contain exclusively methylated or unmethylated HPV-18 L1 DNA, or a mixture of the two molecular species
The L1 gene of HPV-18 is frequently hypo- or unmethylated in asymptomatic infections and precursor lesions, but hypermethylated in carcinomas. Our previous study (Turan et al., 2006) also identified some samples that deviated from this rule, as a small fraction of high-grade precursor lesions and carcinomas contained both hypo- and hypermethylated L1 sequences. Fig. 3 summarizes the published data of samples that were exclusively hypo- or hypermethylated, and new analyses of four samples (L2, L4, T3, T8) with mixtures of hypo- and hypermethylated DNA performed as part of this new study.
Fig. 3. CpG methylation patterms of the L1 gene of HPV-18 in pre-cancerous lesions and carcinomas of the cervix.
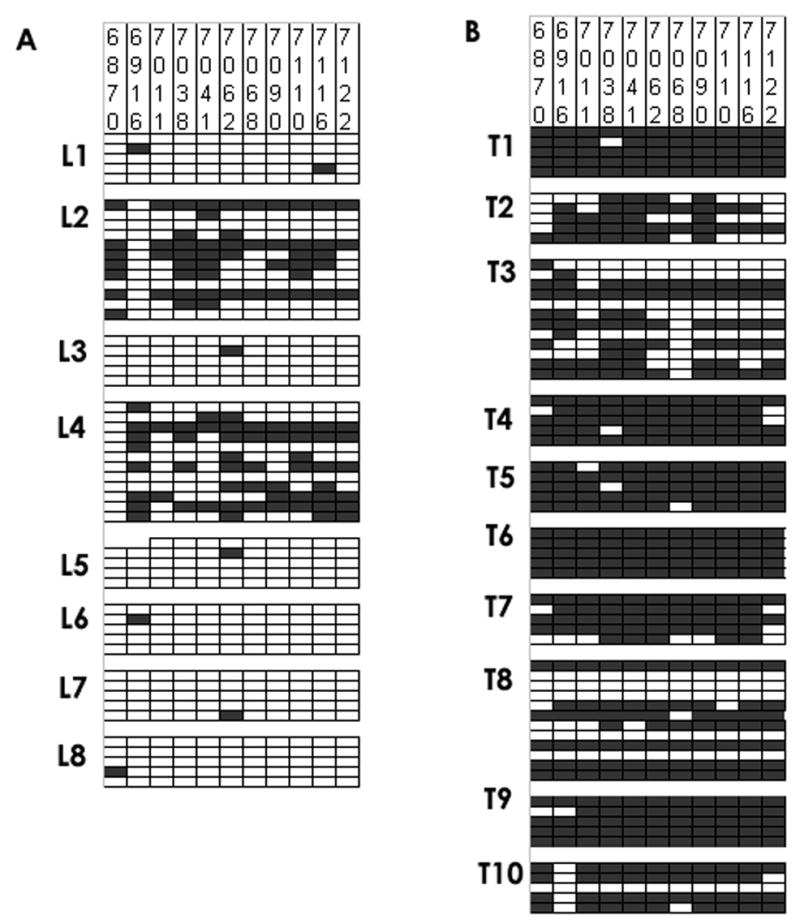
The figure shows the methylation status of individual CpGs in E. coli clones obtained after bisulfite modification and PCR amplification of DNA from precursors lesions (L1 to L8) and malignant cervical carcinomas (tumors T1 to T10). For each lesion, we sequenced five and twelve, respectively, clones to analyze and document intra-lesion heterogeneity of HPV-18 methylation. Vertical columns of rectangles: CpGs at genomic positions as numbered in the top of the figure, horizontal lines of rectangles: individual E. coli clones obtained from each sample. Black rectangles: meCpGs, white rectangles, CpGs. The data for all samples except L2, L4, T3, and T8 have been published before (Turan et al., 2006), while the data sets for L2, L4, T3 and T8 have been expanded to confirm and quantify the heterogeneity of these samples regarding their further analysis (see Fig.s 4 and 5). These data are presented here as they are a prerequisite to understand the outcome of the MSP experiments shown in Fig. 4 and the real-time PCR in Table 3.
Fig. 3A visualizes HPV-18 L1 methylation in eight precursor lesions. Horizontal blocks of five or twelve lines separate clones from individual lesions, while vertical columns identify specific unmethylated CpGs (white rectangles) or meCpGs (black). The data from six lesions with exclusively hypomethylated sequences (L1, L3, L5–L8) have been published (Turan et al., 2006), while we expanded the data sets for two lesions with mixtures of hypo- and hypermethylated sequences (L2, L4). These latter two lesions contained five out of twelve hypermethylated L1 segments (with hypermethylation being defined as six or more meCpGs out of a total of eleven), and seven unmethylated or hypomethylated (defined as five or less mCpGs) L1 segments. Fig. 3B depicts L1 methylation in ten carcinomas. The data from eight carcinomas with exclusively hypermethylated sequences (T1, T2, T4–T7, T9, T10) have been published. The two lesions with mixtures of hypo- and hypermethylated sequences (T3 and T8) contained seven and six hypermethylated molecules, and five and six unmethylated or hypomethylated sequences, respectively.
Characterization of the methylation state of the HPV-18 L1 gene with primers specific for methylated and unmethylated DNA
Fig. 4 shows the MSP analysis of the 18 HPV-18 samples, whose methylation patterns were shown in Fig. 3. As expected, of the eight DNA samples extracted from precursor lesions, amplification with the methylation-specific primers yielded products with the clones L2 and L4 only, while the primer pairs specific for unmethylated DNA yielded clear products for the seven (expected) samples. Clone L2 gave only barely detectable or no signals in these two analyses as well as in the real-time analysis described below. As a likely explanation for this, we identified a G to A mutation at the genomic position 7,130, which overlapped with the reverse primer of both testing methods described in this paper, which could have reduced the efficiency of the PCR amplification. All ten of the DNA samples extracted from cancer material tested positive with the methylation specific primers (Fig. 4A and B, lanes 2–6), while it was only clones T3 and T8 that tested positive with the primers specific for unmethylated DNA. We conclude from these data that the primer pairs can distinguish between methylated and unmethylated HPV-18 L1 sequences in clinical samples. Both DNA species were detectable when they were present, and within the level of sensitivity of the test, no methylated DNA was detected among six out of eight precursor lesions and no unmethylated sequence among eight out of ten carcinomas, as evidenced by the analysis of individual clones.
Fig. 4. Methylation-specific PCR (MSP) analysis of methylated and unmethylated DNA in cervical precursor lesions and tumors.
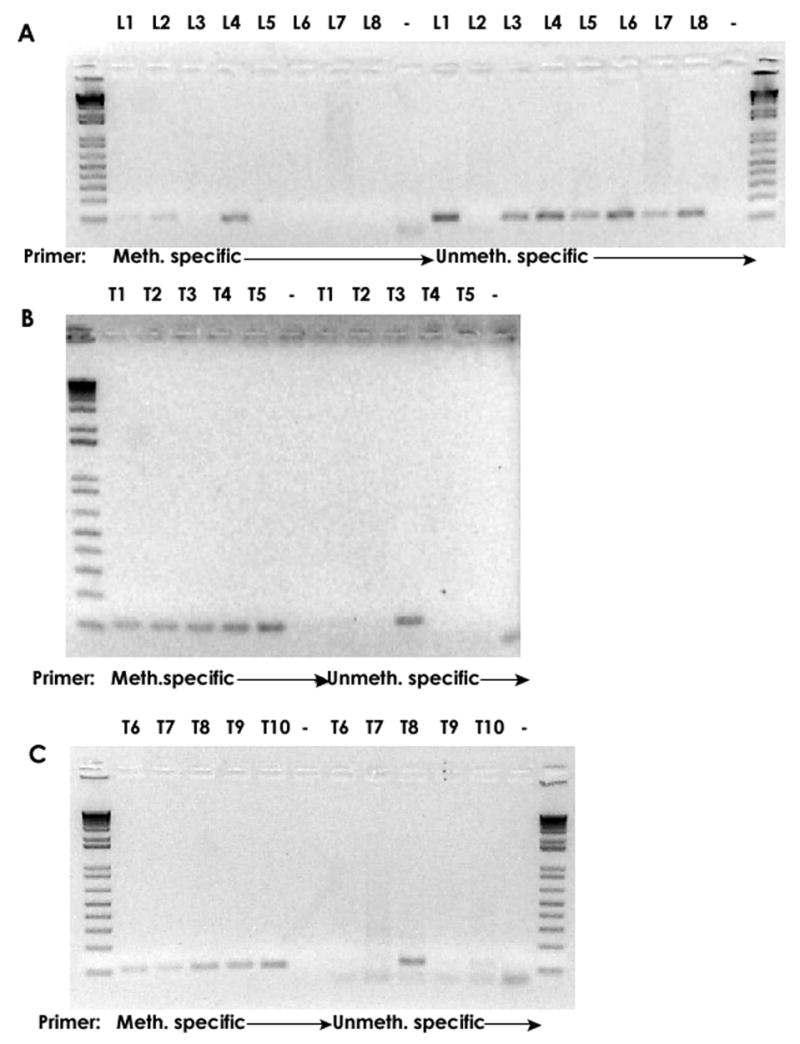
MSP analysis of 18 HPV-18 samples with known methylation patterns (Fig. 3). Analysis of the eight DNAs from precursor lesions (L) with methylation-specific primers gave signals only with the clones L2 and L4, primer pairs specific for unmethylated gave signals for seven samples. A mutation overlapping with the reverse primer likely explains the unsatisfactory outcome with clone L2. Analysis of the tumors (T) with methylation specific primers gave positive signals for all ten samples. Only T3 and T8 gave showed for unmethylated DNA as expected from Fig.3. The DNA of L8 was exhausted and could not be included in this analysis.
Evaluation of mixtures of methylated and unmethylated HPV-18 L1 DNA by real-time PCR
While the labor intensity of the MSP technique is reduced compared to the bisulfite sequencing approach, it still incorporates analysis by agarose gel electrophoresis. It is also only qualitative, as it measures the endpoint of a reaction. Thus we developed a real-time PCR to bypass this step. The method is based on the amplification of DNA treated by bisulfite modification with primers specific for modified but not methylated DNA. Amplification was performed in the presence of TaqMan probes which included CpG motifs to detect sequences that originally contained either meCpGs or TpGs or CpAs for sequences that were originally unmethylated CpGs. The TaqMan probes contain a 5’ fluorescent dye (e.g. 6FAM) and a 3’TAMRA quencher. Probes that hybridize to the amplicon are cleaved by the exonuclease activity of the Taq polymerase, thus separating dye and quencher, generating a resulting fluorescent signal that is proportional to the amount of amplicons. We adjusted signal output to determine HPV-18 methylation via the process described in the methods section. In the following section, we report on the quantitative measurement of methylation by cT-values (cycle threshold values), which indicate the PCR cycle, when a sample passed a defined threshold of fluorescence.
Table 1 shows a cT value of 32.3 with 24 pg of methylated HPV-18 plasmid DNA amplified in the presence of methylation specific probe, and a cT value of 31.9 for the combination of unmethylated plasmid with probe specific for unmethylated DNA. No fluorescence signals were obtained by combining methylated plasmid with the probe specific for unmethylated DNA, and unmethylated plasmid with methylation specific probe, which confirms a specificity of our probes.
Table 1. Real-time PCR of methylated and unmethylated HPV-18 L1 DNA with unspecific primers but specific probes.
The cycle-threshold (cT) show that methylated DNA could only be detected with methylation specific probes and unmethylated DNA with probes specific for unmethylated DNA, although the PCR primers and amplification conditions were the same in all four reactions. 0 = no signal. All cT values of this table as well as of table 2 and 3 are arithmetic means of duplicate experiments.
| Methylated plasmid | unmethylated plasmid | |
|---|---|---|
| Probe for methylated DNA | 32.3 | 0 |
| probe for unmethylated DNA | 0 | 31.9 |
Table 2 shows the potential of this approach for quantification of DNA. When the amount of methylated HPV-18 L1 target DNA was varied from 0.3 to 12 pg, i.e. by a factor of 40, the cT value in the presence of methylation specific probe varied from 31.5 to 26.5, i.e. fivefold, representing a similar factor, namely 25 = 32. When the unmethylated target DNA was varied by a factor of 40, the cT value in the presence of probe specific for unmethylated DNA changed from 34.7 to 26.8, or by 5.9, representing a factor of about 60. Overall, increasing cT values correlated with decreasing product concentration.
Table 2.
Quantitative relationship between methylated an unmethylated HPV-18 L1 input DNA and cT values in real-time PCR experiments.
| Methylated plasmid | cT (Probe for methylated DNA) |
|---|---|
| 0.3 pg | 31.5 |
| 0.6 pg | 29.4 |
| 2.4 pg | 28.4 |
| 12 pg | 26.5 |
| Unmethylated plasmid | cT (Probe for unmethylated DNA) |
| 0.3 pg | 34.7 |
| 0.6 pg | 30.8 |
| 2.4 pg | 28.5 |
| 12 pg | 26.8 |
Table 3 summarizes the analysis of DNA from the same precursor lesions and tumors, whose methylation patterns and MSP analysis were depicted in Fig.s 3 and 4. As a sample and amplification control, we used the single copy endogenous gene myoD1, an amplimer that is free of any CpG dinucleotides (Eads et al., 2000). The myoD cT values of different samples are not the same, as the DNA input was not constant, but a sample aliquot based on volume. As an additional control we included DNA from HeLa cells, which are known to contain about 50 copies of HPV-18 DNA, in which L1 genes are hyper- as well as hypomethylated (Turan et al., 2006).
Table 3. Real-time PCR analysis of 17 HPV-18 DNAs from clinical samples.
The samples are identical with those represented in Fig. 3 and 4, and the detection of methylated and unmethylated DNA correlates with the data of these two figures. For a detailed interpretation, see the main text.
| Lesions | cT (myoD) | cT (HPV-18 L1 meth.) | cT (HPV-18 L1 unmeth.) |
|---|---|---|---|
| HeLa (contr.) | 30.3 | 28.1 | 30.9 |
| L1 | 32.3 | 37.5 | 30.3 |
| L2 | 32.1 | 35.5 | 0 |
| L3 | 30.9 | 0 | 39.3 |
| L4 | 30.1 | 32.4 | 29.3 |
| L5 | 34.0 | 0 | 37.6 |
| L6 | 38.9 | 0 | 37.3 |
| L7 | 30.1 | 0 | 33.7 |
| HeLa(contr.) | 29.1 | 29.3 | 30.0 |
| T1 | 33.9 | 36.8 | 0 |
| T2 | 32.7 | 35.6 | 0 |
| T3 | 32.9 | 34.4 | 38.1 |
| T4 | 29.3 | 34.9 | 0 |
| T5 | 32.0 | 35.3 | 0 |
| T6 | 32.1 | 34.8 | 0 |
| T7 | 34.1 | 36.5 | 0 |
| T8 | 28.6 | 36.9 | 38.2 |
| T9 | 30.9 | 32.9 | 0 |
| T10 | 33.3 | 32.9 | 0 |
In the controls to both data sets, the cT value for methylated HPV-18 L1 DNA in HeLa cells was lower than that for the unmethylated sequence, reflecting the excess of methylated HPV-18 sequences in this cell line. Among the lesions, methylated HPV-18 DNA was found only in L2 and L4, as expected from Fig.s 3 and 4. As expected, unmethylated DNA was detected in the six other lesions, with the exception of lesion L2, which remained repeatedly negative in spite of the observation of unmethylated DNA in the pattern analysis (Fig. 3) and a weak signal in Fig. 4. This was the only deviation from the expected findings in this set of data, and was likely due to a mutation in the sequence overlapping one of the primers (see above). Before entering future large scale medical studies, it will be necessary to exclude that genomic diversity due to the typical variant branches of HPV-18 (Ong et al., 1993) and HPV-16 (Ho et al., 1993; Yamada et al., 1997) overlaps with primer design. Our approach detected in all ten tumor samples methylated DNA (ten cT values between 32.9 and 36.9). As expected from Fig.s 3 and 4, unmethylated DNA could be detected only in the tumors T3 and T8. We conclude that real-time PCR is an efficient and powerful technique to reveal the methylated state of HPV-18 L1 DNA.
DISCUSSION
The studies of the methylation of HPV DNA cited above in addition to the observations of nucleosomal organization of HPV DNA and transcriptional responses to chromatin targeting effectors (Stünkel and Bernard, 1999; Zhao et al., 1999) have provided extensive evidence for the importance of the chromatin organization of HPV-16 and HPV-18 in the viral life cycle and during virus induced pathogenesis. Here, we have presented a technical study based on one of these epigenetic processes, the methylation of the HPV late gene L1 during carcinogenic progression. Our research in this area began with protracted labor intensive approaches, namely PCR amplification combined with methylation specific restriction enzyme digestion and direct sequencing after bisulfite modification (Badal et al., 2003; Badal et al., 2004) in addition to sequencing of cloned DNA subsequent to bisulfite modification (Kalantari et al, 2004; Wiley et al., 2005; Turan et al., 2006). These studies elucidated the complexity of the methylation patterns during HPV infection. Here, we have developed MSP and real-time PCR as technically less demanding tools for the performance of HPV methylation analyses of clinical sample sets. Our techniques are comprehensively described, easily reproducible and amenable to high-throughput. We selected HPV-18 for this study, as previous evidence has pointed to a single type of change in the methylation state in the 2.5 kb genomic segment containing the L1 gene and the LCR, namely hypermethylation of L1 in cancers and a proportion of precursor lesions as opposed to lack of methylation in asymptomatic infection. Our selection of HPV-18 was also justified due to an absence of any methylation of the LCR independent of the severity of the lesion. We propose to confirm this model by testing larger numbers of HPV-18 positive clinical samples crosssectionally. We also wish to analyze longitudinal cohorts to further assess the prognostic value of a methylation-positive result in HPV-18 mediated disease. A further intention is to apply analogous techniques targeting the HPV-16 L1 gene and LCR to analyze the more complex phenomena governing the life cycle and pathogenesis of this virus.
MATERIALS AND METHODS
In vitro methylation of an HPV-18 plasmid
SssI methyltransferase (New England Biolabs) was used to methylate all CpG sites on the HPV-18 plasmid DNA (pBR322-HPV-18, an isolate originally obtained from H. zur Hausen and maintained in our lab for two decades) in vitro. The reaction was carried out in 20 μl volume containing 0.6 mM S-adenosyl methionine, 1x NEB2 buffer (supplied by the manufacturer), 4 units of SssI methyltransferase and 1 to 50 ng of plasmid DNA for 1 hour at 37 ºC. To confirm the complete methylation, one aliquot of the reaction was digested with a methylation sensitive restriction enzyme, HhaI, and run on an agarose gel. We observed that it was necessary to linearize the plasmids (with the single cut restriction enzyme NcoI) before bisulfite treatment in order to achieve complete modification.
Clinical specimens
The project reported here includes the reinvestigation of a subset of clinical specimens introduced in a previous paper (Turan et al., 2006). These DNA samples were obtained from cervical carcinomas from an archival collection of tumors maintained at the Department of Pathology, National Hospital, Oslo, Norway and from an archive of precursor lesion material from the Royal Infirmary of Edinburgh, Edinburgh, Scotland (H.A.C. and K.C.). No patient was sampled for the purpose of the specific research described here.
Bisulfite modification
For bisulfite treatment, 50–1000 ng of sample DNA supplemented with 1 μg of salmon sperm DNA in a total volume of 18 μl in water were denatured with 2 μl of 3 M NaOH and incubated at 37 ºC. After denaturation, 278 μl of 4.8 M sodium bisulfite and 2 μl of 100 mM hydroquinone were added. In some experiments, we used a bisulfite kid supplied by Zymo Research Inc. (EZ bisulfite modification kit). Controls showed identical experimental outcome whether this kit or our own reagents were used. The mixture was incubated in a thermal cycler for 20 cycles of 15 minutes at 55 ºC and 30 seconds at 95 ºC. The modified DNA was desalted with the QIAquick PCR purification protocol and desulfonated thereafter by adding 5.5 μl of 3 M NaOH and 5 μg glycogen prior to 15 min incubation at 37 ºC. The DNA was precipitated with 5.6 μl of 3 M sodium acetate and 150 μl of 100% ethanol, followed by centrifugation. The pellet was washed with 70% ethanol and dissolved in 30–50 μl TE buffer (10mM Tris-HCl pH 8, 1 mM EDTA).
Polymerase chain reactions, primers, T/A cloning and DNA sequencing
Since bisulfite treated DNA is partially degraded, large amplicons cannot be generated, and we report here on the detailed analysis of a fragment of the HPV-18 L1 gene between the genomic positions 6845–7186. The sequences of the primers were designed according to the genomic sequence of HPV-18 (Myers et al., 1994) assuming conversion of all cytosine residues into uracils. The genomic segment 6845–7186 encompassed the 3’ part of the L1 gene and was amplified with the primers Bsp6Fa (for bisulfite specific primer), AATTATTAGTTTGGTGGATATATAT (positions 6845–6869) and Bsp6R, AAAACATACAAACACAACAATAAATA (positions 7186–7161).
PCR was carried out in a 25 μl volume containing 0.2 mM of each of the four dNTPs, 10 pmol of each primer, 2 mM MgCl2 and 1 unit of AmpliTaqGold (Perkin-Elmer). The PCR conditions were 94ºC for 9 min, followed by 40 cycles of 94 ºC for 10 sec, 54 ºC for 30 sec and 68 ºC for 1 min with a final extension at 68 ºC for 7 min. The presence of PCR products was verified by agarose gel electrophoresis, and confirmed amplicons were cloned with the TOPO TA cloning kit for sequencing (Invitrogen). Cloned DNAs were sequenced by Big Dye terminator v3.1 Cycle Sequencing (Applied Biosystems).
Amplification of bisulfite modified DNA with primers selective for methylated or unmethylated HPV-18 L1 DNA (methylation-specific PCR (MSP) amplification
Selective amplification of methylated or unmethylated DNA after bisulfite treatment can be achieved by designing primers spanning CpG dinucleotides, potential methylation targets. Primers containing CpGs recognize sequences that were originally methylated after bisulfite treatment, while primers specific for unmethylated DNA need replacement of these CpGs by TpGs or CpAs, respectively. Here, we used the methylation-specific forward (M5F) and reverse (M5R) primers to amplify the methylated L1 gene between the genome positions 7017 and 7140 of HPV-18.
M5F, genomic position 7022–7041: 5’-GGTTTAGGTTGGATTGCGTC-3’, identifying meCpGs at the positions 7038 and 7041 (as underlined; only the C of the CpG at position 7041 is included in the primer), and M5R, genomic position 7111–7131: 5’-TCCTAACACGTACACGCACAC-3’, identifying meCpGs at the positions 7116 and 7122 (underlined). The equivalent primers to selectively amplify unmethylated sequences were U3F: 5’-TTTTTGGTTTAGGTTGGATTGTGTTGT-3’, and U3R: 5’-CATATTACTTCCTAACACATACACACACACAC-3’ (underlined are TpGs and CpAs that were derived from CpGs). PCR conditions were: 9 min denaturation at 94ºC, and 35 cycles of 94ºC (30 sec, denaturation) and 68ºC (75 sec, annealing and extension at the same temperature), prior to a final extension step of 7 min at 68 ºC.
Detection of methylated HPV-18 L1 DNA by real-time PCR
We designed a quantitative PCR approach to detect methylated and unmethylated HPV-18 L1 DNA in the same reaction without need for agarose gel electrophoresis. For other genes this approach has been described by Eads et al., 2000 and was labeled with the term “MethyLight”. This general approach allows four different strategies, and here we used a modification where HPV-18 DNA is amplified after bisulfite modification with primers that target methylated as well as unmethylated DNA. Quantitative data were generated by probes that differentially hybridize to bisulfite modified DNA that had originally been methylated or unmethylated. The principles of the probe technology, frequently referred to as TaqMan probes, have been published (Morris et al., 1996; Clement et al., 2005). Each of the two oligonucleotides was tagged with a different 5’ fluorescent reporter dye (6FAM, and VIC) and a 3’ quencher (TAMRA). The reaction was quantified with an ABIprism 7900HT instrument. To normalize the DNA amount after bisulfite modification, we used primers and a probe specific for myoD gene. The reaction was carried out in 25 μl volume containing 1x Taqman Universal PCR Mastermix (Applied Biosystems), 0.6 μM of each primer and 0.2 μM of Taqman probe. The thermal cycler conditions were 10 min denaturation at 94 ºC, and 45 cycles of 15 sec at 94 ºC and 1 min at 58 ºC. The results were analyzed using AbiPrism’s SDS v2.1 software.
MyoD1 primers and probes were designed according to Eads et al., 2000. Amplification primers: CCAACTCCAAATCCCCTCTCTAT and TGATTAATTTAGATTGGGTTTAGATAAGGA; probe: 6FAM5’-TCCCTTCCTATTCCTAAATCCAACCTAAATACCTAA-3’TAMRA. Primers for HPV-18 L1 gene were the following: 18mspL1-F: TATATATACACACACACATATTACTTCCTAACA (positions 6955–6979) and 18mspL1-R: GGAATGTGGATTTAAAGGAAAAGTT (positions 7124–7156). Probes for methylated and unmethylated HPV-18 L1 were: 18L1-Mpb: 6FAM5’-CGAAAACCTATAATAAACTTACGACGCAATCCAAC-3’TAMRA (genomic positions 7029–7063) and 18L1-Unpb: VIC5’-CAAAAACCTATAATAAACTTACAACACAATCCAACC-3’TAMRA (genomic positions 7028–7063). The probes spanned the CpG sites at the HPV-18 genome positions 7038, 7041 and 7062.
Acknowledgments
Our research was supported by NIH grant ROI CA-91964 and by funds from the Chao Family Comprehensive Cancer Center of the University of California Irvine to H.U.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arias-Pulido H, Peyton CL, Torrez-Martinez N, Anderson DN, Wheeler CM. Human papillomavirus type 18 variant lineages in United States populations characterized by sequence analysis of LCR-E6, E2, and L1 regions. Virology. 2005;338:22–34. doi: 10.1016/j.virol.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Arias-Pulido H, Peyton CL, Joste NE, Vargas H, Wheeler CM. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J Clin Microbiol. 2006;44:1755–1762. doi: 10.1128/JCM.44.5.1755-1762.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badal S, Badal V, Calleja-Macias IE, Kalantari M, Chuang LSH, Li BFL, Bernard HU. The human papillomavirus-18 genome is efficiently targeted by cellular DNA methylation. Virology. 2004;324:483–492. doi: 10.1016/j.virol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Badal V, Chuang LSH, Badal S, Tang E, Villa LL, Wheeler CM, Li BFL, Bernard HU. CpG methylation of human papillomavirus-16 DNA in cervical cancer cell lines and in clinical specimens: genomic hypomethylation correlates with carcinogenic progression. J Virol. 2003;77:6227–6234. doi: 10.1128/JVI.77.11.6227-6234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee B, Sengupta S. CpG methylation of HPV 16 LCR at E2 binding site proximal to P97 is associated with cervical cancer in presence of intact E2. Virology. 2006 doi: 10.1016/j.virol.2006.06.018. 2006 epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Burnett TS, Sleeman JP. Uneven distribution of methylation sites within the human papillomavirus la genome: possible relevance to viral gene expression. Nucleic Acids Res. 1984;12:8847–8860. doi: 10.1093/nar/12.23.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement G, Benhattar J. A methylation specific dot blot assay (MS-DBA) for the quantitative analysis of DNA methylation in clinical samples. J Clin Pathol. 2005;58:155–158. doi: 10.1136/jcp.2004.021147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford GM, Smith JS, Plummer M, Munoz N, Franceschi S. Human papillomavirus types in invasive cervical cancer worldwide: a meta-analysis. Br J Cancer. 2003;88:63–73. doi: 10.1038/sj.bjc.6600688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel B, Mukherjee G, Seshadri L, Vallikad E, Krishna S. Changes in the physical state and expression of human papillomavirus type 16 in the progression of cervical intraepithelial neoplasia lesions analysed by PCR. J Gen Virol. 1995;76:2589–2593. doi: 10.1099/0022-1317-76-10-2589. [DOI] [PubMed] [Google Scholar]
- Demeret C, Desaintes C, Yaniv M, Thierry F. Different mechanisms contribute to the E2 mediated transcriptional repression of human papillomavirus type 18 viral oncogenes. J Virol. 1997;71:9343–9349. doi: 10.1128/jvi.71.12.9343-9349.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology. 2004;324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- Doerfler W, Remus R, Muller K, Heller H, Hohlweg U, Schubbert R. The fate of foreign DNA in mammalian cells and organisms. Dev Biol (Basel) 2001;106:89–97. [PubMed] [Google Scholar]
- Dyson N, Howley P, Munger K, Harlow E. The human papillomavirus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28:E32. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F. DNA methylation and histone modifications: teaming up to silence genes. Curr Opin Genet Dev. 2005;15:490–495. doi: 10.1016/j.gde.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Chan SY, Burk RD, Das BC, Fujinaga K, Icenogle JP, Kahn T, Kiviat N, Lancaster W, Mavromara P, Labropoulou V, Mitrani-Rosenbaum S, Norrild B, Pillai MR, Stoerker J, Syrjaenen K, Syrjaenen S, Tay SK, Villa LL, Wheeler CM, Williamson AL, Bernard HU. The genetic drift of human papillomavirus type 16 is a means of reconstructing prehistoric viral spread and movement of ancient human populations. J Virol. 1993;67:6413–6423. doi: 10.1128/jvi.67.11.6413-6423.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human papillomaviruses. Vol. 64. IARC; Lyon: 1995. International Agency for Research on Cancer Monograph on the evaluation of carcinogenic risks to humans. [Google Scholar]
- Kalantari M, Calleja-Macias IE, Tewari D, Hagmar B, Barrera-Saldana HA, Wiley DJ, Bernard HU. Conserved methylation patterns of human papillomavirus-16 DNA in asymptomatic infection and cervical neoplasia. J Virol. 2004;78:12762–12772. doi: 10.1128/JVI.78.23.12762-12772.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Garner-Hamrick PA, Fisher C, Lee D, Lambert PF. Methylation patterns of papillomavirus DNA, its influence on E2 function, and implications in viral infection. J Virol. 2003;77:12450–12459. doi: 10.1128/JVI.77.23.12450-12459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris T, Robertson B, Gallagher M. Rapid reverse transcription-PCR detection of hepatitis C virus RNA in serum by using the TaqMan fluorogenic detection system. J Clin Microbiol. 1996;34:2933–2936. doi: 10.1128/jcm.34.12.2933-2936.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz N, Bosch FX, de Sanjosé S, Herrero R, Castellsagué X, Shah KV, Snijders PJF, Meijer CJLM. Epidemiological classification of human papillomavirus types associated with cervical cancer. New Engl J Med. 2003;348:518–527. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- Myers G, Bernard HU, Delius H, Favre M, Iconogle J, van Ranst M, Wheeler C, editors. Human papillomaviruses 1994 Compendium. Los Alamos National Laboratory; Los Alamos, New Mexico, USA: pp. 1-C-4–1-A-7. [Google Scholar]
- O’Connor MJ, Stünkel W, Koh CH, Zimmermann H, Bernard HU. The differentiation-specific factor CDP/Cut represses transcription and replication of human papillomaviruses. J Virol. 2000;74:401–410. [PMC free article] [PubMed] [Google Scholar]
- Ong CK, Chan SY, Campo MS, Fujinaga K, Mavromara P, Labropoulou V, Pfister H, Tay SK, ter Meulen J, Villa LL, Bernard HU. Evolution of human papillomavirus type 18: An ancient phylogenetic root in Africa and intratype diversity reflect coevolution with human ethnic groups. J Virol. 1993;67:6424–6431. doi: 10.1128/jvi.67.11.6424-6431.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz E, Freese UK, Gissmann L, Mayer W, Roggenbuck B, Stremlau A, zur Hausen H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature. 1985;314:111–114. doi: 10.1038/314111a0. [DOI] [PubMed] [Google Scholar]
- Stanley MA, Browne HM, Appleby M, Minson AC. Properties of a non-tumorigenic human cervical keratinocyte cell line. Int J Cancer. 1989;43:672–676. doi: 10.1002/ijc.2910430422. [DOI] [PubMed] [Google Scholar]
- Stünkel W, Bernard HU. The chromatin structure of the long control region of human papillomavirus type 16 represses viral oncoprotein expression. J Virol. 1999;73:1918–1930. doi: 10.1128/jvi.73.3.1918-1930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stünkel W, Huang Z, Tan SH, O’Connor M, Bernard HU. Nuclear matrix attachment regions of human papillomavirus-16 repress or activate the E6 promoter depending on the physical state of the viral DNA. J Virol. 2000;74:2489–2501. doi: 10.1128/jvi.74.6.2489-2501.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara K, Fujinaga K, Yamashita T, Ito Y. Integration and methylation of shope papilloma virus DNA in the transplantable Vx2 and Vx7 rabbit carcinomas. Virology. 1983;131:88–99. doi: 10.1016/0042-6822(83)90536-6. [DOI] [PubMed] [Google Scholar]
- Tan SH, Leong LEC, Walker PA, Bernard HU. The human papillomavirus type 16 transcription factor E2 binds with low cooperativity to two flanking binding sites and represses the E6 promoter through displacement of Sp1 and TFIID. J Virol. 1994;68:6411–6420. doi: 10.1128/jvi.68.10.6411-6420.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turan T, Kalantari M, Calleja-Macias IE, Villa LL, Cubie HA, Cuschieri K, Skomedal H, Barrera-Saldana HA, Bernard HU. Methylation of the Human Papillomavirus-18 L1 Gene: A Biomarker of Neoplastic Progression? Virology. 2006;349:175–183. doi: 10.1016/j.virol.2005.12.033. [DOI] [PubMed] [Google Scholar]
- Van Tine BA, Kappes JC, Banerjee NS, Knops J, Lai L, Steenbergen RD, Meijer CL, Snijders PJ, Chatis P, Broker TR, Moen PT, Jr, Chow LT. Clonal selection for transcriptionally active viral oncogenes during progression to cancer. J Virol. 2004;78:11172–11186. doi: 10.1128/JVI.78.20.11172-11186.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werness B, Levine A, Howley P. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–79. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- Wettstein FO, Stevens JG. Shope papilloma virus DNA is extensively methylated in non-virus-producing neoplasms. Virology. 1983;126:493–504. doi: 10.1016/s0042-6822(83)80007-5. [DOI] [PubMed] [Google Scholar]
- Wiley DJ, Huh J, Chang C, Kalantari M, Rao JY, Goetz M, Msongsong E, Poulter M, Bernard HU. Methylation of human papillomavirus DNA in samples of HIV-1 infected men screened for anal cancer. J Acqu Immunodef Syndr. 2005;39:143–151. [PubMed] [Google Scholar]
- Yamada T, Manos MM, Peto J, Greer CE, Munoz N, Bosch FX, Wheeler CM. Human papillomavirus type 16 sequence variation in cervical cancers: a worldwide perspective. J Virol. 1997;71:2463–2472. doi: 10.1128/jvi.71.3.2463-2472.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Noya F, Chen WY, Townes TM, Chow LT, Broker T. Trichostatin A up-regulates human papillomavirus type 11 upstream regulatory region-E6 promoter activity in undifferentiated primary human keratinocytes. J Virol. 1999;73:5026–5033. doi: 10.1128/jvi.73.6.5026-5033.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]