

Abstract
GnRH binds its cognate G protein-coupled GnRH receptor (GnRHR) located on pituitary gonadotropes and drives expression of gonadotropin hormones. There are two gonadotropin hormones, comprised of a common α- and hormone-specific β-subunit, which are required for gonadal function. Recently we identified that Fanconi anemia a (Fanca), a DNA damage repair gene, is differentially expressed within the LβT2 gonadotrope cell line in response to stimulation with GnRH. FANCA is mutated in more than 60% of cases of Fanconi anemia (FA), a rare genetically heterogeneous autosomal recessive disorder characterized by bone marrow failure, endocrine tissue cancer susceptibility, and infertility. Here we show that induction of FANCA protein is mediated by the GnRHR and that the protein constitutively adopts a nucleocytoplasmic intracellular distribution pattern. Using inhibitors to block nuclear import and export and a GnRHR antagonist, we demonstrated that GnRH induces nuclear accumulation of FANCA and green fluorescent protein (GFP)-FANCA before exporting back to the cytoplasm using the nuclear export receptor CRM1. Using FANCA point mutations that locate GFP-FANCA to the cytoplasm (H1110P) or functionally uncouple GFP-FANCA (Q1128E) from the wild-type nucleocytoplasmic distribution pattern, we demonstrated that wild-type FANCA was required for GnRH-induced activation of gonadotrope cell markers. Cotransfection of H1110P and Q1128E blocked GnRH activation of the αGsu and GnRHR but not the β-subunit gene promoters. We conclude that nucleocytoplasmic shuttling of FANCA is required for GnRH transduction of the αGSU and GnRHR gene promoters and propose that FANCA functions as a GnRH-induced signal transducer.
BINDING OF GnRH to the GnRH receptor (GnRHR) stimulates the biosynthesis of the gonadotropin common α-, glycoprotein hormone α-subunit (αGSU), and β-subunits. To form αβ-gonadotropin hormone heterodimers, biosynthesis of αGSU must exceed that of the LH and FSH β-subunits. Thus, there are complex regulatory mechanisms that ensure differential transcriptional regulation of the subunit gene promoters, which rely on variation in GnRH pulsatility, GnRHR number, and the activation of key intracellular second messenger signaling pathways (1-3). However, further levels of differential control must exist because there is considerable overlap in the GnRH-activated MAPK second-messenger signaling pathways that regulate each subunit gene promoter (4-7). Furthermore, although GnRHR and FSH β-subunit are regulated by activin A, the time frame and mechanism for each appears to be different (8, 9), so there does appear to be some targeted selectivity in activation of GnRHR gene expression. This selective signal transduction could be provided by cell compartmentalization of signal transduction components, which when linked to the conformational state of GnRHR can activate disparate pathways (10) and by using scaffold proteins to anchor and sequester proteins that activate specific target promoters. Indeed, we have shown that the scaffold protein proline-rich protein tyrosine kinase-2 transduces a focal adhesion complex/ERK signal to the LH β-subunit gene promoter (submitted). Thus, how GnRH disparately activates intracellular signaling pathways to transduce specific transcriptional activation of αGSU, LH, and FSH β-subunit and GnRHR is likely to be a key event among the biomolecular processes required for differential gene activation.
We previously identified that a DNA damage repair gene, Fanconi anemia a (Fanca), was acutely regulated and differentially expressed in response to GnRH within gonadotropes (11). FANCA is mutated in more than 60% of cases of Fanconi anemia (FA; OMIM 227650), a genetically heterogeneous, rare autosomal recessive disorder characterized by progressive bone marrow failure, endocrine tissue cancer susceptibility, and infertility (12). FANCA is functionally conserved across mammalian species because murine FANCA can correct sensitivity to the DNA cross-linker mitomycin C in lymphoblastoid cells derived from FANCA patients (13). It is a relatively large protein (∼160 kDa) with an N-terminal nuclear localization signal (NLS) required for interactions with other FA proteins (14-16). Nuclear localization of FANCA promotes nuclear accumulation of the FA protein complex (17, 18). FA proteins are encoded by several distinct genes [FANCA, FANCB, FANCC, FANCD1(BRCA2), FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, and FANCM] and FANCA forms a nuclear core complex with FANCB, -C, -E, -F, -G, -L, and -M to repair interstrand DNA cross-links (for review see Ref.19). The nuclear core complex is essential for monoubiquitination of a further protein named FANCD2, a key event in the FA DNA-damage response pathway, but it has been reported that FANCA can biochemically associate with non-FA and FA proteins in different cellular compartments (20, 21).
Thus, although FANCA has an established role in DNA repair, a number of questions still remain unresolved. The genetic heterogeneity observed in FANCA patients could not be recapitulated in Fanca−/− gene-targeted mice, although the mice were infertile and had limited anemia (22, 23). Further targeted disruption of the first six exons of Fanca did expand this phenotype because growth, cranial and eye development, and maintenance of primordial germ cells were all affected (24), but a disparity still remains. Detailed study of FANCA mutant proteins does not match the location or type of defect with the phenotype (25). For instance, deletion of exon 43 causes severe but variable pathogenicity (26), whereas other analyses have attempted to subclassify FANCA point mutations according to their severity (27). The data suggest that FANCA may be required for a number of disparate molecular processes, so further work is clearly required to identify what additional role the molecule assumes.
Here we report that GnRH induces a rapid, transient nuclear localization of functional FANCA protein, which is required for GnRH-induced activation of the αGSU and GnRHR gene promoters but not the LH or FSH β-subunit gene promoters.
Materials and Methods
Cell culture and reagents
LβT2 cells (obtained from P. Mellon, University of California, San Diego, CA) and HeLa cells (European Collection of Cell Cultures, Porton Down, UK) were cultured as described (11). GnRH (Peninsula, St. Helens, UK) was used at the concentrations stated in the figure legends, and GnRH antagonist Cetrorelix (supplied by Robert Millar, Medical Research Council, Human Reproductive Sciences Unit, Edinburgh, Scotland, UK) was added before GnRH at a concentration of 1 μm/ml. Recombinant activin A was used at 25 ng/ml and obtained from R&D Systems (Abingdon, Oxon, UK). The chemical inhibitors cycloheximide (CHX) and actinomycin D (AMD) were used at 15 μg/ml and 5 μg/ml, respectively, and purchased from Calbiochem (Nottingham, UK). Leptomycin B (LMB; Sigma, Dorset, UK) was added at a final concentration of 2 ng/ml.
Immunofluorescence and confocal microscopy
Cells were plated on glass chamber slides (Nunc, Hereford, UK) and left untreated or treated with 1 μm GnRH for 2 h, fixed in methanol (BDH, Poole, UK), and then permeabilized. After 30 min incubation with Avidin, followed by biotin (Vector Laboratories, CA), cells were incubated overnight at 4 C with Fanca antisera (1:200 dilution, supplied by Fre Arwert, University of Amsterdam, Amsterdam, The Netherlands). The next day, biotin-conjugated goat-antirabbit secondary antibody (1:500 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) was added for 30 min, and excess removed before incubating 1 h, in the dark, with streptavidin coupled Alexa 488 fluorescent probe (Cambridge Biosciences, Cambridge, UK). Topro-3 (1:2000 dilution in PBS, Cambridge Biosciences) counterstain identified the nucleus before addition of Permafluor mounting medium (Immunotech, High Wycombe, UK). Analysis of indirect immunofluorescence was performed using a Axiovert 100-m confocal microscope and LSM510 scanning module with an oil immersion ×40 objective (Zeiss, Welwyn Garden City, UK). To visualize fluorescence, excitation was performed using the Argon 488 nm (fluorescein isothiocyanate/green) and HeNe 633-nm lasers (Cy5/blue). Single scans and z-stack analysis, 12-16 optical slices of 0.2-μm increments through the cell, were performed at 1024 × 1024 pixels resolution.
Western blotting analysis
Whole-cell protein extracts were prepared and Western blotting analysis was performed as previously described (11). Cytoplasmic and nuclear extracts were isolated as described (28). The integrity of these extracts was confirmed by stripping off anti-FANCA in 2% sodium dodecyl sulfate, 50 mm Tris (pH 6.8), 0.7% β-mercaptoethanol at 55 C for 30 min before washing and reprobing with either antimouse β-tubulin (Santa Cruz) or antirat Sp1 antisera (Autogen Bioclear, Wiltshire, UK) at a dilution of 1:500. green fluorescent protein (GFP)-tagged proteins were detected using goat anti-Aequorea victoria GFP (Abcam, Cambridge, UK) at a dilution of 1:1000.
Generation of FANCA mutant constructs
Mutagenesis was performed using the QuikChange XL site directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer’s instructions. The enhanced GFP (EGFP)-FANCA plasmid was a kind gift from Manuel Buchwald (The Hospital for Sick Children, Toronto, Canada). Mutagenesis was performed using synthetic oligonucleotide primers containing patient-derived mutations (see Table 1) and was confirmed by DNA sequencing.
TABLE 1.
Primer sequences
| Primer | Mutation | DNA sequence (5′-3′) | Position (bp) |
|---|---|---|---|
| Sense | H1110P | GCGAGCAGTTCTTCCCCTTGGTCAACTCTG | 3313-3343 |
| Antisense | H1110P | CAGAGTTGACCAAGGGGAAGAACTGCTCGC | 3343-3313 |
| Sense | Q1128E | GAGGTGCCCTGACAGAGGACATGACTGCCC | 3367-3397 |
| Antisense | Q1128E | GGGCAGTGATGTCCTCTGTCAGGGCACCTC | 3397-3367 |
Transient transfection and visualization of EGFP expression in cultured cells
LβT2 or HeLa cells were plated on two-well glass slides (Nunc) at a density of 100,000 cells/ml; 16 h later cells were transfected with 1.5 μg plasmid DNA using Fugene 6 transfection reagent (Roche, Lewes, UK) and incubated for a further 48 h. Before visualizing EGFP expression, cells were fixed with methanol (BDH) for 10 min at −20 C, washed with PBS, and nuclei stained by incubating for 2 min, in the dark, with Topro-3 (1:2000 dilution in PBS; Cambridge Biosciences) before washing in PBS and mounting coverslips using Permafluor (Immunotech, Beckman Coulter, Fullerton, CA). Fluorescence cells were detected using a Zeiss Axiovert 100-m microscope as described previously.
Transient transfection of constructs for reporter gene assays
Reporter constructs were generated by fusing −692 sheep LH β-subunit gene promoter upstream of a luciferase reporter gene (LHβ-Luc) or were obtained as promoter-luciferase fusion constructs; −480 mouse α-subunit gene (D. Gordon, University of Colorado, Denver, CO), −1135/−26 rat GnRH receptor (J.-N. Laverriere, Université Pierre et Marie Curie, Paris, France) and −4741/+759 sheep FSH β-subunit (W. Miller, North Carolina State University, Raleigh, NC). LβT2 cells were seeded into six- and 12-well plates and incubated overnight at 37 C before transfecting with a total DNA amount of 2 μg (12 well) or 7.5 μg (six well) with GFP-FANCA-WT or GFP-FANCA-H1110P or −Q1124E plasmids and Fugene 6 (Roche Diagnostics). In six-well plates, 250 ng CMV-β-galactosidase expression vector was included as a measure of transfection efficiency, and in 12-well plates, either 100 or 33 ng of renilla vector (Promega Biosciences, Southampton, UK) were added. LHβ-Luc and αGSU-Luc transfections were treated with 100 nm of GnRH for 6 h or 1 nm GnRH for 8 h, respectively, and then harvested and assayed as described (28) or assayed using a dual-light luciferase assay kit (Promega) in a FLUOStar Optima luminometer (BMG Lab Technologies, Aylesbury, UK). Luciferase activity was normalized for either renilla or CMV-β-galactosidase activity to correct for transfection efficiency. For FSHβ-Luc transfections, we used the procedure described elsewhere (4). For GnRHR. cells were incubated in DMEM with 2% fetal calf serum with no antibiotics before transfecting 7.5 μg of control pGL3 (Promega) or GnRHR-Luc. After transfection cells were incubated for the indicated times followed by a further 4-h exposure to 100 nm GnRH before harvesting. FSHβ-Luc and GnRHR-Luc reporter gene activity was assayed as described (28), and luciferase levels were adjusted for transfection efficiency by normalizing for CMV-β-galactosidase activity. Statistical significance was established as P < 0.05 by one-way ANOVA analysis.
Results
FANCA protein exhibits a nucleocytoplasmic localization pattern in LβT2 cells
Previously we had determined that a 2-h time frame was optimal for GnRH induction of FANCA protein (11). This analysis was extended to include various GnRH concentrations ranging from 0.1 nm to 1 μm. We found that there was a lower but no upper limit in the range tested and that a threshold GnRH concentration of 1 nm was required to induce a 2-fold increase in FANCA protein expression after 2 h (data not shown). This was dependent on GnRH activation of the GnRHR because a 30-min preincubation with a GnRHR antagonist, Cetrorelix, blocked induction of FANCA protein (Fig. 1A). We used these optimized conditions to investigate the localization pattern of endogenous FANCA protein in untreated and GnRH-treated LβT2 gonadotrope cells (Fig. 1B). Comparing the localization pattern of FANCA immunofluorescence with nuclear Topro-3 staining suggested that FANCA appeared to be distributed throughout the cytoplasm and nucleus in both untreated and GnRH-treated cells (Fig. 1B). This was confirmed by confocal microscope z-stack sectioning through the middle of a representative cell (Fig. 1B; C and G, nontreated; F and H, treated). Comparison of the levels of FANCA protein expression in untreated and GnRH-treated cells suggested that there was an increase in fluorescence intensity, i.e. expression, after addition of hormone. Western blotting analysis of subcellular fractions was used to confirm this distribution and increase of FANCA protein across both cytoplasmic and nuclear cellular compartments (Fig. 1C). Further analysis with cytoplasmic and nuclear markers indicated that there was little cross-contamination of the cytoplasmic and nuclear protein fractions and that FANCA protein levels increased 2-fold in both the cytoplasmic and nuclear protein fraction after a 2-h treatment with hormone (Fig. 1C). Thus, we concluded that although FANCA carries a functional NLS, capable of targeting it to the nucleus, the endogenous protein is actually distributed across the cytoplasmic and nuclear compartments in gonadotrope cells and that GnRH increases levels of FANCA in both.
FIG. 1.
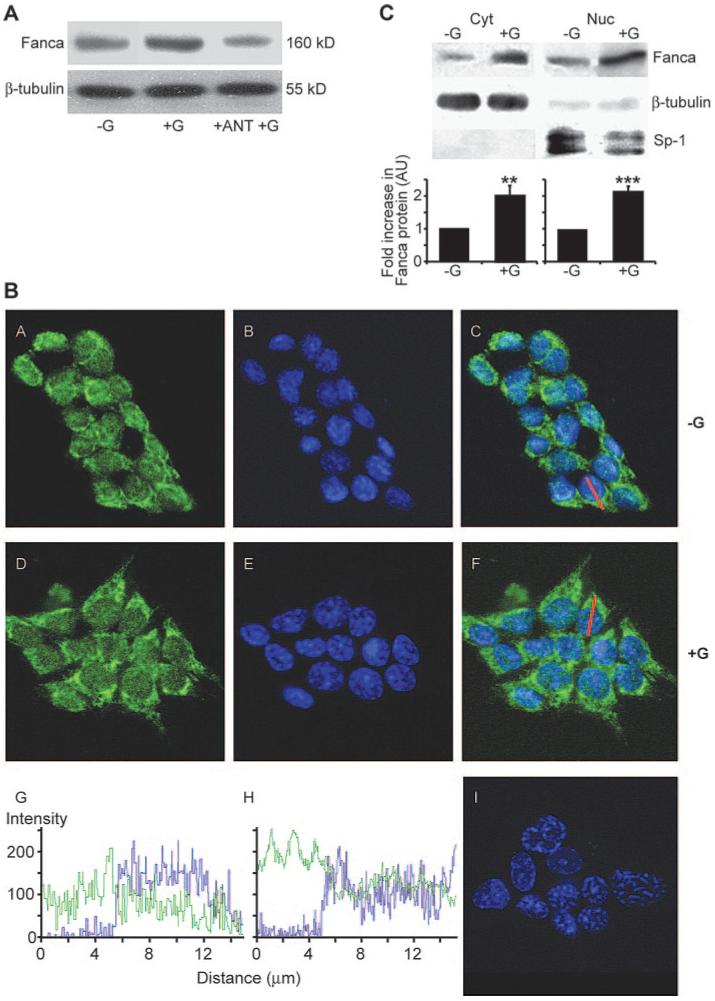
Endogenous FANCA is located across the nucleocytoplasmic compartment in LβT2 cells. A, LβT2 cells were pretreated with GnRHR antagonist 1 μm Cetrorelix (ANT) before treatment for 2 h with 1 μm GnRH. Western blotting analysis of wholecell extracts identified that addition of ANT blocked the GnRH-induced increase in FANCA. A representative gel, of two performed, is shown. B, Confocal microscopy analysis of untreated (−G) and GnRH-treated (+G; 1 μm for 2 h) LβT2 cells. Anti-FANCA antibody immunostaining, followed by biotin-conjugated goat antirabbit is shown in green in panels A and D, nuclei were stained blue with 1:2000 Topro-3 (panels B and E). FANCA immunostaining merged with Topro-3 is shown in panels C and F. Panel I shows the pattern if anti-FANCA antibody is omitted. A cross-sectional line was drawn through a representative cell in panels C and F. The intensity of the pixels that fall on the line were plotted against distance (micrometers) and are shown for each channel, FANCA in green and Topro-3 in blue, in panels G(−G) and H (+G). Zero equals no signal, whereas 256 is the maximum detectable level. C, Detection of FANCA in nuclear (Nuc) and cytoplasmic (Cyt) compartments using Western blotting analysis. Cells were either left untreated (−G) or treated (+G) with 1 μm GnRH for 2 h. Seventy-five micrograms of Nuc or Cyt protein extract were immunoblotted for FANCA and then reprobed for nuclear marker, Sp1, and cytoplasmic marker, β-tubulin. A representative blot is shown; the graph represents the average ± sem increase in FANCA calculated from three separate experiments. Protein levels were quantified using a Storm PhosphoImager and untreated extracts were assigned a value of 1. One-way ANOVA established statistical significance of GnRH treated vs. untreated as **, P < 0.01 and ***, P < 0.001.
GnRH-induced, CRM1-dependent nucleocytoplasmic shuttling of FANCA in LβT2 cells
The GnRH-induced increase in FANCA within the cytoplasmic and nuclear compartments of LβT2 cells could be explained by reduced protein turnover or an increase in de novo translation followed by intracellular redistribution. To investigate these possibilities further, LβT2 cells were treated with a translational inhibitor, CHX, 30 min before addition of GnRH, followed by Western blotting analysis of the isolated cytoplasmic extract (Fig. 2A). Addition of CHX did not affect basal levels; they were still significantly different from GnRH stimulated, but addition of CHX and GnRH had a varying effect on cytoplasmic FANCA levels. They were not significantly different from basal (untreated or CHX treated) or GnRH treated.
FIG. 2.
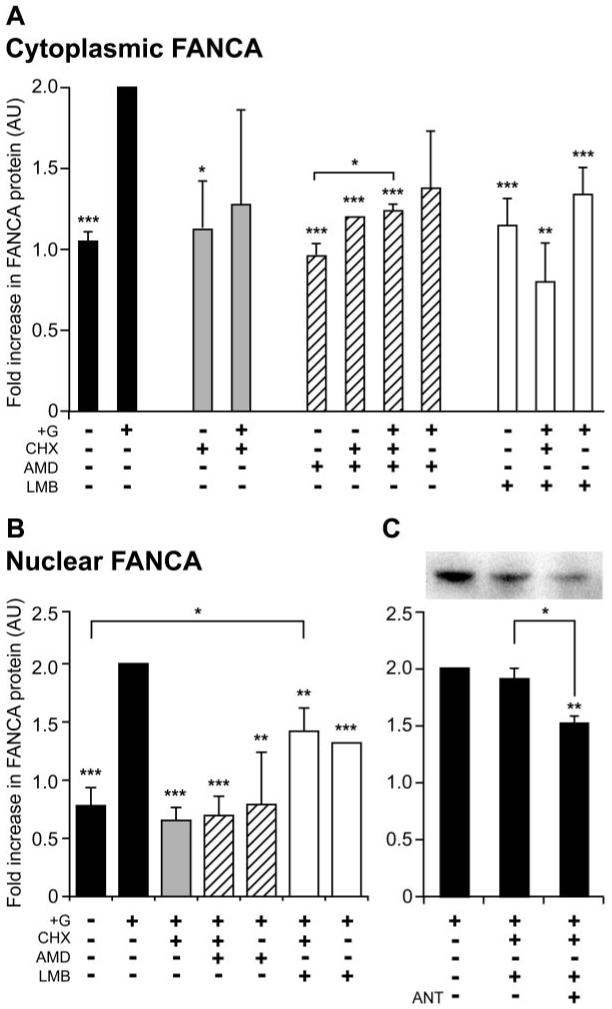
FANCA undergoes nucleocytoplasmic shuttling in LβT2 cells. Cytoplasmic (A) and nuclear (B) location of FANCA after treatment with trafficking inhibitors and hormone. The following were added to LβT2 cells: no GnRH (−); GnRH was added at 1 μm for 2 h (+G); cells had a 30-min pretreatment of 15 μg/ml CHX, 15 μg/ml AMD, and 2 ng/ml LMB. Cytoplasmic and nuclear extracts were isolated from the same experiment, Western blotted, and probed with anti-FANCA antibody. C, Effect of addition of GnRHR inhibitor Cetrorelix on nuclear accumulation of FANCA. Cells were either left untreated or pretreated with 1 μm Cetrorelix (ANT) for 30 min before adding CHX and LMB for 30 min and 1 μm GnRH for 2 h. Seventy-five micrograms of nuclear extract were analyzed for levels of FANCA and a representative Western blot is shown. The average ± sem change in FANCA levels from three separate experiments is shown graphically, with +G assigned a value of 2. One-way ANOVA identified that ***, P < 0.001, **, P < 0.01, and *, P < 0.05 were significantly different from +G, unless indicated otherwise. AU, Arbitrary units.
This variation may be due to GnRH redistributing the nucleocytoplasmic location of existing FANCA. To block this, we added inhibitors of nuclear import, AMD, and nuclear export, LMB (Fig. 2A). AMD acts as a broad range blocker of nuclear import by inhibiting RNA polymerase II transcription, which inhibits the nucleocytoplasmic shuttling of pre-mRNA proteins (hnRNP) bound to mRNA (29), whereas LMB blocks a specific nuclear export pathway by covalently modifying Cys-539 within the docking site of nuclear export receptor CRM1 (30). Addition of either AMD or LMB in the absence of hormone had no effect on cytoplasmic levels of FANCA. In comparison, addition of CHX with either AMD or LMB combined with hormone significantly blocked the GnRH-induced increase (Fig. 2A), but LMB reduced FANCA levels more than AMD. Dual treatment with AMD and GnRH did not appear to effect cytoplasmic levels of FANCA, whereas treatment with LMB and GnRH significantly reduced cytoplasmic FANCA. This suggested that there was movement of FANCA into the nucleus, so measuring levels of FANCA in nuclear extracts treated with a similar pattern of inhibitors should confirm this. Nuclear levels of FANCA have a converse pattern from cytoplasmic levels (Fig. 2B). Taken together this analysis identified that addition of CHX, GnRH, and AMD or GnRH and AMD treatment may have promoted cytoplasmic accumulation (Fig 2A) because nuclear extracts were depleted of FANCA (Fig 2B). CHX, GnRH and LMB or GnRH and LMB both reduced cytoplasmic levels (Fig 2A) but caused an increase in nuclear levels (Fig 2B). This suggested that nucleocytoplasmic shuttling of FANCA did occur and we hypothesized that it may be dependent on GnRH stimulation. Cells were treated with CHX and LMB, as before, but were also pretreated with Cetrorelix. Addition of antagonist significantly blocked nuclear accumulation of FANCA (Fig. 2C). This result demonstrated that CRM1 was required for nuclear export and suggested that GnRH induced nuclear import of FANCA.
We then investigated what happened to the distribution of a GFP-tagged FANCA protein after hormone treatment to establish whether GnRH effected a change in nucleocytoplasmic distribution of FANCA. A control GFP- or GFP-FANCA expressing construct was transfected into LβT2 cells, which were treated for various time intervals with GnRH, fixed, and then the distribution patterns of untagged or tagged GFP were visualized and compared. Comparison of untreated cells with 5 min of GnRH treatment showed that GFP-FANCA redistributed to become mainly confined within the nucleus (Fig. 3, compare B, C, and D, untreated, with F, G, and H, treated). After 10 min of GnRH treatment, GFP-FANCA reappeared in the cytoplasm (Fig. 3, J, K, and L), and by 15 min, cytoplasmic accumulation had progressed even further (Fig. 3, M, N, and O). We concluded that FANCA protein responds to a GnRH-induced translocation signal, because control GFP did not redistribute in response to hormone (Fig. 3, A, E and I), and the shuttling profile of FANCA was blocked by 30 min pretreatment with Cetrorelix (Fig. 3, P-U). Taken together, these data indicate that the nuclear accumulation of FANCA within gonadotrope cells is a dynamic, rapid, GnRH-driven event, and recycling of nuclear protein is dependent on nuclear export receptor CRM1.
FIG. 3.
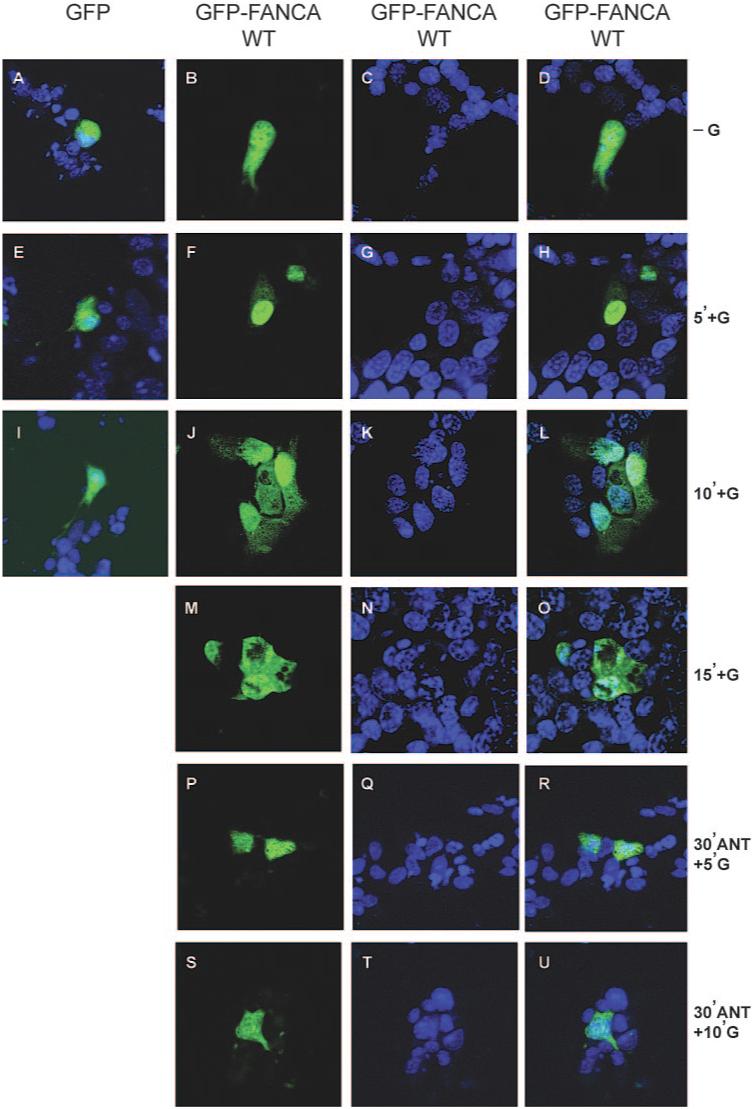
GnRH induces rapid nuclear accumulation of GFP-FANCAWT. Visualization of GFP-tagged FANCA after GnRH treatment of transfected cells are shown. Cells were either transfected with GFP-empty vector or GFP-tagged FANCA and then left untreated or treated with 100 nm GnRH for the indicated time periods. Cells were fixed before staining nuclei with Torpro-3. A, E, and I, Merged GFP-empty vector and Torpro-3-stained nuclei −G (A) and +G (E and I). B-D, −G GFP-FANCAWT (B), Torpro-3 staining (C), and merged images are shown in the right-hand panel (D). F-H and J-O after various time intervals +G, the localization of GFP-FANCAWT was compared with the Torpro-3 nuclear staining pattern (G, K, and N). Images are shown merged (H, L, and O). P-U, Localization of GFP-FANCAWT after 30 min pretreatment with 1 μm of GnRHR antagonist Cetrorelix (ANT) and then +G. Overlay of GFP-FANCAWT images are shown in the right-hand panels, and numbers refer to length of exposure +G in minutes. These are representative examples of images taken from at least three different, separate experiments.
Analysis of the localization pattern of GFP-tagged mutant proteins in HeLa and LβT2 cells
A number of FANCA mutant proteins have been described with aberrant nucleocytoplasmic distribution profiles that interact poorly with other FA proteins and have varying phenotypic penetrance (25, 27). We chose two of these mutants for further study; the first was a severe mutant with an amino acid conversion of histidine to proline at position 1110 (H1110P) (14, 27), and the second had replacement of a glutamine with glutamic acid at amino acid 1128 (Q1128E) and has been biochemically classified as a mild mutation (27). First, we used site-directed mutagenesis to engineer the point mutations into a GFP-tagged FANCA clone, creating GFP-FANCAH1110P and GFP-FANCAQ1128E. We checked that the wild-type (WT) and mutated proteins were expressed as GFP-FANCA fusion proteins by transiently transfecting LβT2 cells for 48 h followed by Western blotting analysis of whole-cell protein extracts (Fig. 4A). A protein band corresponding to the expected size of GFP-FANCA, approximately 187 kDa, was detected only in extracts from transfected cells; no band was visible in mock-transfected cells. Figure 4B shows a schematic presentation of FANCA with the location of these point mutations and their biochemical properties.
FIG. 4.
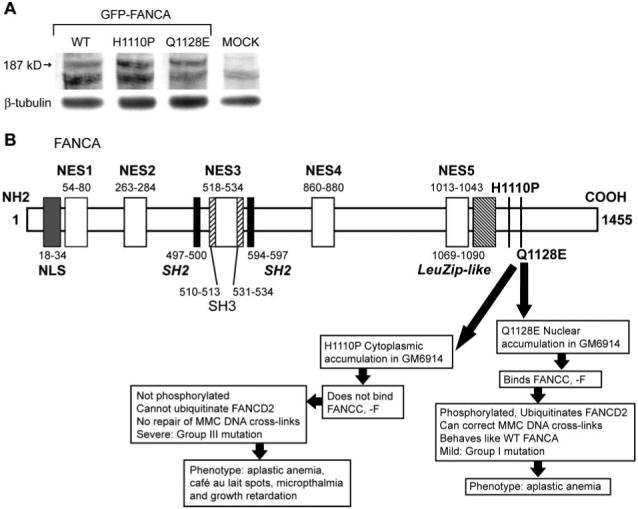
Expression analysis and schematic of FANCA point mutations. A, Western blotting analysis of protein extracts made from untransfected (MOCK) and cells transfected with GFP-tagged WT and mutant (H1110P and Q1128E) proteins identified a 187-kDa GFP-FANCA fusion protein. Blots were stripped and reprobed with β-tubulin to control for protein loading. B, Schematic representation of WT and the two mutant forms of FANCA. WT FANCA is 1455 amino acids with a NLS at the NH2-terminus (15) and five nuclear export signals (NES) (36). Putative domains include Src-homology domains (SH2, SH3) and a leucine zipper-like motif (LeuZip-like) and are shown in italics (13). The biochemical properties and phenotype of the point mutations H1110P and Q1128E are also summarized (14, 25, 27). The numbers orientate the domains on the amino acid backbone. MMC refers to ability of FANCA to repair DNA cross-links generated by mitomycin C.
Transient transfection of the tagged constructs into HeLa and LβT2 cells followed by visualization with a confocal microscope identified that WT and mutant GFP-tagged FANCA proteins localized to different cellular compartments (Fig. 5, A and B). GFP-FANCA was located in the cytoplasm and nucleus of both cell types (Fig. 5, A and B, panels B, F, and J). Confocal z-stack sectioning through a representative cell confirmed that the expression pattern was predominantly nuclear (Fig. 5, A and B, panels B and N). The GFP-FANCAH1110P mutant was restricted to the cytoplasm in both HeLa and LβT2 cells (Fig. 5, A and B, panels C, G, K, and O), confirming the aberrant nucleocytoplasmic localization pattern of this mutation (27). In contrast, GFP-FANCAQ1128E predominantly located to the nucleus of HeLa cells with very little cytoplasmic staining (Fig. 5A, panels D, H, L, and P), but in LβT2 cells, the distribution pattern resembled empty vector GFP (Fig. 5B, panels A, D, E, H, I, L, M, and P) rather than the biased nuclear localization pattern seen for GFP-FANCA.
FIG. 5.
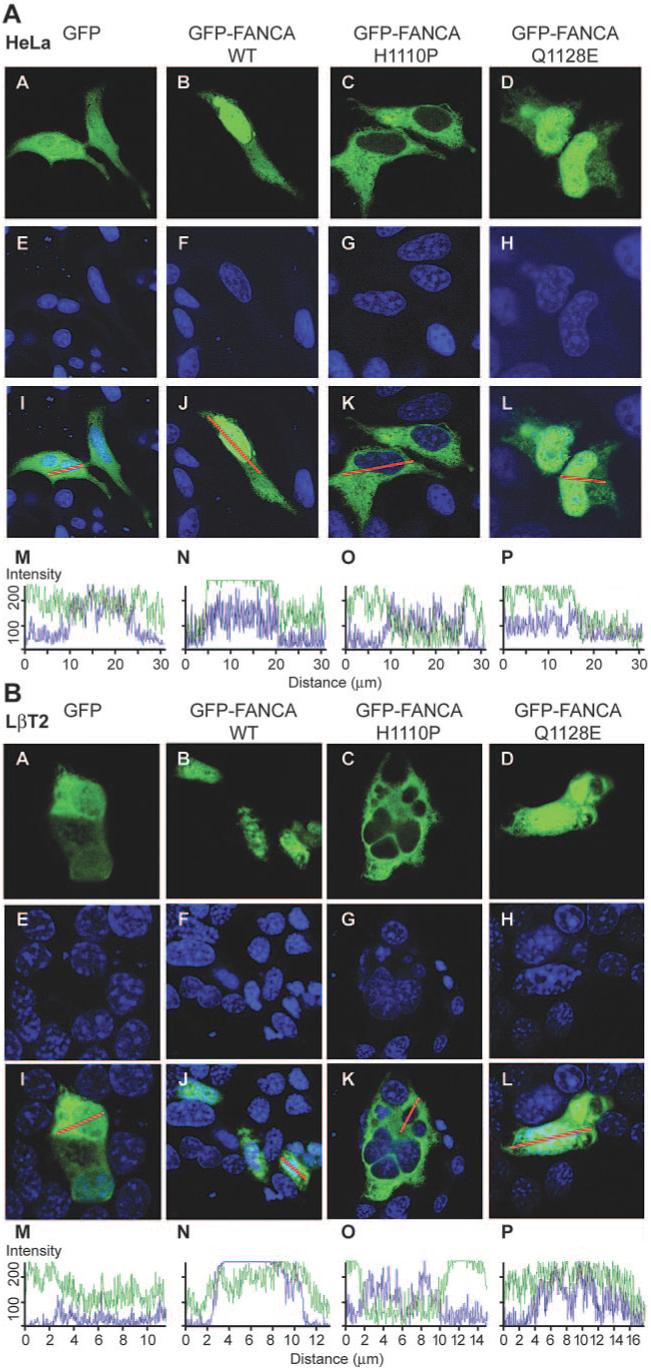
Effect of amino acid substitutions on the nucleocytoplasmic localization pattern of GFP-FANCA. HeLa (A) and LβT2 (B) cells were transfected with GFP-empty vector (A), GFP-FANCAWT (B), GFP-FANCAH1110P (C), or GFP-FANCAQ1128E (D) and then examined using confocal microscopy. Torpro-3 staining was used to visualize the nuclei of fixed cells (E-H), and the merged images show an overlay of the two (I-L). A representative merged cell was chosen (I-L), transected, and the intracellular location of GFP (green) and Torpro-3 (blue) fluorescent intensities shown graphically (M-P). The intensity ranges from zero to a maximum of 256 and measures how many pixels hit the line for each individual channel.
We next determined whether the point-mutated fusion proteins could redistribute within the nucleocytoplasmic compartment in response to GnRH. LβT2 cells were transfected with GFP, GFP-FANCA, GFP-FANCAH1110P, or GFP-FANCAQ1128E and treated with GnRH (Fig. 6). Addition of GnRH had no effect on the location of either GFP-FANCAH1110P or GFP-FANCAQ1128E (Fig. 6, panels G, H, K, L, O, and P) or on GFP (Fig. 6, panels E, I, and M). There was no difference in the cross-sectional analysis of GnRH treated (Fig. 6, panels S and T) with untreated (Fig. 5B, panels O and P). In contrast, GnRH-induced nuclear accumulation of GFP-FANCA (Fig. 6, panels F, J, and N), and this was blocked by a 30-min pretreatment with antagonist, confirming previous results. These differences in localization of mutant FANCA indicate that the molecule may interact differently with the trafficking apparatus in disparate cell types and demonstrates that the point mutations block hormone-induced trafficking.
FIG. 6.
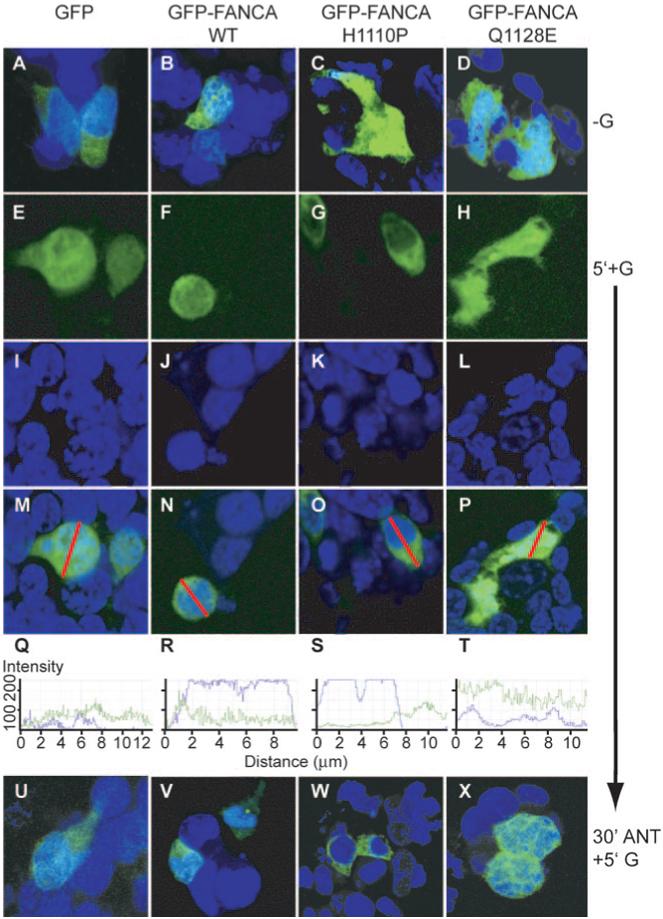
Point mutations block GnRH-induced nucleocytoplasmic shuttling of FANCA. Cells were transfected with either GFP-empty vector (GFP) or GFP-FANCA (WT) or the point-mutated GFP-FANCA mutants (H1110P or Q1128E) and then left untreated or treated with 100 nm GnRH for 5 min. Cells were fixed before staining nuclei with Torpro-3. A-D, Merged untreated (−G). GFP-(E-H) and Torpro-3-stained nuclei (I-L) are merged (M-P) and demonstrate the differential locations of untagged and tagged GFP after addition of GnRH for 5 min (5′+G). A line was drawn through the merged cell (M-P), and the intensity of the fluorescence, ranging from zero to a maximum of 256, was measured by calculating how many pixels hit the line for each individual channel, GFP in green, Topro-3 in blue, and are shown graphically in Q-T. U-W, Localization of untagged and GFP-tagged proteins after 30 min pretreatment with 1 μm of GnRHR antagonist Cetrorelix (30′ANT), then 5′+G. These are representative examples of images taken from at least three separate experiments.
FANCA has differential effects on basal and GnRH-induced LHβ and FSHβ promoter activity
Given that we had identified a cell type-specific difference in nucleocytoplasmic localization pattern of mutant FANCA proteins and that GnRH induced a redistribution of both endogenous and exogenously introduced protein, we hypothesized that FANCA may be important for gonadotrope cell function, possibly influencing GnRH-driven biosynthesis of gonadotropin hormones. Therefore, we tested what effect expression of GFP-FANCA and mutant proteins had on GnRH induction of promoter activity. First, we determined the optimal concentration and time frame required for GnRH induction of gene expression of the ovine LH β-subunit gene promoter (−692LHβ-Luc) fused to a luciferase reporter (Fig. 7A). Maximal induction of −692LHβ-Luc was observed after treatment with either 100 nm or 1 μm GnRH for 6 h (Fig. 7A). Cotransfection of GFP-FANCA or mutant constructs with −692LHβ-Luc followed by GnRH treatment identified that GnRH-induced luciferase expression and that a slight nonsignificant decrease occurred when WT and mutant GFP-FANCA constructs were cotransfected (Fig. 7B). Cotransfection of GFP-empty vector did not have any effect on the promoter (data not shown), suggesting that the differences were due to FANCA. The ovine −4741-bp FSH β-subunit gene promoter requires stimulation with 25 ng/ml activin A and 100 nm GnRH for 8 h to induce maximal levels of luciferase (4), so we cotransfected −4741FSHβ-Luc with or without GFP-FANCA and treated with 25 ng/ml activin A and 100 nm GnRH for 8 h (Fig. 7C). This had no effect on basal or GnRH-induced activity of the promoter. However, cotransfection of GFP-FANCAH1110P or GFP-FANCAQ1128E resulted in a small but significant increase in the basal activity of the promoter, which decreased the fold increase in promoter activity (Fig. 7C). We concluded that FANCA did not appear to be directly involved in basal or GnRH-induced LH β-subunit gene transcription but did have a small stimulating effect on basal FSH β-subunit gene transcription that dampened the GnRH-induced increase in luciferase activity.
FIG. 7.
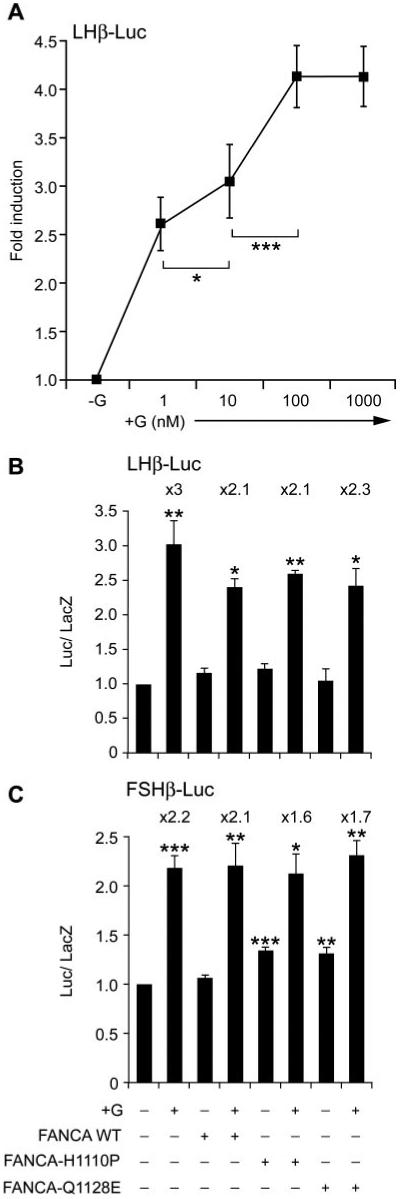
FANCA has minimal effects on GnRH-induced activity of the β-subunit gene promoters. A, Optimal concentration of GnRH required for stimulation of the −692LHβ-Luc gene promoter was determined. LβT2 cells were transfected with −692LHβ-Luc 48 h before stimulating with increasing concentrations of GnRH (nm) for 6 h and then harvested and luciferase levels measured. B, Effect of FANCA on −692LHβ-Luc. Expression plasmids containing either GFP-FANCA WT or −H1110P or Q1128E were cotransfected with −692LHβ-Luc at a ratio of reporter to effector of 6:1; 48 h later they were stimulated with 100 nm GnRH for 6 h and then assayed for luciferase. C, Effect of FANCA on FSHβ-Luc. Either GFP-FANCA WT or −H1110P or −Q1128E were cotransfected with −4741FSHβ-Luc at a ratio of reporter to effector of 6:1; 40 h later they were stimulated with 100 nm GnRH and 25 ng/ml activin A for 8 h and then assayed for luciferase. A-C, Fold stimulation above basal is indicated, corrected against the levels of β-galactosidase, which was used as an internal control, and each experiment was done in triplicate and repeated at least twice. Results shown are average ± sem. One-way ANOVA established that for A ***, P < 0.001 +G vs. −G (not shown), 100 vs. 10 nm, and *, P < 0.05 10 vs. 1 nm. B and C, ***, P < 0.001, **, P < 0.01, and *, P < 0.05 when comparing +G vs. −G and when comparing −G cotransfected FANCA WT vs. −G cotransfected FANCA-H1110P and −Q1128E.
FANCA is required for GnRH induction of the αGSU and GnRHR gene promoters
The small differential effect of cotransfecting FANCA with the gonadotropin β-subunits hinted that perhaps a separate regulatory pathway was being perturbed. GnRH transcriptionally up-regulates its own receptor (31), and regulation of the levels of GnRHR ultimately drives the differential regulation of αGSU and β-subunits (32). Thus, we decided to examine the effect of FANCA on αGSU and the GnRHR gene promoter. First the concentration and duration of GnRH treatment required to induce maximal levels of αGSU gene promoter activity was determined. LβT2 cells were transiently transfected with a −480-bp murine αGSU gene promoter and treated with increasing concentrations of GnRH for various time periods (Fig. 8A). Addition of 100-1000 nm GnRH for 6-8 h induced a reproducible doubling of gene expression, but maximal levels of luciferase were induced by treatment with 1-10 nm GnRH for 8-10 h. Indeed, increasing the dose of GnRH actually appeared to down-regulate αGSU promoter activity over time. Having identified the time frame and concentration of GnRH crucial for induction of promoter expression, we tested whether FANCA had any effect on basal or GnRH induction of αGSU gene transcription (Fig. 8B). Cotransfection with GFP-FANCA had no effect on the GnRH-induced activity of this promoter, but cotransfection with either GFP-FANCAH1110P or GFP-FANCAQ1128E did significantly reduce GnRH-induced activity after addition of 1 nm GnRH (Fig. 8B). Cotransfection of mutated FANCA constructs blocked the levels of GnRH induction of αGSU-Luc by 50% after treatment with 1 nm GnRH.
FIG. 8.
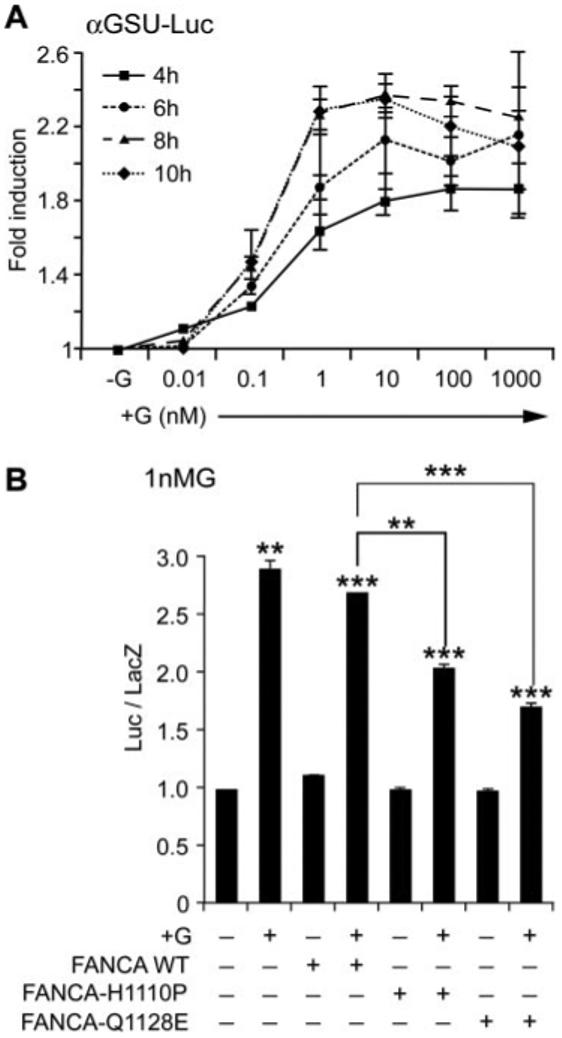
FANCA suppresses GnRH-induced activity of the αGSU gene promoter. A, Optimal time course and concentration of GnRH required for stimulation of the −480αGSU-Luc gene promoter was determined. LβT2 cells were transfected with −480αGSU-Luc 48 h before stimulating with increasing concentrations of GnRH and then harvested 4, 6, 8, and 10 h later and luciferase levels measured. B, Effect of FANCA on −480αGSU-Luc after stimulation with 1 nm GnRH. Expression plasmids containing either GFP-FANCA WT or −H1110P or Q1128E were cotransfected with −480αGSU-Luc at a ratio of reporter to effector of 6:1; 48 h later they were stimulated with 1 nm GnRH for 8 h and then assayed for luciferase. A and B, Fold stimulation above basal is indicated and corrected against the levels of renilla (A) or β-galactosidase (B), which were used as internal controls, and each experiment was done in triplicate and repeated at least twice; results shown are average ± sem. One-way ANOVA established that ***, P < 0.001, **, P < 0.01, and *, P < 0.05 when comparing treated vs. −G and when comparing +G cotransfected FANCA WT vs. +G cotransfected FANCA-H1110P and −Q1128E.
Given the small but measurable effect on the β-subunit promoters, we investigated whether FANCA had any effect on GnRHR gene expression. As already established for the other subunits, we optimized the transfection regime for the GnRHR promoter construct and included a transfection time course because previous reports indicated that the stimulatory effects of GnRH on the GnRHR promoter dampen after prolonged transfection (8). We identified that transfection of −1135GnRHR-Luc for 14 h followed by 4 h stimulation with 100 nm GnRH efficiently activated the promoter and had no stimulatory effect on the control plasmid (Fig. 9A). Using this regimen, we cotransfected GFP-FANCA WT and the two mutant constructs with −1135GnRHR-Luc for 14 h then induced the promoter with GnRH (Fig. 9B). No differences in basal unstimulated levels were measured, but GnRH induction of the GnRHR promoter was completely silenced by addition of GFP-FANCAH1110P or Q1128E, indicating that FANCA was required for GnRH induction of GnRHR gene expression. These results provide evidence for involvement of FANCA in transduction of the GnRH inductive signal in LβT2 cells and suggest target gene selectivity in this process because only GnRH induction of the αGSU and GnRHR gene promoters were perturbed.
FIG. 9.
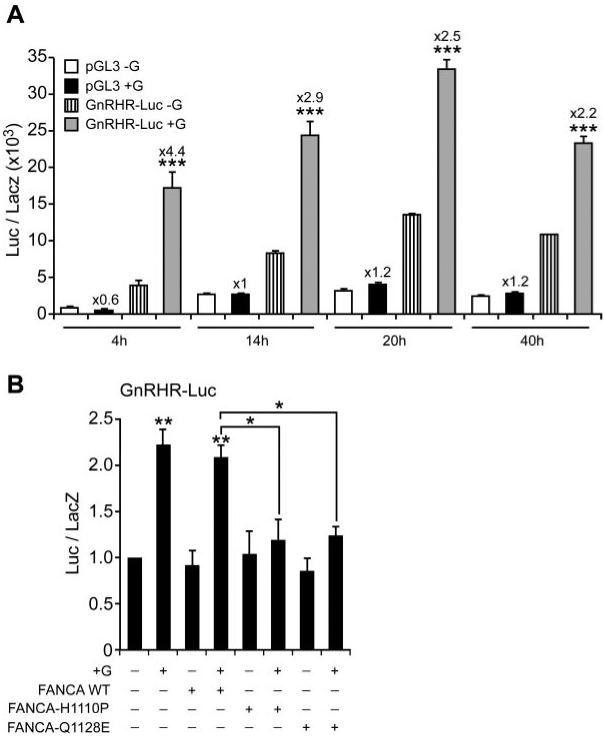
FANCA blocks GnRH-induction of the GnRHR gene promoter. A, Optimal transfection time course required for maximal GnRH stimulation of the −1135GnRHR-Luc gene promoter was determined. LβT2 cells were either transfected with control pGL3 or −1135Gn-RHR-Luc for 4, 14, 20, and 40 h before stimulating with 100 nm GnRH and then harvested 4 h later and luciferase levels measured. B, Effect of FANCA on −1135GnRHR-Luc after stimulation with 100 nm GnRH. Expression plasmids containing either GFP-FANCA WT or −H1110P or −Q1128E were cotransfected with −1135GnRHR-Luc at a ratio of reporter to effector of 24:1; 14 h later they were stimulated with 100 nm GnRH for 4 h and then assayed for luciferase. A and B, Fold stimulation above basal is indicated and corrected against the levels of β-galactosidase, with each experiment done in triplicate, and the results are average ± sem. Representative experiment of two is shown for A, whereas B is the average of three separate experiments. One-way ANOVA established that ***, P < 0.001, **, P < 0.01, and *, P < 0.05 when comparing +G vs. −G and when comparing +G cotransfected FANCA WT vs. +G cotransfected FANCA-H1110P and −Q1128E.
Discussion
Reports on the nucleocytoplasmic location of FANCA agree that nuclear accumulation of FANCA is necessary for function (15, 16, 33-35). FANCA protein encodes a NLS (15, 16) and CRM1 export motifs, which when perturbed promote cytoplasmic accumulation (36). However, dynamic hormone regulated nucleocytoplasmic shuttling, with an impact on transcription, has not been reported. In this study, we observed both nuclear and cytoplasmic FANCA by indirect immunofluorescence and Western blotting analysis of subcellular fractions. Rather than the nucleocytoplasmic distribution of FANCA being benign, we found that hormone induced a rapid nuclear accumulation and that significant levels of FANCA were exported back to the cytoplasm in LβT2 cells. Indeed, addition of AMD and GnRHR antagonists blocked nuclear accumulation and LMB blocked CRM1-dependent nuclear export. This dynamic redistribution of a protein-signaling complex in specialized hormone producing cells is appealing, especially because FA patients have numerous endocrinopathies (17, 37). Alternatively, FANCA may show rapid nuclear accumulation in response to diverse stimuli, as a consequence of differential phosphorylation (38), in a variety of cell types.
Hormone-dependent nuclear accumulation of FANCA suggests that the molecule may act as a signal transducer in gonadotropes, presumably by binding FANC proteins and translocating them to the nucleus. However, to our knowledge, a signal transduction role has not been previously proposed for this molecule but fits well with the structural motifs identified by bioinformatics, including Src-homology domains and leucine zipper motifs (13). FANCA and other FA proteins are orphan proteins with no homology to each other or to other proteins (20), except FANCG, which encodes a putative scaffold motif (39). Given that FANCA and FANCG associate and their nuclear accumulation is required for function (40), then it is possible that FANCG is the molecular signal transducer in gonadotropes. However, our use of FANCA mutants that bind FANCG but still have aberrant localization and function suggests that this is unlikely. Nevertheless, individual targeted disruption of Fanca, Fancc, and Fancg in mice generates an infertile phenotype, implicating the proteins in a common pathway (22, 23, 41, 42). We propose that a protein scaffold function would explain why no mutational hot spot has been identified for FANCA (43) because there are probably numerous protein attachment sites, that, if disrupted, could impact on different intracellular signaling pathways. Indeed, a large number of FANCA-interacting proteins have been identified by functional and biochemical assays (44), but it is more likely that a FANCA protein complex fulfills different cellular functions by differential recruitment of FANC-proteins. Using RT-PCR, we have so far identified transcripts for FANCC, -D2, -E, -F, -G, and -L in LβT2 cells and have yet to screen for -B, D1, -I, -J, and -M (Larder, R., unpublished results).
Due to the rapid nucleocytoplasmic shuttling of FANCA, we chose to investigate the subcellular distribution and function of two different FANCA mutants. The first mutant, FANCAH1110P, locates to the cytoplasm in lymphoblastoid and HeLa cells, fails to bind FANCC and FANCF, is nonphosphorylated, was originally isolated from a FA patient, and failed to rescue mitomycin C sensitivity of FANCA mutant cells and has been classified as a biochemically severe group III mutation (14, 27). The second FANCA mutant, FANCAQ1128E (25), was classified in group I, because this protein behaved in some respects like FANCA WT including the crucial monoubiquitination of FANCD2 (27). First, we demonstrated cytoplasmic accumulation of GFP-FANCAH1110P in LβT2 cells; second, we noted that GFP-FANCAQ1128E differed from the predicted nuclear location pattern demonstrated in HeLa cells and instead distributed evenly across both nuclear and cytoplasmic compartments in LβT2 cells. Furthermore, addition of GnRH did not promote subcellular redistribution of either mutant, indicating that the mutations uncoupled FANCA trafficking.
We then studied these proteins in our assay of gonadotrope cell function, specifically GnRH activation of transcription. Cotransfection of each mutant with the highly tissue-specific LH and FSH β-subunit gene promoters into LβT2 cells highlighted only marginal effects on basal FSH β-subunit gene transcription and GnRH activation of LH β-subunit. Given that Fanca gene targeted mice are initially fertile but become infertile after repeated ovarian cycles and that immunohistochemical examination of their pituitaries failed to reveal any obvious differences in LH and FSH content (23), then we would argue that there does not appear to be any direct regulatory effect of FANCA on transcriptional regulation of these gene promoters. Instead, our results suggest that active FANCA is required for GnRH stimulation of the αGSU and GnRHR gene promoters. Cotransfection of mutant FANCA molecules abrogated αGSU gene expression. However, the fold stimulation of αGSU gene expression varied in LβT2 cells. Previously, studies using the less differentiated αGSU-expressing αT3-1 gonadotrope cell line have shown that 1 nm GnRH induces maximal levels of αGSU expression and that prolonged stimulation with high concentrations of GnRH desensitizes the transcriptional apparatus (6, 45).
The results from this study indicate that these effects appear to be consistent between the two cell lines. Induction of αGSU gene expression requires interaction between the basal and GnRH-induced transcriptional apparatus (46), and a number of transcription factors and signal transduction pathways are known to be required (5, 6, 47, 48). However, because basal and induced αGSU in vivo expression levels are always in excess over the β-subunits (49), then we propose that the likely pituitary gonadotrope component of the observed phenotype in Fanca gene targeted mice arises from misregulation of the GnRHR gene promoter. Cotransfection of mutant FANCA with a GnRHR-Luc construct completely blocked GnRH stimulation. Indeed, the minimal fluctuations that we measured in LH and FSH β-subunit gene activation, from one pulse of GnRH, likely reflect the upstream perturbation of GnRHR gene activation. Recently others demonstrated that against a background of differing levels of GnRHR expression repeated GnRH stimulation of the β-subunit gene promoters produced minimal effects on LH β-subunit, but induction of FSH β-subunit was reduced (32). The biased disruption of GnRH-induced αGSU and GnRHR gene activation by FANCA mutants indicates that either a known signal transduction pathway common to both promoters is being perturbed or that FANCA is a new novel signal transmission molecule.
It was unexpected that cotransfection of FANCA-Q1128E with the GnRHR or αGSU gene promoters followed by GnRH stimulation would be as equally effective at blocking stimulation as cotransfection with FANCAH1110P. Previously, functional assays had failed to identify any negative impact of the Q1128E mutation on FANC-mediated DNA repair or FANCD2 monoubiquitination (27). However, the Q1128E mutation was isolated from a patient who died from bone marrow failure (Pasquini, R., personal communication). Indeed, linking the genotype to the phenotype in FANCA patients is an ongoing problem (50). Our results with the Q1128E mutant indicate that the FANC pathway can impact on different cellular mechanisms in addition to DNA repair. Indeed, FANCA interacts with the SWI/SNF chromatin regulatory complex, which is recruited to specific gene promoters to activate transcription (51, 52).
Others have hypothesized that the gonadal phenotype in Fanca−/− mice may arise due to direct impact of FANCA on gonadal development, particularly primordial germ cell formation, evidenced by hypogonadism and an increased incidence of ovarian cysts (24). Rather, we propose that targeted disruption of Fanca causes a progressive loss of fertility because FANCA is required for both primordial germ cell function and regulated gonadotropin hormone production via the GnRHR. Furthermore, evidence for misregulation of gonadotropins has been obtained from studies of FA patients (53), and children with FA enter puberty late, men are generally azoospermic, and women have early menopause (54). Testing of the hypothalamic-pituitary-gonadal axis revealed a blunted or prolonged LH response indicating dysregulation (37, 54).
In conclusion, we have identified two new, novel functions for FANCA. First, FANCA acts as a signal transduction component molecule in gonadotrope cells because we observed rapid nucleocytoplasmic shuttling in response to GnRH stimulation. Second, we have identified that this signaling function appears to specifically target the αGSU and GnRHR gene promoters in gonadotrope cells. This suggests FANCA is a signal transducer and is important for differential regulation of gene transcription in the reproductive system.
Acknowledgments
We thank Professor Pamela Mellon (University of California, San Diego, CA) for supplying LβT2 cells and Dr. Fre Arwert (University of Amsterdam, Amsterdam, The Netherlands) for the generous gift of the antimouse Fanca-specific antisera.
This work was supported by Medical Research Council (MRC) research studentship (to R.L.). R.L. was a MRC Ph.D. research student during the course of this work.
Abbreviations
- AMD
Actinomycin D
- CHX
cycloheximide
- EGFP
enhanced GFP
- FA
Fanconi anemia
- FANCA
Fanconi anemia a
- GFP
green fluorescent protein
- GnRHR
GnRH receptor
- GSU
glycoprotein hormone subunit
- LMB
leptomycin B
- NLS
nuclear localization signal
- WT
wild type
Footnotes
Disclosure statement: the authors have nothing to declare.
References
- 1.Charlton HM, Halpin DM, Iddon C, Rosie R, Levy G, McDowell I, Megson A, Morris J, Bramwell A, Speight A, Ward B, Broadhead J, Davey-Smith G, Fink G. The effects of daily administration of single and multiple injections of gonadotropin releasing hormone on pituitary and gonadal function in the hypogonadal (HPG) mouse. Endocrinology. 1983;113:535–544. doi: 10.1210/endo-113-2-535. [DOI] [PubMed] [Google Scholar]
- 2.Kaiser UB, Sabbagh E, Katzenellenbogen RA, Conn PM, Chin WW. A mechanism for the differential regulation of gonadotropin subunit gene expression by gonadotropin-releasing hormone. Proc Natl Acad Sci USA. 1995;92:12280–12284. doi: 10.1073/pnas.92.26.12280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shacham S, Harris D, Ben-Shlomo H, Cohen I, Bonfil D, Przedecki F, Lewy H, Ashkenazi IE, Seger R, Naor Z. Mechanism of GnRH receptor signaling on gonadotropin release and gene expression in pituitary gonadotrophs. Vitam Horm. 2001;63:63–90. doi: 10.1016/s0083-6729(01)63003-6. [DOI] [PubMed] [Google Scholar]
- 4.Bonfil D, Chuderland D, Kraus S, Shahbazian D, Friedberg I, Seger R, Naor Z. Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone β-subunit promoter. Endocrinology. 2004;145:2228–2244. doi: 10.1210/en.2003-1418. [DOI] [PubMed] [Google Scholar]
- 5.Roberson MS, Misra-Press A, Laurance ME, Stork PJS, Maurer RA. A role for mitogen-activated protein kinase in mediating activation of the glycoprotein hormone α-subunit promoter by gonadotropin-releasing hormone. Mol Cell Biol. 1995;15:3531–3539. doi: 10.1128/mcb.15.7.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris D, Chuderland D, Bonfil D, Kraus S, Seger R, Naor Z. Extracellular signal-regulated kinase and c-Src, but not Jun N-terminal kinase, are involved in basal and gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone α-subunit promoter. Endocrinology. 2003;144:612–622. doi: 10.1210/en.2002-220690. [DOI] [PubMed] [Google Scholar]
- 7.Harris D, Bonfil D, Chuderland D, Kraus S, Seger R, Naor Z. Activation of MAPK cascades by GnRH: ERK and Jun N-terminal kinase are involved in basal and GnRH-stimulated activity of the glycoprotein hormone LHβ-subunit promoter. Endocrinology. 2002;143:1018–1025. doi: 10.1210/endo.143.3.8675. [DOI] [PubMed] [Google Scholar]
- 8.Norwitz ER, Xu S, Jeong KH, Bedecarrats GY, Winebrenner LD, Chin WW, Kaiser UB. Activin A augments GnRH-mediated transcriptional activation of the mouse GnRH receptor gene. Endocrinology. 2002;143:985–997. doi: 10.1210/endo.143.3.8663. [DOI] [PubMed] [Google Scholar]
- 9.Pernasetti F, Vasilyev VV, Rosenberg SB, Bailey JS, Huang HJ, Miller WL, Mellon PL. Cell-specific transcriptional regulation of follicle-stimulating hormone-β by activin and gonadotropin releasing hormone in the LβT2 pituitary gonadotrope cell model. Endocrinology. 2001;142:2284–2295. doi: 10.1210/endo.142.6.8185. [DOI] [PubMed] [Google Scholar]
- 10.Caunt CJ, Hislop JN, Kelly E, Matharu AL, Green LD, Sedgley KR, Finch AR, McArdle CA. Regulation of gonadotropin-releasing hormone receptors by protein kinase C: inside out signalling and evidence for multiple active conformations. Endocrinology. 2004;145:3594–3602. doi: 10.1210/en.2004-0092. [DOI] [PubMed] [Google Scholar]
- 11.Larder R, Chang L, Clinton M, Brown P. Gonadotropin-releasing hormone regulates expression of the DNA damage repair gene, Fanconi anemia A, in pituitary gonadotroph cells. Biol Reprod. 2004;71:828–836. doi: 10.1095/biolreprod.104.030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Auerbach AD, Buchwald M, Joenje H. Fanconi anemia. In: Vogelstein B, Kinzler KW, editors. Genetic basis of human cancer. New York: McGraw-Hill; 1997. pp. 317–332. [Google Scholar]
- 13.van de Vrugt HJ, Cheng NC, de Vries Y, Rooimans MA, de Groot J, Scheper RJ, Zhi Y, Hoatlin ME, Joenje H, Arwert F. Cloning and characterization of murine fanconi anemia group A gene: Fanca protein is expressed in lymphoid tissues, testis, and ovary. Mamm Genome. 2000;11:326–331. doi: 10.1007/s003350010060. [DOI] [PubMed] [Google Scholar]
- 14.Kupfer G, Naf D, Garcia-Higuera I, Wasik J, Cheng A, Yamashita T, Tipping A, Morgan N, Mathew CG, D’Andrea AD. A patient-derived mutant form of the Fanconi anemia protein, FANCA, is defective in nuclear accumulation. Exp Hematol. 1999;27:587–593. doi: 10.1016/s0301-472x(99)00022-3. [DOI] [PubMed] [Google Scholar]
- 15.Lightfoot J, Alon N, Bosnoyan-Collins L, Buchwald M. Characterization of regions functional in the nuclear localization of the Fanconi anemia group A protein. Hum Mol Genet. 1999;8:1007–1015. doi: 10.1093/hmg/8.6.1007. [DOI] [PubMed] [Google Scholar]
- 16.Naf D, Kupfer GM, Suliman A, Lambert K, D’Andrea AD. Functional activity of the fanconi anemia protein FAA requires FAC binding and nuclear localization. Mol Cell Biol. 1998;18:5952–5960. doi: 10.1128/mcb.18.10.5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joenje H, Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet. 2001;2:446–457. doi: 10.1038/35076590. [DOI] [PubMed] [Google Scholar]
- 18.Kupfer GM, Naf D, Suliman A, Pulsipher M, D’Andrea AD. The Fanconi anaemia proteins, FAA and FAC, interact to form a nuclear complex. Nat Genet. 1997;17:487–490. doi: 10.1038/ng1297-487. [DOI] [PubMed] [Google Scholar]
- 19.Thompson LH. Unraveling the Fanconi anemia-DNA repair connection. Nat Genet. 2005;37:921–922. doi: 10.1038/ng0905-921. [DOI] [PubMed] [Google Scholar]
- 20.Gregory RC, Taniguchi T, D’Andrea AD. Regulation of the Fanconi anemia pathway by monoubiquitination. Semin Cancer Biol. 2003;13:77–82. doi: 10.1016/s1044-579x(02)00102-5. [DOI] [PubMed] [Google Scholar]
- 21.Thomashevski A, High AA, Drozd M, Shabanowitz J, Hunt DF, Grant PA, Kupfer GM. The Fanconi anemia core complex forms four complexes of different sizes in different subcellular compartments. J Biol Chem. 2004;279:26201–26209. doi: 10.1074/jbc.M400091200. [DOI] [PubMed] [Google Scholar]
- 22.Noll M, Battaile KP, Bateman R, Lax TP, Rathbun K, Reifsteck C, Bagby G, Finegold M, Olson S, Grompe M. Fanconi anemia group A and C double-mutant mice: functional evidence for a multi-protein Fanconi anemia complex. Exp Hematol. 2002;30:679–688. doi: 10.1016/s0301-472x(02)00838-x. [DOI] [PubMed] [Google Scholar]
- 23.Cheng NC, van de Vrugt HJ, van der Valk MA, Oostra AB, Krimpenfort P, de Vries Y, Joenje H, Berns A, Arwert F. Mice with a targeted disruption of the Fanconi anemia homolog Fanca. Hum Mol Genet. 2000;9:1805–1811. doi: 10.1093/hmg/9.12.1805. [DOI] [PubMed] [Google Scholar]
- 24.Wong JC, Alon N, McKerlie C, Huang JR, Meyn MS, Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. 2003;12:2063–2076. doi: 10.1093/hmg/ddg219. [DOI] [PubMed] [Google Scholar]
- 25.Levran O, Erlich T, Magdalena N, Gregory JJ, Batish SD, Verlander PC, Auerbach AD. Sequence variation in the Fanconi anemia gene FAA. Proc Natl Acad Sci USA. 1997;94:13051–13056. doi: 10.1073/pnas.94.24.13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koc A, Pronk JC, Alikasifoglu M, Joenje H, Altay C. Variable pathogenicity of exon 43del (FAA) in four Fanconi anaemia patients within a consanguineous family. Br J Haematol. 1999;104:127–130. doi: 10.1046/j.1365-2141.1999.01156.x. [DOI] [PubMed] [Google Scholar]
- 27.Adachi D, Oda T, Yagasaki H, Nakasato K, Taniguchi T, D’Andrea AD, Asano S, Yamashita T. Heterogeneous activation of the Fanconi anemia pathway by patient-derived FANCA mutants. Hum Mol Genet. 2002;11:3125–3134. doi: 10.1093/hmg/11.25.3125. [DOI] [PubMed] [Google Scholar]
- 28.Quirk J, Brown P. Hesx1 homeodomain protein represses transcription as a monomer and antagonises transactivation of specific sites as a homodimer. J Mol Endocrinol. 2002;28:193–205. doi: 10.1677/jme.0.0280193. [DOI] [PubMed] [Google Scholar]
- 29.Pinol-Roma S, Dreyfuss G. Shuttling of pre-mRNA binding proteins between nucleus and cytoplasm. Nature. 1992;355:730–732. doi: 10.1038/355730a0. [DOI] [PubMed] [Google Scholar]
- 30.Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 31.Turgeon JL, Kimura Y, Waring DW, Mellon PL. Steroid and pulsatile gonadotropin-releasing hormone (GnRH) regulation of luteinizing hormone and GnRH receptor in a novel gonadotrope cell line. Mol Endocrinol. 1996;10:439–450. doi: 10.1210/mend.10.4.8721988. [DOI] [PubMed] [Google Scholar]
- 32.Bedecarrats GY, Kaiser UB. Differential regulation of gonadotropin subunit gene promoter activity by pulsatile gonadotropin-releasing hormone (GnRH) in perifused LβT2 cells: role of GnRH receptor concentration. Endocrinology. 2003;144:1802–1811. doi: 10.1210/en.2002-221140. [DOI] [PubMed] [Google Scholar]
- 33.Kruyt FA, Youssoufian H. The Fanconi anemia proteins FAA and FAC function in different cellular compartments to protect against cross-linking agent cytotoxicity. Blood. 1998;92:2229–2236. [PubMed] [Google Scholar]
- 34.Kruyt FA, Waisfisz Q, Dijkmans LM, Hermsen MA, Youssoufian H, Arwert F, Joenje H. Cytoplasmic localization of a functionally active Fanconi anemia group A-green fluorescent protein chimera in human 293 cells. Blood. 1997;90:3288–3295. [PubMed] [Google Scholar]
- 35.Walsh CE, Yountz MR, Simpson DA. Intracellular localization of the Fanconi anemia complementation group A protein. Biochem Biophys Res Commun. 1999;259:594–599. doi: 10.1006/bbrc.1999.0768. [DOI] [PubMed] [Google Scholar]
- 36.Ferrer M, Rodriguez JA, Spierings EA, de Winter JP, Giaccone G, Kruyt FA. Identification of multiple nuclear export sequences in Fanconi anemia group A protein that contribute to CRM1-dependent nuclear export. Hum Mol Genet. 2005;14:1271–1281. doi: 10.1093/hmg/ddi138. [DOI] [PubMed] [Google Scholar]
- 37.Wajnrajch MP, Gertner JM, Huma Z, Popovic J, Lin K, Verlander PC, Batish SD, Giampietro PF, Davis JG, New MI, Auerbach AD. Evaluation of growth and hormonal status in patients referred to the International Fanconi Anemia Registry. Pediatrics. 2001;107:744–754. doi: 10.1542/peds.107.4.744. [DOI] [PubMed] [Google Scholar]
- 38.Yagasaki H, Adachi D, Oda T, Garcia-Higuera I, Tetteh N, D’Andrea AD, Futaki M, Asano S, Yamashita T. A cytoplasmic serine protein kinase binds and may regulate the Fanconi anemia protein FANCA. Blood. 2001;98:3650–3657. doi: 10.1182/blood.v98.13.3650. [DOI] [PubMed] [Google Scholar]
- 39.Blom E, van de Vrugt HJ, de Vries Y, de Winter JP, Arwert F, Joenje H. Multiple TPR motifs characterize the Fanconi anemia FANCG protein. DNA Repair (Amst) 2004;3:77–84. doi: 10.1016/j.dnarep.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 40.Waisfisz Q, De Winter JP, Kruyt FAE, de Groot J, van der Weel L, Dijkmans LM, Arwert F, Scheper RJ, Youssoufian H, Hoatlin ME, Joenje H. A physical complex of the Fanconi anemia proteins FANCG/XRCC9 and FANCA. Proc Natl Acad Sci USA. 1999;96:10320–10325. doi: 10.1073/pnas.96.18.10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koomen M, Cheng NC, van de Vrugt HJ, Godthelp BC, van der Valk MA, Oostra AB, Zdzienicka MZ, Joenje H, Arwert F. Reduced fertility and hypersensitivity to mitomycin C characterize Fancg/Xrcc9 null mice. Hum Mol Genet. 2002;11:273–281. doi: 10.1093/hmg/11.3.273. [DOI] [PubMed] [Google Scholar]
- 42.Yang Y, Kuang Y, De Oca RM, Hays T, Moreau L, Lu N, Seed B, D’Andrea AD. Targeted disruption of the murine Fanconi anemia gene, Fancg/Xrcc9. Blood. 2001;98:3435–3440. doi: 10.1182/blood.v98.12.3435. [DOI] [PubMed] [Google Scholar]
- 43.Levran O, Diotti R, Pujara K, Batish SD, Hanenberg H, Auerbach AD. Spectrum of sequence variations in the FANCA gene: an International Fanconi Anemia Registry (IFAR) study. Hum Mutat. 2005;25:142–149. doi: 10.1002/humu.20125. [DOI] [PubMed] [Google Scholar]
- 44.Reuter TY, Medhurst AL, Waisfisz Q, Zhi Y, Herterich S, Hoehn H, Gross HJ, Joenje H, Hoatlin ME, Mathew CG, Huber PA. Yeast two-hybrid screens imply involvement of Fanconi anemia proteins in transcription regulation, cell signaling, oxidative metabolism, and cellular transport. Exp Cell Res. 2003;289:211–221. doi: 10.1016/s0014-4827(03)00261-1. [DOI] [PubMed] [Google Scholar]
- 45.Kay TW, Chedrese PJ, Jameson JL. Gonadotropin-releasing hormone causes transcriptional stimulation followed by desensitization of the glycoprotein hormone α promoter in transfected αT3 gonadotrope cells. Endocrinology. 1994;134:568–573. doi: 10.1210/endo.134.2.7507827. [DOI] [PubMed] [Google Scholar]
- 46.Brown P, McNeilly AS. Transcriptional regulation of pituitary gonadotrophin subunit genes. Rev Reprod. 1999;4:117–124. doi: 10.1530/ror.0.0040117. [DOI] [PubMed] [Google Scholar]
- 47.Tremblay JJ, Lanctot C, Drouin J. The pan-pituitary activator of transcription, Ptx1 (pituitary homeobox 1), acts in synergy with SF-1 and Pit1 and is an upstream regulator of the Lim-homeodomain gene Lim3/Lhx3. Mol Endocrinol. 1998;12:428–441. doi: 10.1210/mend.12.3.0073. [DOI] [PubMed] [Google Scholar]
- 48.Horn F, Windle JJ, Barnhart K, Mellon PL. Tissue-specific gene expression in the pituitary: the glycoprotein hormone α-subunit gene is regulated by a gonadotrope-specific protein. Mol Cell Biol. 1992;12:2143–2153. doi: 10.1128/mcb.12.5.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown P, McNeilly JR, Evans JG, Crawford GM, Walker M, Christian HC, McNeilly AS. Manipulating the in vivo mRNA expression profile of FSH β to resemble that of LH β does not promote a concomitant increase in intracellular storage of follicle-stimulating hormone. J Neuroendocrinol. 2001;13:50–62. doi: 10.1046/j.1365-2826.2001.00594.x. [DOI] [PubMed] [Google Scholar]
- 50.Magdalena N, Pilonetto DV, Bitencourt MA, Pereira NF, Ribeiro RC, Jeng M, Pasquini R. Frequency of Fanconi anemia in Brazil and efficacy of screening for the FANCA 3788-3790del mutation. Braz J Med Biol Res. 2005;38:669–673. doi: 10.1590/s0100-879x2005000500003. [DOI] [PubMed] [Google Scholar]
- 51.Otsuki T, Furukawa Y, Ikeda K, Endo H, Yamashita T, Shinohara A, Iwamatsu A, Ozawa K, Liu JM. Fanconi anemia protein, FANCA, associates with BRG1, a component of the human SWI/SNF complex. Hum Mol Genet. 2001;10:2651–2660. doi: 10.1093/hmg/10.23.2651. [DOI] [PubMed] [Google Scholar]
- 52.Peterson CL, Workman JL. Promoter targeting and chromatin remodeling by the SWI/SNF complex. Curr Opin Genet Dev. 2000;10:187–192. doi: 10.1016/s0959-437x(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 53.Berkovitz GD, Zinkham WH, Migeon CJ. Gonadal function in two siblings with Fanconi’s anemia. Horm Res. 1984;19:137–141. doi: 10.1159/000179880. [DOI] [PubMed] [Google Scholar]
- 54.Alter BP, Bagby G. Fanconi anemia. Standards for clinical care. Fanconi Anemia Standards for Clinical Care Conference; Chicago, IL. 2003; pp. 1–190. [Google Scholar]