

Abstract
A recombinant HSV-1 virus expressing EGFP from the HCMV major immediate early promoter (KOS-CMVGFP) was constructed to monitor viral replication and spread in vitro and in mice. KOS-CMVGFP replicated as efficiently as wild-type virus, strain KOS, in single cycle growth experiments in Vero cells indicating that the recombinant virus has no significant growth defects in vitro. Following ocular inoculation of mice, KOS-CMVGFP exhibited slight but statistically significant reductions in mouse tear film titers relative to wild-type virus. Progression of virus infection of the eyes, periocular tissue, and snout was readily followed by fluorescence microscopy. Insertion of the EGFP expression cassette into the KOS genome had no effect on the efficiency of establishment of latency as determined by quantitative competitive PCR of viral genomes in latently infected TG. KOS-CMVGFP reactivated with wild-type kinetics and efficiency by explant cocultivation, but exhibited a significant delay in the kinetics and a modest reduction in the efficiency of reactivation compared to KOS in the more sensitive TG cell culture model. Notably, EGFP expression preceded the detection of infectious virus by greater than 24 h in both ex vivo models and thus is a useful marker of the early stages in the induction of reactivation.
Keywords: Herpes simplex virus, in vivo replication, mice, GFP, latency, reactivation, recombinant virus
Introduction
Because of the broad host range of HSV-1, several animal models are available to study viral infection in vivo. Two such models, the rabbit and guinea pig, recreate to a limited extent all aspects of HSV-1 infection in humans (i.e. acute replication, establishment of latency, and reactivation), but at considerable expense to the investigator. The mouse model is more widely used because it is relatively inexpensive in that sufficient numbers of animals can be purchased at a lower cost to achieve statistically significant data. Unlike rabbits and guinea pigs, however, spontaneous reactivation in mice has not been demonstrated (Feldman et al., 2002; Gebhardt and Halford, 2005). Therefore, reactivation is most commonly induced by explanting trigeminal ganglia (TG), which contain the cell bodies of latently infected neurons. In this case the stress associated with explant induces reactivation.
In the mouse ocular model, mice are inoculated on the surface of the eye following corneal scarification. Virus replication occurs in corneal epithelial cells, keratocytes, and endothelial cells. Virus then enters the termini of sensory neurons that innervate the cornea and travels intraaxonally to neuronal cell bodies in TG resulting in either (i) productive infection, characterized by extensive viral gene expression, viral DNA replication, production of infectious virus and cell death, or (ii) latency, characterized by limited viral gene expression, the absence of viral DNA replication, the failure to produce infectious virus, and neuronal survival. Although the factors that determine which program is operative are not known, these opposing biological endpoints, productive infection and latency, reflect the fact that the neuron plays a central role in determining the outcome of HSV-1 infection (Margolis, Dawson, and Lavail, 1992; Margolis et al., 1992). Virus reaches TG between 1 and 2 days post-inoculation and viral latency is established within two weeks of corneal inoculation as determined by the absence of infectious virus and the presence of viral DNA.
Traditionally, to examine the role of viral proteins or cis-acting elements in the mouse ocular model, mutant viruses are compared to wild-type virus with regard to (i) the efficiency of viral replication in eyes and TG, (ii) the efficiency of the establishment of latency, and (iii) the kinetics and frequency of reactivation following TG explant. This and other similar animal models have the disadvantage that infected animals must be euthanized at specified times post-infection in order to harvest tissue needed to determine the distribution and titers of virus. Thus, the ability to monitor both the spread and ultimate outcome of viral infection in the same animal is precluded. As a means of circumventing this problem, recombinant viruses expressing various reporter genes visible by eye have been used to monitor the kinetics and extent of virus spread in animals.
LacZ (Escherichia coli), firefly luciferase (Photinus pyralis), and enhanced green fluorescent protein (EGFP; Aequorea victoria) (Cormack, Valdivia, and Falkow, 1996) are among the reporter genes used to generate recombinant viruses. Relevant properties of these reporter genes include the following: (i) to visualize β–galactosidase, the product of the lacZ gene, chemical fixation of cells prior to staining is required such that additional experimentation with the same infected mouse cannot be performed; (ii) in tests of noninvasive bioluminescent imaging in mice infected by the ocular route with a recombinant HSV-1 expressing firefly luciferase, luminescence produced in TG was indistinguishable from overlying periocular tissues in the intact animal (Luker et al., 2002), thus rendering the firefly luciferase reporter system undesirable for some purposes in the mouse ocular model; (iii) unlike LacZ and firefly luciferase, EGFP requires no cofactors or substrates for detection; (iv) EGFP is resistant to heat, alkaline pH (Patterson et al., 1997), detergents, chaotropic salts, organic solvents, and most proteases; and (v) EGFP intensity in living cells can be quantified by FACS (Soboleski, Oaks, and Halford, 2005), confocal scanning laser microscopy, and fluorometric assays. For these reasons we have constructed a recombinant of HSV-1, strain KOS, expressing EGFP from the human cytomegalovirus major immediate early promoter (HCMV-MIEP), termed KOS-CMVGFP. We describe the construction and in vitro and in vivo characterization of KOS-CMVGFP and demonstrate the utility of this virus for studies of viral infection and pathogenesis in mice.
Results
Construction and isolation of a recombinant of HSV-1, strain KOS, expressing EGFP
The strategy described by Cai et al. was used to generate a recombinant of HSV-1, strain KOS, expressing high levels of EGFP (Fig. 1) (Cai et al., 1993). This strategy involved insertion of the EGFP gene into the minimally and convergently transcribed region between the UL26/26.5 and UL27 genes in the HSV-1 genome (Fig. 1C). Insertion at this location was thought to have little or no effect on expression of adjacent viral genes (Bzik et al., 1984; Holland et al., 1984; Liu and Roizman, 1991). As the first step in generating the virus, plasmid pGFP53K was constructed (Fig. 1D and E). This plasmid contains an expression cassette consisting of the HCMV-MIEP (Brewer and Roth, 1991) driving EGFP. The SV40 late polyadenylation signal was added to ensure efficient polyadenylation of the EGFP transcript. Viral DNA restriction fragments consisting of an ~1 kb SalI-FspI fragment, which spans nucleotides 52,192 to 53,150 and contains a portion of UL26 and UL26.5, and an ~2 kb DraI-SalI fragment, spanning nucleotides 52,764 to 54,826 and containing a portion of UL27, were ligated to the 5’ and 3’ ends of the EGFP expression cassette, respectively, as flanking sequences to facilitate recombination with the HSV-1 genome (Fig. 1D). Because nucleotide sequences downstream of canonical viral AAUAAA polyadenylation signals have been shown to enhance the efficiency of polyadenylation, the 386 bp DraI-FspI sequence (spanning nt 52,764 to 53,150) was included as a direct repeat on each side of the EGFP expression cassette (shown as thick black lines in Fig. 1E).
Figure 1. Construction of an EGFP-expressing recombinant of HSV-1, strain KOS.
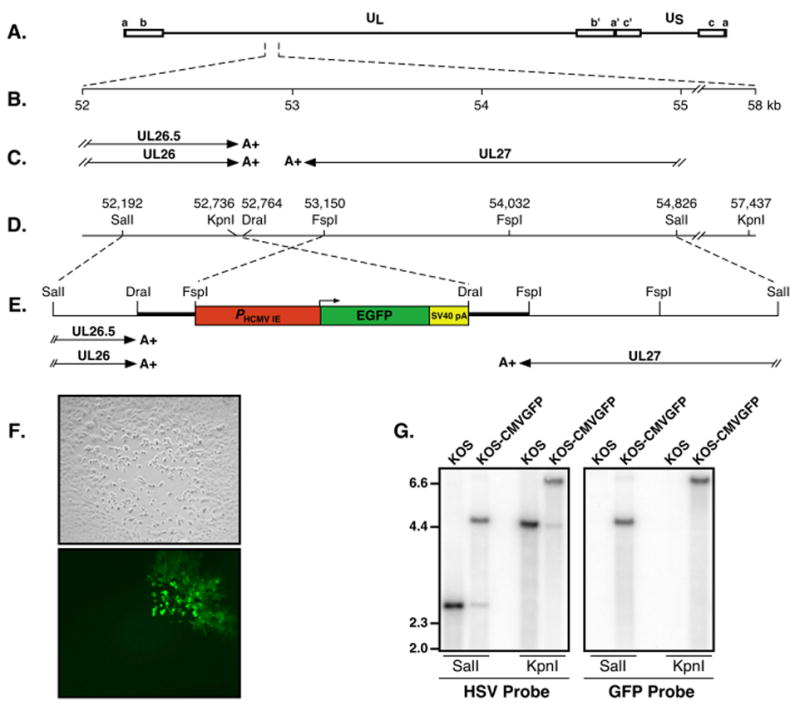
(A) Diagram of the HSV-1 genome showing the UL region flanked by inverted repeats ab and b’a’ and the US region flanked by inverted repeats a’c’ and ca. (B) Scale in kilobases of the UL26/26.5 and UL27 region. (C) Transcripts of known genes in the 52 to 55 kb region of the HSV-1 genome. Arrows indicate the locations and directions of transcription of individual transcripts; double slash lines indicate breaks in transcripts, which are initiated outside of the 52 to 55 kb region of the HSV-1 genome; A+ indicates the location of polyadenylation signals for each transcript. (D) Restriction map of the 52 to 58 kb region of the genome. Numbers are map units in kb. (E) Location of the EGFP expression cassette within the minimally transcribed intergenic region between UL26/26.5 and UL27. The CMV major immediate early promoter, the EGFP gene and the SV40 polyadenylation signal (late orientation) are indicated in red, green and yellow, respectively. The predicted transcripts including polyadenylation signals for UL26/UL26.5 and UL27 are shown beneath the diagram. (F) Loss of fluorescence in ¾ of a sectioned plaque of KOS-CMVGFP on Vero cell monolayers. Vero cell monolayers were infected at a multiplicity of 0.05 PFU/cell of KOS-CMVGFP for one hour and overlaid with 2% methylcellulose. At 4 d p.i., plaques were examined by phase contrast microscopy (upper panel) and fluorescence microscopy (lower panel). (G) Southern blot analysis of KOS and KOS-CMVGFP DNA digested with the restriction enzymes indicated beneath each blot. Total cellular DNA from Vero cells infected at a multiplicity of 5 PFU/cell was analyzed using either a 32P-labeled HSV-1 DraI to SalI fragment (nt 52,764 to 54,826; left panel) or an AgeI to XbaI of plasmid pEGFP-N1, which contains the EGFP ORF, (right panel) as probe. Molecular weight markers in kb are indicated on the left.
To generate a recombinant virus expressing EGFP using the standard marker transfer procedure (Sacks et al., 1985), the SalI-SalI fragment containing the EGFP cassette was isolated and co-transfected with infectious wild-type viral DNA. Recombinant green fluorescent plaques (GFP+) were identified by fluorescence microscopy, picked and plaque purified five times. Notably, GFP-negative (GFP−) white plaques were observed throughout the purification process at frequencies ranging from 0.3 to 0.9% indicating that the EGFP gene was continually repressed or lost, presumably by recombination between the direct repeats (DraI-Fspl sequence) flanking the EGFP expression cassette (thick black lines, Fig. 1E). Supporting this hypothesis, sectoring of plaques was observed on rare occasions (Fig. 1F).
The recombinant viral genome was analyzed by Southern blot analysis (Fig. 1G). Total cellular DNA from Vero cell monolayers infected with KOS and KOS-CMVGFP was digested with SalI (to verify the presence or absence of the EGFP cassette) or KpnI (to verify the site of the insertion). DNA fragments were separated on an agarose gel, transferred to a membrane, and hybridized either to (i) P32-labeled DraI-SalI fragments from HSV-1 genomic DNA (nt 52,764 to 54,826; HSV probe, Fig. 1G) or (ii) P32-labeled EGFP fragments (GFP probe, Fig. 1G).
Using the HSV probe, KOS DNA digested with either SalI or KpnI produced the expected 2.6 and 4.7 kb fragments, respectively, whereas digestion of KOS-CMVGFP DNA with each enzyme produced fragments of 4.8 and 6.9 kb, respectively (Fig. 1G). In addition, weak hybridization to 2.6 and 4.7 kb fragments was observed in SalI and KpnI KOS-CMVGFP lanes indicating the presence of low levels of revertant (wild-type), viral genomes. These findings were confirmed using a GFP probe (Fig. 1G), which hybridized only to the larger 4.8 and 6.9 kb fragments of KOS-CMVGFP and not to KOS DNA. These results are consistent with insertion of the 2.2 kb EGFP expression cassette into the viral genome between UL26/26.5 and UL27 and indicate that a low level of revertant, GFP− virus that had lost the EGFP cassette was present in the KOS-CMVGFP stock.
In vitro replication of KOS-CMVGFP
To examine the effect of the presence of the EGFP expression cassette on viral replication in vitro, single cycle growth experiments were performed in Vero cells at a calculated multiplicity of infection of 0.1 PFU per cell (Fig. 2). KOS and KOS-CMVGFP replicated with nearly identical efficiencies and kinetics. Similar results were observed at a multiplicity of 2.5 PFU/cell indicating that the recombinant virus is not significantly impaired for growth in Vero cells (data not shown).
Figure 2. Single cycle growth curves of KOS and KOS-CMVGFP in Vero cells.
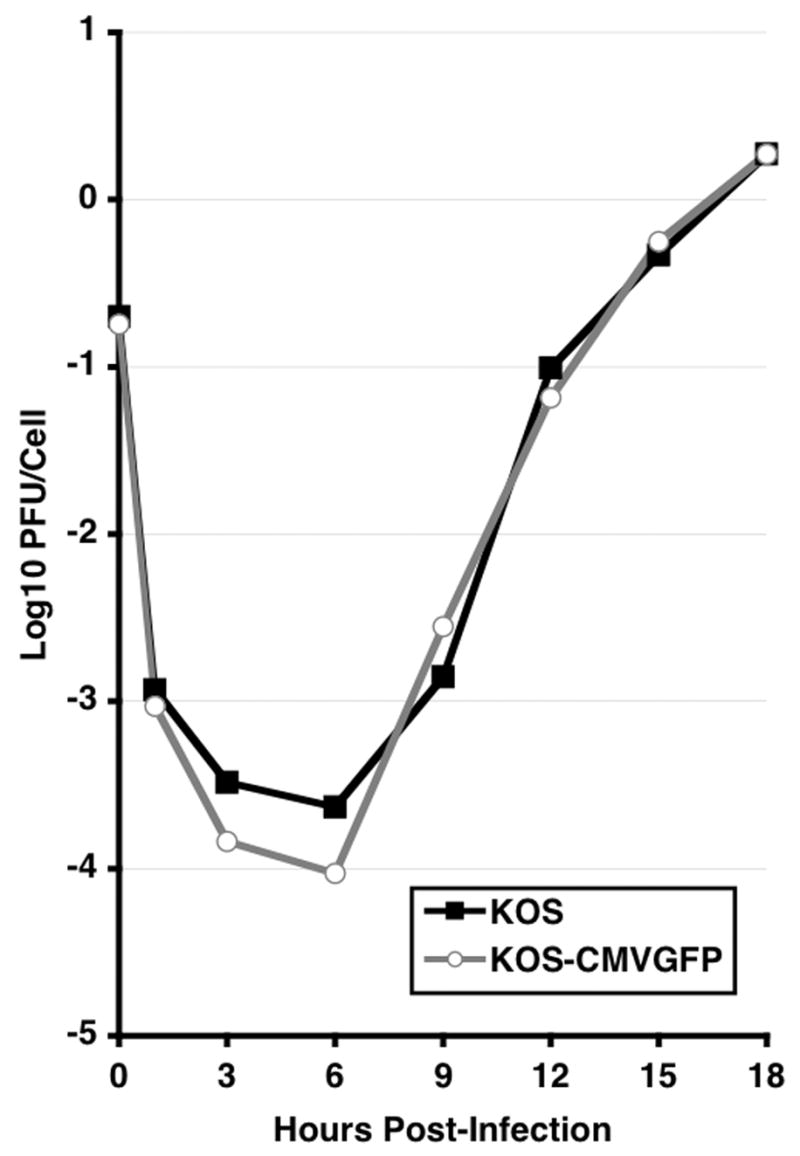
Monolayers of Vero cells were infected at a multiplicity of 0.1 PFU/cell of KOS or KOS-CMVGFP. Infected cells were incubated at 37°C and harvested at the indicated times. Infectious virus was quantified by standard plaque assays on Vero cell monolayers. The results of one of two independent assays are shown.
Acute infection of mice with KOS-CMVGFP
To observe the spread of KOS-CMVGFP in live mice during the acute phase of infection, animals were infected with 2 X 105 PFU/eye of recombinant virus via the ocular route following corneal scarification. Mice were anesthetized daily post-infection (p.i.) and the eyes, periocular region, and snout were examined by fluorescence microscopy (Fig. 3). On day 1 p.i., fluorescence was observed in eyes as distinct small foci on the cornea. Fluorescence spread throughout the eye between days 1 and 3 p.i., and foci grew larger and coalesced resulting in large dendritic corneal lesions. Maximal intensity and spread of fluorescence were consistently observed on day 2 p.i.. The intensity and extent of spread of fluorescence declined rapidly in the eye after day 3 with few or no small ocular foci observed between days 4 (data not shown) or 5 and 11 p.i.
Figure 3. Expression of EGFP during acute infection of mouse corneas.
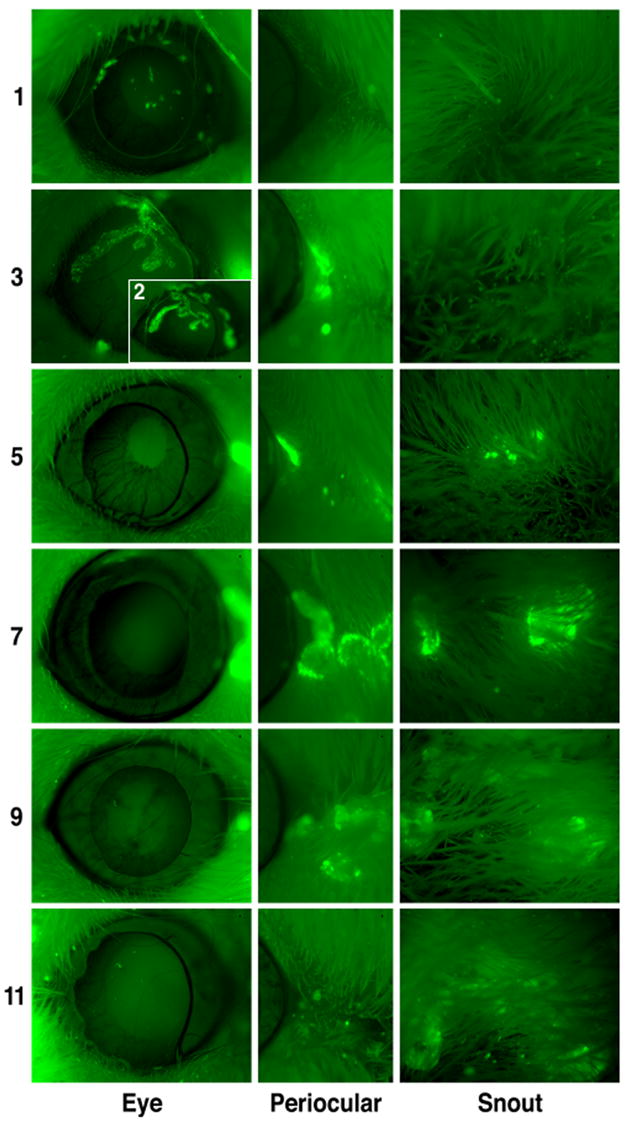
Two-5 to 6 week old male ICR mice were infected in both eyes with 2 X 105 PFU/eye of KOS-CMVGFP following corneal scarification. The mice were anesthetized daily p.i. and examined by fluorescence microscopy at 40X magnification. The results of one of two independent experiments are shown. Days p.i. and area of the mouse photographed are indicated to the left and bottom, respectively, of the photomicrographs.
During the course of acute infection, fluorescent foci in periocular tissue and the snouts of mice were also observed (Fig. 3). Foci in periocular tissue were first observed on day 3 p.i., expanded radially from the initial site of fluorescence until day 7 p.i. and diminished in intensity thereafter. Foci were initially observed on the snout on day 5 p.i. These foci expanded radially from the initial site until day 7 or 8 p.i. and diminished thereafter. Although the general pattern of spread was consistent, the level and extent of spread of fluorescence varied considerably among mice. Collectively, these tests confirm the accepted model that HSV-1 spreads from the cornea to adjacent sites and that viral spread is time-dependent.
Effects of the presence of the EGFP cassette on acute viral replication and pathogenesis
(i) Gross pathology
Physical examination of mice acutely infected with KOS and KOS-CMVGFP revealed little difference in gross pathology throughout the 30-day test period. Mice in both experimental groups exhibited moderate to severe symptoms of blepharoconjuctivitis, ulcerative lesions, and periocular hair loss.
(ii) Viral titers in tear film
To examine the efficiency of acute viral replication at the site of inoculation in mice, viral titers in tear film were determined daily for 9 days following corneal inoculation (Fig. 4A). KOS-CMVGFP replicated to levels slightly lower than wild-type KOS for the first 2 days p.i. and to similar levels between day 2 and day 4 p.i. From day 4 through day 6 p.i., the levels of KOS-CMVGFP in tear film were slightly but significantly reduced with levels 3.5- to 4.5-fold lower than wild type [P=0.002–0.007, two-tailed t test]. On days 7 through 9, the levels of KOS-CMVGFP were similar to KOS. Overall, a minor but significant difference in the patterns of replication of the two viruses [P=0.016, two-way ANOVA] was evident, primarily as a slight reduction in the levels of KOS-CMVGFP in tear films. These findings demonstrate that insertion of the EGFP cassette between the UL26/26.5 and UL27 genes had a slight but statistically significant effect on the levels of virus in tear film.
Figure 4. Tear film titers of KOS and KOS-CMVGFP during acute infection of mice.
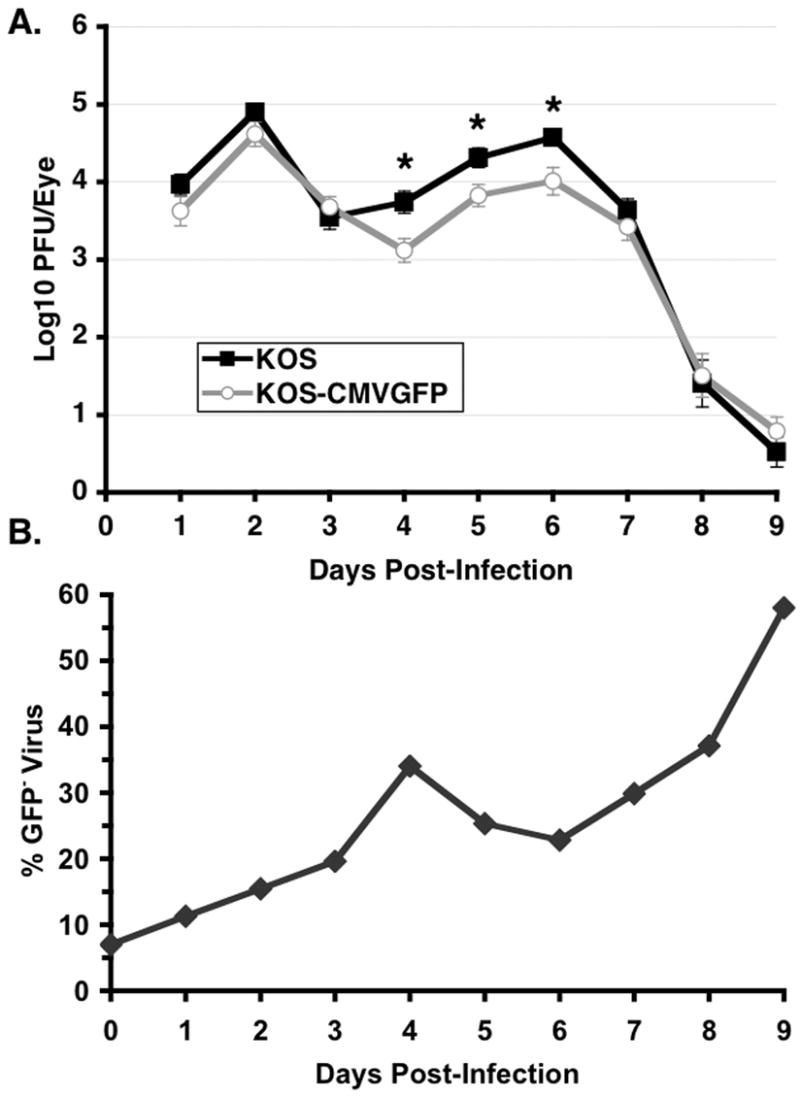
Thirty-three mice per group were infected as described in the legend to Fig. 3. On the days indicated, eyes were swabbed and viral titers were determined by standard plaque assays on Vero cell monolayers. (A) Viral titers are shown as the means ± standard error of the means (error bars). Asterisks indicate significant differences between KOS and KOS-CMVGFP. (B) The percent of GFP− (revertants) virus over time in tear film are shown as the means ± standard error of the means (error bars).
In addition to determining the absolute levels of virus recovered from mouse tear film, the percentage of revertant (GFP−) virus in KOS-CMVGFP samples was determined by standard plaque assays (Fig. 4B). The levels of GFP− virus increased 3.0-fold between days 1 and 4 p.i. [P=0.007, two-tailed t test], decreased 1.5-fold between days 4 and 6 p.i., and then increased 2.5-fold through day 9 p.i., when GFP− virus represented nearly 60% of the virus sample. These observations indicate that either EGFP activity or the EGFP cassette was gradually lost during acute replication. Although the decrease in the amount of GFP− between days 4 and 6 p.i. was not statistically significant, the timing and magnitude of the decrease were similar in two independent experiments. Because the ratios of GFP− to GFP+ virus decreased (Fig. 4B) during a period of increase in overall viral titer (days 4–6, Fig. 4A and B) it is possible that some of the virus in tear film on these days was derived from a distal site (i.e. TG neurons).
Effects of the EGFP cassette on the efficiency of establishment and reactivation of latency
To determine the effects of the presence of the EGFP cassette in the viral genome on the establishment and reactivation of latency, mice latently infected with KOS and KOS-CMVGFP were euthanized between days 30 and 35 p.i. TG were removed and the efficiency of establishment of latency was determined by measuring viral genome loads (Fig. 5). The kinetics and efficiency of reactivation were determined by measuring the presence of infectious virus in TG explants (Fig. 6) and in TG cell cultures (Fig. 7).
Figure 5. Viral genome loads of KOS and KOS CMVGFP in latently infected TG.
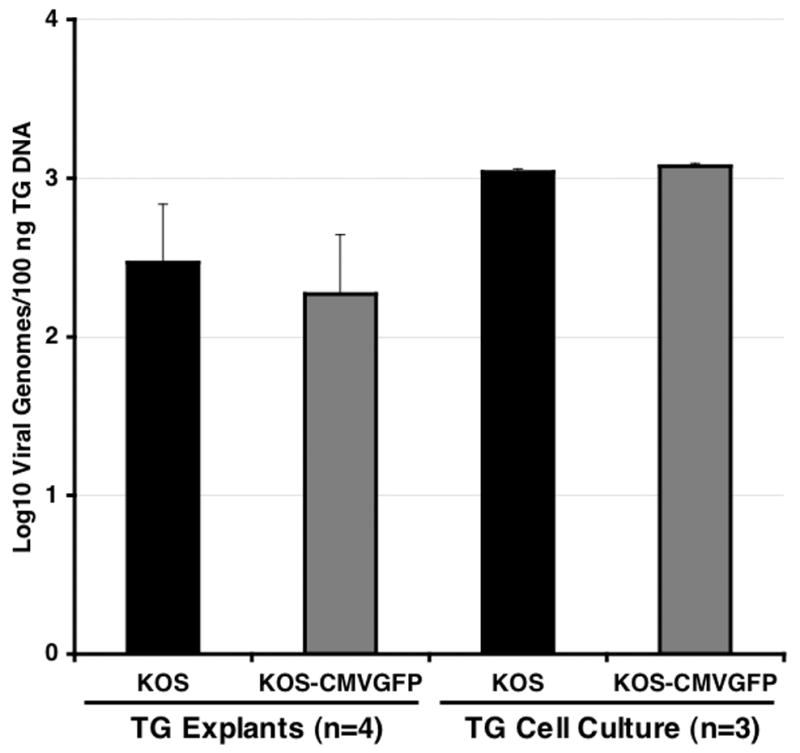
One hundred nanograms of DNA from individual TG or pooled TG cell suspensions (used to make TG cell culture in Fig. 7) obtained between days 30 and 35 p.i. were analyzed by competitive PCR and compared to competitive PCR products of HSV-1 genome standards ranging from 1 to 6 X 104 genome copies per 100 ng of TG DNA. The numbers (n) of explanted TG or replicate TG cell suspensions analyzed are shown below the graph. The results shown are the means ± standard errors of the means (error bars).
Figure 6. Reactivation of KOS and KOS-CMVGFP in explanted TG.
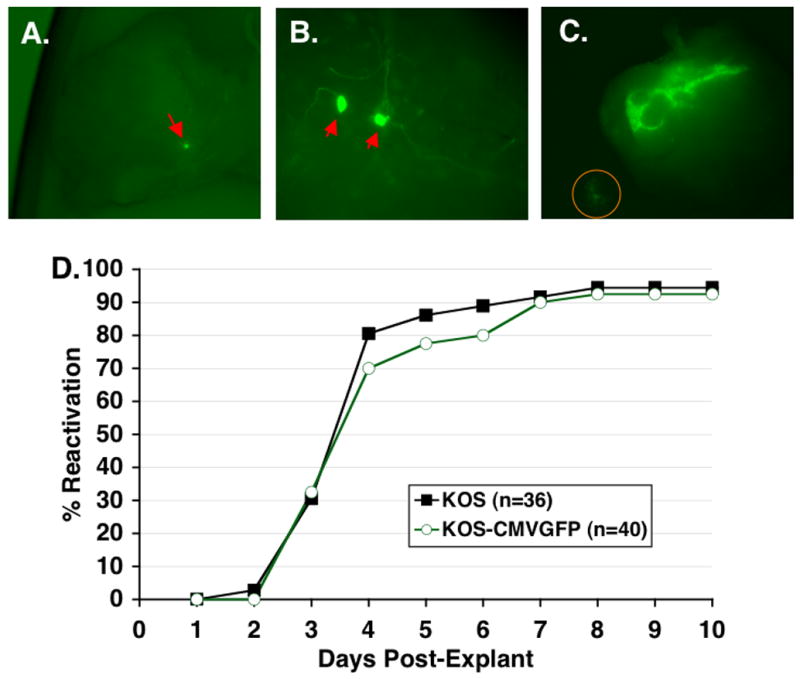
TG from mice infected 30 to 35 d p.i. with KOS or KOS-CMVGFP were cut into six to eight pieces and cocultured on Vero cell monolayers. Explant cultures of KOS-CMVGFP were examined by fluorescence microscopy at 100X (A and C) or 400X (B). (A) Explanted TG on day 1 p.e. A single GFP+ ganglionic cell is indicated by the red arrow. (B) Explanted TG on day 2 p.e. showing morphology of GFP+ cells. Red arrows point to apparent cell bodies of GFP+ ganglionic neurons. (C) Reactivating TG explant on day 4 post-explant. Orange circle shows GFP+ Vero cells. (D) Efficiencies of reactivation from explant cultures of TG latently infected with KOS and KOS-CMVGFP. Explant culture medium was assayed daily for the presence of infectious virus as indicated by cytopathic effects in indicator plates of Vero cells. Each point represents the cumulative percentage of reactivating TG from two experiments. The number (n) of TG per group is indicated.
Figure 7. Reactivation of KOS and KOS-CMVGFP in TG cell cultures.
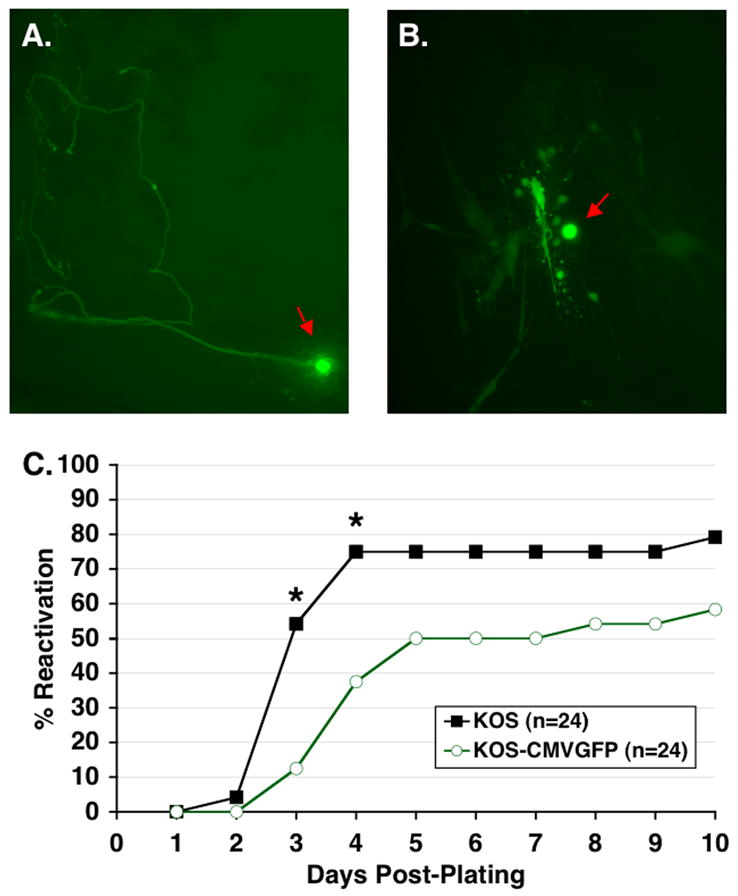
TG cell cultures were prepared by pooling TG infected with KOS-CMVGFP 30 to 35 d p.i., gently teasing them apart, treating with collagenase to generate single-cell suspensions, and distributing the resulting cell suspensions into 24-well plates. Cultures were examined by fluorescence microscopy at 100X at (A) day 1 or (B) day 4 post-plating. Apparent neuronal cell bodies are indicated by red arrows. (C) Efficiencies of reactivation of KOS and KOS-CMVGFP from TG cell cultures. TG cell cultures were prepared by pooling 12 TG per virus group as described above. Culture medium was assayed daily for the presence of infectious virus as indicated by cytopathic effects in indicator plates of Vero cells. Each point represents the cumulative percentage of reactivating wells from two experiments. Asterisks indicate significant differences between KOS and KOS-CMVGFP as determined by Fisher’s exact test. The number (n) of TG cell culture wells is indicated.
(i) Viral genome loads
The efficiency of establishment of latency was determined by measuring viral genome loads in individual explanted TG and in replicate samples of dissociated TG cells by competitive PCR (Halford and Schaffer, 2000). As shown in Fig. 5, KOS-CMVGFP established latency as efficiently as wild type virus as assayed in both explanted TG (2.3–2.5 X 102 genomes/100 ng TG DNA) and dissociated TG cells (1.1–1.2 X 103 genomes/100 ng TG DNA) [P=0.66 and 0.29, two tailed t test of data from explanted TG and dissociated TG cell samples, respectively]. Based on these findings, the presence of the EGFP cassette in the viral genome did not significantly affect the efficiency of establishment of latency.
(ii) Reactivation by explant co-cultivation
Examination of TG latently infected with KOS-CMVGFP by fluorescence microscopy immediately following explant revealed no detectable EGFP expression, indicating either that the EGFP expression cassette was silenced during latency or that expression of EGFP was below the limit of detection. EGFP expression in explanted TG was first detected 14 h post-explant (p.e.) and the intensity of single cell fluorescence increased through 24 h p.e. (Fig. 6A). High levels of EGFP expression were maintained thereafter. Notably, cells expressing EGFP appeared to be neurons as determined by cell morphology (i.e. large round cell bodies with processes extending outward) (Fig. 6B). Spread of fluorescence, and hence, infectious virus, from TG to the Vero cell monolayer was observed between days 2 and 10 p.e. (Fig. 6C). Only very rarely were GFP- plaques observed in the Vero cell monolayer suggesting loss of GFP expression during viral spread through the monolayer.
The kinetics and efficiency of reactivation of wild-type and EGFP-containing virus were assayed by explant cocultivation of latently infected TG (Fig. 6D). The mean time to initial detection of reactivated virus in culture medium was determined by averaging the time in days post-explant when infectious virus was first detected in each reactivation culture. The mean time (mean ± SD) to initial detection of reactivated virus in culture medium was similar for KOS (4.0 ± 1.2 days p.e.) and KOS-CMVGFP (4.2 ± 1.4 days p.e.) [P=0.49, two-sided t test]. KOS-CMVGFP also reactivated with wild-type frequency in that 34 of 36 (92%) explanted KOS-CMVGFP TG reactivated compared to 37 of 40 (94%) of KOS TG by 8 days p.e. [P=1.0, Fisher’s exact test].
(iii) Reactivation in TG cell cultures
Reactivation efficiencies were also assessed in TG cell cultures. An interesting feature of this assay is that differences in the kinetics and efficiency of reactivation observed by the TG explant assay are often enhanced in the TG cell culture assay (Balliet and Schaffer, 2006; Halford and Schaffer, 2001). As for explanted TG, examination of dissociated TG cells latently infected with KOS-CMVGFP by fluorescence microscopy immediately after plating revealed no detectable EGFP expression. EGFP expression was first detected at 8 h post-plating (p.p.) with peak fluorescence intensity observed on day 2 p.p. (Fig. 7A). In addition, the greatest number of single fluorescing neurons expressing EGFP (identified by their morphology) was observed on day 2 p.p. with 14 ± 4.0 GFP+ and 7.3 ± 3.1 GFP+ neurons/well (mean ± SD) in two independent experiments (n=12 wells/experiment), respectively. A steady reduction in the number of GFP+ neurons occurred after day 2 p.p., likely due to cell death. Notably, however, although 7–14 neurons expressed EGFP per well, only one to three multicell fluorescent foci (i.e. foci of reactivated virus) containing a single neuronal cell body were observed between days 2 and 10 p.p. (Fig. 7B). This finding indicates that not all neurons that initiate transcription of the viral genome are competent to complete the viral lytic cascade resulting in the production of infectious virus. Similar to explant cultures, very rarely were GFP− plaques observed suggesting loss of GFP expression during viral spread through the culture.
Although, based on morphology, the only cells expressing GFP following plating appeared to be neurons, immunofluorescence tests were performed to test this hypothesis using a monoclonal antibody that recognizes the neuron-specific β3 isoform of tubulin, TuJ1 (Ferreira and Caceres, 1992). Dissociated TG cells were plated in the presence of 200 μM acyclovir, an inhibitor of viral replication and spread that does not affect the activity of the HCMV MIEP. Cells were fixed and stained with the neuronal marker, TuJ1 on days 2 or 4 p.p. (Fig. 8). In these tests, all cells expressing EGFP were also TuJ1+. In no instance were EGFP+/TuJ1− cells observed in this test. These results are consistent with the long held view (Goodpasture, 1929) that ganglionic neurons are the cell type that harbors latent viral genomes (Maggioncalda et al., 1996; Mehta et al., 1995; Sawtell, 1997; Shimeld, Efstathiou, and Hill, 2001; Stevens, 1989).
Figure 8. Identification of GFP+ cells in TG cell cultures by immunofluorescence.
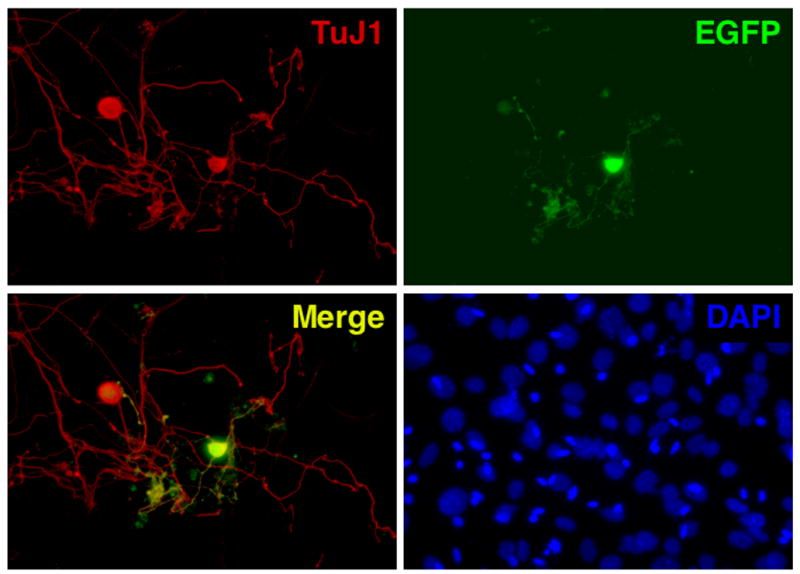
TG cell cultures latently infected with KOS-CMVGFP were prepared as described in Fig. 7 and plated on coverslips in the presence of 200 μM acyclovir to repress reactivation. On days 2 (data not shown) and 4 p.p., cells were washed, fixed, permeabilized, and probed with anti-TuJ1 antibody, which is specific for post-mitotic neurons. Cultures were examined by fluorescence microscopy at 400X for staining with TuJ1 antibody and for EGFP expression. Staining of cellular DNA with DAPI was performed to confirm specificity of TuJ1 antibody.
Consistent with previous findings, assays for viral reactivation in TG cell cultures indicated that the time of initial detection of reactivated virus was similar in these cultures and in explant cultures for both viruses (compare Fig. 6D with 7C) (Balliet and Schaffer, 2006). In TG cell culture, however, KOS reactivated more rapidly than KOS-CMVGFP (Fig. 7C). On days 3 and 4 p.p., 13 of 24 (54%) and 18 of 24 (75%) of the KOS cultures, respectively, reactivated compared to 3 of 24 (12.5%) and 9 of 24 (37.5%) of KOS-CMVGFP cultures. These differences in reactivation frequencies were statistically significant [P=0.004 and 0.02, respectively]. Consistent with this observation, the mean time to reactivation of KOS was 3.6 ± 1.65 days p.p. compared to 4.8 ± 1.9 days p.p. for KOS-CMVGFP, representing a 1.2 day delay relative to KOS. Between days 5 and 10 p.p., the percentage of KOS cultures that reactivated was greater than the percentage of KOS-CMVGFP cultures culminating with 19 of 24 (79%) of KOS cultures reactivating by 10 days p.p. compared to 14 of 24 (58%) of KOS-CMVGFP cultures. Although a trend towards KOS-CMVGFP reactivating somewhat less efficiently than KOS was evident, the differences in reactivation frequencies were not statistically significant [P=0.485 (day 5 p.p.) to 0.65 (day 10 p.p.)] reflecting the limited power of the test.
Collectively, these findings indicate that EGFP expression in latently infected ganglionic cells is a reliable marker of neurons that are able to activate viral lytic promoters, potentially culminating in viral reactivation. They also show that insertion of the EGFP cassette between the UL26/26.5 and UL27 genes affects the kinetics and efficiency of reactivation from latency minimally in the TG explant model of reactivation but moderately in the more rigorous TG cell culture model.
Discussion
To examine the nature of virus-host interactions and to study the mechanisms underlying viral latency and reactivation, a recombinant HSV-1 virus that expresses EGFP during productive infection (KOS-CMVGFP) was generated and characterized. The results of these tests demonstrate that insertion of the EGFP expression cassette between UL26/26.5 and UL27 genes did not grossly impair viral replication in vitro or in vivo nor did it affect the efficiency of establishment of latency. Only under rigorous ex vivo conditions used to assay viral reactivation was a phenotype for KOS-CMVGFP apparent. These studies also demonstrate the usefulness of the fluorescent reporter as an early marker of neurons capable of supporting HSV-1 reactivation.
Strategy for constructing KOS-CMVGFP
The strategy for the construction of the EGFP-expressing recombinant virus was based on two criteria. First, the expression cassette must constitutively express EGFP to high levels. Second, insertion of the EGFP expression cassette into the viral genome must not adversely affect the functions of viral genes or cis-acting elements.
We chose the HCMV-MIEP because of its high level of constitutive expression and because its activity is unaffected by common antiviral compounds (e.g. acyclovir). Specifically, we selected the HCMV-MIEP from the expression vector pCB6 (Brewer and Roth, 1991). This choice was based on findings from transient transfection assays comparing the levels of viral and cellular proteins transiently expressed from pCB6 and the widely used, commercially available expression vector, pcDNA3 (Invitrogen, Carlsbad, CA). Consistently, 2- to 3-fold higher levels of viral and cellular proteins were expressed in cells transiently transfected with pCB6 constructs than in cells transfected with pcDNA3 constructs (J.W.B. and P. Bates, unpublished observations). This is likely due to the fact that the HCMV-MIEP in pCB6 is considerably larger than the IEP in pcDNA3. In addition, to ensure that transcripts from the EGFP expression cassette were efficiently processed and polyadenylated, we appended the highly efficient and well-characterized SV40 late polyadenylation signal to the EGFP ORF (Hans and Alwine, 2000, and references therein).
We chose to insert the EGFP expression cassette between the UL26/26.5 and UL27 genes because (i) this region of the viral genome appears to be transcriptionally inert (Bzik et al., 1984; Holland et al., 1984; Liu and Roizman, 1991) and (ii) insertion of the ICP0 gene at this site resulted in stable expression of functional ICP0 in mutant virus 171 (Cai et al., 1993). Although in that study insertion at this location was stable, the EGFP cassette in our recombinant virus was continually lost at a low but measurable rate. The difference in stabilities of the inserted genes in 171 (ICP0) and KOS-CMVGFP (EGFP) is likely due to differences in the effects of insertion of the cassette on the fitness of the recombinant virus. Insertion of the ICP0 gene conferred a selective advantage on 171 relative to the parental ICP0− double mutant virus whereas insertion and expression of the nonviral EGFP gene likely conferred a selective disadvantage on KOS-CMVGFP relative to the wild-type virus. Loss of EGFP expression in a recombinant HSV-1 virus is not unique to KOS-CMVGFP. Infrequent loss of EGFP expression was also observed when an EGFP expression cassette was inserted between UL53 and UL54 in the KOS genome (Foster, Chouljenko, and Kousoulas, 1999). In this case, however, while the EGFP cassette was retained, expression of EGFP was repressed (T. Foster, personal communication).
Effects of the EGFP expression cassette on acute infection of mice
Tear film titers of mice infected with KOS-CMVGFP were slightly but significantly lower than wild-type virus indicating that insertion of the EGFP expression cassette into the viral genome had a minor but detectable effect on virus replication during the acute phase of infection. Like wild-type virus, however, titers of KOS-CMVGFP in tear film exhibited a biphasic curve. The first peak is thought to consist of virus produced primarily in the eye, the site of primary infection. The second peak is thought to, but has not been demonstrated directly, consist of “round trip” virus produced in the TG and transported back to the eye and periocular tissue.
Two findings from this study support the round trip model. First, the finding that peak spread and intensity of fluorescence of infected ocular cells correlated with peak viral titers on day 2 but not on day 6 p.i., the timing and occurrence of the second peak of infectious virus, strongly suggesting that virus produced in TG is transported to and accumulates in the eye. Second, after a steady increase in the percentage of GFP− virus in tear film during the first 4 days of infection, an appreciable drop in the percentage of GFP− virus was observed between days 5 and 6 p.i. indicating an increase in the amount of GFP+ KOS-CMVGFP virus. Because it is improbable that revertant GFP− viruses incorporated the EGFP expression cassette by genetic recombination, the additional GFP+ virus was likely produced at a distal site.
The most likely distal source of virus is cells of the TG. Consistent with the role of ganglionic cells as the source of GFP+ virus found in the eye at day 6 p.i., a correlation exists between (i) the number of GFP+ HSV-1-infected cells in the TG and the second peak of virus in tear film ((Shimeld, Efstathiou, and Hill, 2001); J.W.B. and P.A.S., unpublished observations), and (ii) the time of peak titers of virus in TG and the second peak of virus in tear film (Balliet and Schaffer, 2006).
During the course of acute infection, significant spread of virus from the eyes to periocular tissue and skin of the snout and nose was observed, demonstrating the usefulness of KOS-CMVGFP as a tool for studying zosteriform spread of HSV-1 in mice. Summers et al. have elegantly demonstrated zosteriform spread of virus from eyes to periocular tissue using recombinant HSV-1 viruses expressing luciferase (Summers, Margolis, and Leib, 2001); however, virus spread to the skin of the snout and nose following ocular inoculation has not been described previously. While mechanical spread of virus by transfer of tear film by scratching cannot be ruled out, zosteriform spread is likely a significant mechanism of virus spread to these distal sites. This notion is supported by the observations that (i) fluorescent foci consisted of expanding circles rather than scratch-induced streaks; (ii) EGFP expression in the skin of the snout and nose was observed consistently at 4 or 5 d p.i. Thus, the delay in the appearance of virus reflected the amount of time required for transport of virus from the eye to the TG and then to epidermal cells of the snout and nose; (iii) uniform distribution of fluorescent foci on the snout was evident as shown in Fig. 3 and virus spread was restricted only to sites innervated by the maxillary nerve branch; and (iv) virus replication and spread from central foci appeared to occur beneath the skin.
Effects of the EGFP expression cassette on the establishment and reactivation of latency
KOS-CMVGFP established latency in ICR mice as efficiently as wild-type virus, consistent with our findings in C57BL/6 and BALB/c mice (Halford, Balliet, and Gebhardt, 2004). Moreover, no significant difference in the kinetics or efficiency of reactivation in latently infected, explanted TG between KOS-CMVGFP and wild type virus was observed. Using a more sensitive ex vivo model of reactivation, however, KOS-CMVGFP reactivation was delayed compared to wild-type virus on days 3 and 4 post-plating of TG cells and exhibited a moderate 33 and 26% reduction in reactivation efficiency on days 5 and 10 post-plating, respectively. Thus, the EGFP expression cassette had a noticeable effect on viral reactivation under stringent assay conditions (i.e. in the TG cell culture model).
Regardless of the reactivation model, GFP fluorescence was not observed during latency, but was observed 8 to 14 h following plating of TG cells or explant of TG, respectively. This observation is consistent with previous findings that the HCMV-MIEP is silenced during viral latency (Ecob-Prince et al., 1995; Lachmann, Brown, and Efstathiou, 1996; Wilson et al., 1999) although the degree of repression may be dependent on where the promoter/reporter gene cassette is inserted into the viral genome (Palmer et al., 2000). Notably, EGFP expression in both ex vivo models preceded detection of infectious virus by more than 24 h and can thus serve as a sensitive marker of latently infected neurons some of which are in the early stages of initiating viral lytic gene expression, which by definition, precedes reactivation. Moreover, when first detected, EGFP expression was localized to sensory neurons of the TG, supporting the previously published conclusion that HSV-1 latency and reactivation occur only in sensory neurons (Maggioncalda et al., 1996; Mehta et al., 1995; Sawtell, 1997; Shimeld, Efstathiou, and Hill, 2001; Stevens, 1989). It is intriguing that on average 7 to 14 neurons out of an average of 500 neurons per well (Halford, Gebhardt, and Carr, 1996; Halford and Schaffer, 2001) expressed EGFP within the first three days post-plating in TG cell cultures. Of these, only one to three EGFP+ neurons were able to support reactivation as determined by spread of fluorescence from a single GFP+ neuron to surrounding support cells. Thus, although transcription of the viral genome is initiated in a number of latently infected neurons, only a subset of these neurons is competent to support the extensive program of viral gene expression that leads to reactivation. The reason for this is currently unknown but may be due to differences in the activities of cellular stress-induced signaling pathways induced or repressed in cells that support or do not support reactivation. Alternatively, only distinct types of sensory neurons may be competent to support efficient viral reactivation from latency (LaVail, Johnson, and Spencer, 1993; Margolis, Dawson, and Lavail, 1992; Yang, Voytek, and Margolis, 2000).
In this study KOS-CMVGFP yielded novel insights into acute phase replication and viral reactivation from latency. In addition, it has proven useful in understanding mouse strain-specific resistance to HSV-1 (Halford, Balliet, and Gebhardt, 2004). Currently, we are testing compounds that inhibit or induce viral reactivation using TG cell cultures latently infected with KOS-CMVGFP. We are also examining the basis for the reduced pathogenicity and impaired reactivation efficiency of the DoriL-ILR mutation in the KOS-CMVGFP background (Balliet and Schaffer, 2006), a virus containing point mutations in HSV-1 oriL that inhibit initiation of viral DNA replication from this origin (Balliet et al., 2005).
Materials and Methods
Cells and viruses
Vero [ATCC CCL-81(Yasmura and Kawakita, 1963)] and 293 cells [ATCC CRL-1573 (Graham et al., 1977)] were propagated in Dulbecco's modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), penicillin (100 units/ml), streptomycin (100 μg/ml), and 2 mM L-glutamine. The wild-type virus used in this study was HSV-1 strain KOS at p11 from original isolation (Smith, 1964).
Plasmids
Cloning vectors used in the construction of plasmid pGFP53K, which contains the backbone for introducing the EGFP expression cassette (Fig. 1E) into the viral genome by marker transfer (Sacks et al., 1985), were generated as follows. Plasmid pBUC-2 was generated by ligation of annealed oligonucleotides OJB5 [5’-AAT TCG TCG ACA GGC CTA GAT CTT CTA GAT ACG TAT GGC CAG AGC TCA-3] and OJB6 [5’-AGC TTG AGC TCT GGC CAT ACG TAT CTA GAA GAT CTA GGC CTG TCG ACG-3’] into pUC-8 digested with EcoRI and HindIII. In this way, the polylinker of pUC8 was replaced with a different polylinker containing the following unique restriction enzyme cleavage sites (5’ to 3’): EcoRI, SalI, StuI, BglII, XbaI, SnaBI, MscI, SacI, and HindIII. Likewise, plasmid pBUC-3 was generated by ligation of annealed oligonucleotides OJB8 [5’-AAT TCA GAT CTT CTA GAA GGC CTT GGC CAG TCG ACA-3] and OJB9 [5’-AGC TTG TCG ACT GGC CAA GGC CTT CTA GAA GAT CTG-3’] into pUC-8 digested with EcoRI and HindIII. The polylinker of pBUC-3 contains the following unique restriction enzyme cleavage sites (5’ to 3’): EcoRI, BglII, XbaI, StuI, MscI, SalI, and HindIII.
Plasmid pGFP53K was constructed as follows. The CMV promoter from plasmid pEGFP-N1 (Clontech, San Francisco, CA) was removed to produce plasmid pEGFPΔCMV by self-ligation after digestion with AseI and NheI and blunt-ending both ends using Klenow. A 0.75 kb TspRI (blunt-ended with T4 DNA polymerase) to XmnI fragment containing the CMV promoter from plasmid pCB6 (Brewer and Roth, 1991) was ligated into pEGFP CMV digested with SmaI to generate plasmid pEGFP6. Plasmid pSK (Cai et al., 1993) was digested with SalI and FspI (nt 52,192-53,150, Fig. 1D and E) to generate a 0.96 kb fragment which was cloned into plasmid pEGFP6 digested with ApaI (blunt-ended with T4 DNA polymerase) and SalI to generate plasmid pUL26EGFP6. A 0.27kb XbaI to EcoRI (blunt-ended with Klenow) containing the SV40 late polyadenylation signal from plasmid pUPAS (Carswell and Alwine, 1989; Schek, Cooke, and Alwine, 1992) was ligated into pBUC-3 digested with XbaI and StuI. This plasmid was then digested with MscI and SalI and ligated with a 2.1 kb DraI to SalI (nt 52,764-54,826, Fig. 1D and E) fragment from plasmid pSK to generate plasmid pBUCpAUL27. Plasmid pGFP53K was assembled by three-way ligation of a 2.5 kb SalI to XbaI fragment containing HSV-1 UL26 sequences, the CMV promoter, and the EGFP gene from plasmid pUL26EGFP6, and a 2.3 kb XbaI to HindIII fragment containing the SV40 polyadenylation signal and HSV-1 UL27 sequences from plasmid pBUCpAUL27 into vector pBUC-2 digested with SalI and HindIII.
Construction of mutant and rescuant viruses
Recombinant KOS-CMVGFP virus was generated by marker transfer of a fragment containing the EGFP expression cassette in pGFP53K into infectious wild-type HSV-1 DNA (Sacks et al., 1985). Specifically, 1 X 106 293 cells in a 100 mm dish were co-transfected with the 4.8 kb SalI to SalI fragment (Fig. 1E) from pGFP53K and infectious KOS DNA using Lipofectamine 2000 (Invitrogen) as per the manufacturer’s instructions. Transfected monolayers were frozen at −80°C 48 h after transfection. Cultures were later thawed, cells were scraped into medium, cell suspensions were transferred into tubes, sonicated, and clarified by centrifugation (800 x g for 10 min). Serial dilutions of infectious virus in the resulting supernatant fluid were used to infect Vero cell monolayers (2 X 105 cells) in 35 mm dishes for 1 h. Vero cell monolayers were washed once with PBS and overlaid with 2% methylcellulose. Four days post-infection, plaques were examined by fluorescence microscopy with a Nikon Eclipse TE300 inverted fluorescence microscope (Nikon Instruments, Lewisville, TX) and individual GFP+ plaque isolates were picked, amplified, and screened by Southern blotting (described below) for the presence of the EGFP expression cassette. Among several candidate KOS-CMVGFP viruses, a single virus was plaque purified five times and used in functional studies.
Southern blot analysis
Southern blots were performed as described previously (Balliet et al., 2005). Briefly, 8 X 105 Vero cells in 60 mm dishes were infected at a multiplicity of 5 PFU per cell. When cytopathic effects were generalized, cells were scraped into medium, cell suspensions were transferred to tubes, and centrifuged. Cell pellets were lysed and the DNA in the lysate was isolated. Three micrograms of total infected cell DNA was digested with restriction enzymes. The resulting fragments were separated on 0.8% agarose gels and transferred to nylon membranes, prehybridized for 1 h at 68°C in ExpressHyb solution (Clontech, San Francisco, CA) and hybridized for 1 h at 68°C using 32P-labeled random-primed probes specific for the DraI-SalI fragment of HSV-1 DNA (nt 52,764 to 54,826) or for the AgeI-XbaI fragment of plasmid pEGFP-N1, which contains the EGFP ORF. The blots were washed as per the manufacturer’s instructions and the hybridized membrane was exposed on a PhosphorImager cassette (Molecular Dynamics, Sunnyvale, CA) overnight.
Viral growth curves
Vero cells (2 X 105) were plated in 35 mm dishes. The cells were counted 24 h after plating and infected at a calculated multiplicity of 0.1 PFU per cell of KOS or KOS-CMVGFP. Following 1 h adsorption at 37°C, the inoculum was removed, cells were washed 3X with PBS, and growth medium was added back to each culture. The actual titers of viral inocula were determined by standard plaque assays on Vero cell monolayers and are shown as the 0 h time point in Fig. 2. Viral inocula did not differ by more than 2-fold between viruses. Infected monolayers were frozen at −80°C at the indicated times p.i. Cultures were later thawed, cells were scraped into medium, cell suspensions were transferred into tubes, sonicated, and clarified by centrifugation (800 x g for 10 min). Levels of infectious virus in supernatant fluids were determined by standard plaque assays on Vero cell monolayers.
Inoculation of mice
Five to six week old (~30 g) male ICR mice (Harlen Sprague-Dawley, Indianapolis, IN) were handled in accordance with The Guide for the Care and Use of Laboratory Animals (NIH Publication no. 85-23, revised 1996) and the guidelines of the Institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center. Mice were distributed randomly into two groups and anesthetized by intraperitoneal (i.p.) administration of xylazine (9 mg/kg of body weight) and ketamine (100 mg/kg). Mouse corneas were scarified with a 26-gauge needle, tear film was blotted, and 2 X 105 PFU of virus in 3 μl of complete DMEM was placed on each eye as described previously (Halford and Schaffer, 2000). The titers of virus in inocula were confirmed by standard plaque assays on Vero cell monolayers and were found to differ by less than 2-fold.
Analysis of viral replication and spread in mice
Examination of mice by fluorescence microscopy was performed as follows. Two mice infected with KOS-CMVGFP were anesthetized at the same time each day from 1 through 11 d p.i. Images of infected areas of mice were captured at 40X magnification with a Nikon Eclipse TE300 inverted fluorescence microscope, photographed with an RT Slider digital camera (Diagnostic Instruments, Sterling Heights, MI) and assembled with Photoshop software (Adobe Systems, Inc., Mountain View, CA).
To determine viral titers in tear film, tear film samples were collected from both eyes using a single cotton-tipped applicator. The cotton tip was transferred to 500 μl complete DMEM and frozen at −80°C. Frozen samples were later thawed, thoroughly mixed, and infectious virus was quantified by standard plaque assays on Vero cell monolayers. The total number of PFU/cotton tip was determined and divided by 2 to calculate the average viral titer per eye.
Measurement of viral genome loads in TG by competitive PCR
The competitive PCR assay used in these tests has been described previously (Halford and Schaffer, 2000). Briefly, total DNA was isolated from individual latently infected TG harvested 30 to 35 days post-inoculation using the Qiamp DNA mini kit (Qiagen, Hilden, Germany) as per the manufacturer’s instructions. Oligonucleotide primers specific for the HSV-1 ribonucleotide reductase (RR) gene (UL39) and competitor templates were used, amplifying 243- and 322-bp fragments, respectively, from each template. Included in each assay were control samples containing known numbers of viral genomes. The PCR products were vacuum slot blotted in duplicate, hybridized to oligonucleotide probes specific for either HSV RR or competitor templates, and the relative yields of the two PCR products were determined by PhosphorImager analysis. A standard curve was plotted as the ratio of HSV RR to competitor as a function of the number of viral genomes. The viral genome load per TG was calculated as the number of genomes per 0.1 μg of TG DNA.
Measurement of ex vivo reactivation efficiency
To measure reactivation by explant cocultivation, mice were euthanized 30 to 35 days post-inoculation by CO2 asphyxiation. Latently infected TG were removed immediately, cut into eight equal-sized pieces, and cocultivated with 3 X 104 Vero cells in 1.5 ml of complete DMEM per well in 24 well plates. Each day post-explant (p.e.), 150 μl of medium was removed and assayed for the presence of infectious virus. Fresh complete DMEM (500 μL) was added to TG explant cultures every third day. The presence of infectious virus and hence, reactivation, was detected by transferring the 150 μl sample of explant medium to fresh Vero cell monolayers and scoring daily for cytopathic effects.
Measurement of reactivation in primary, latently infected TG cell cultures was performed as described previously (Halford, Gebhardt, and Carr, 1996; Halford and Schaffer, 2001). Briefly, on day 30 post-inoculation, latently infected mice were euthanized and TG were removed (n=3 mice or 6 TG per virus). Individual TG were teased apart longitudinally using a scalpel. All 6 teased TG latently infected with each virus were pooled, incubated in 1.2 ml of 0.1% collagenase (Sigma) in HBSS for 40 min at 37°C, and then 7 ml TG growth medium (MEM supplemented with 10% FCS, 0.15% HCO3−, penicillin (100 units/ml), streptomycin (100 μg/ml), 2 mM L-glutamine, and 10 ng/ml NGF) was added. Dissociated TG cells were washed three times, resuspended in TG medium, and plated in 24 well collagen-coated plates seeded the previous day with 3 X 104 uninfected TG feeder cells. Six TG yield 12 ml of TG cell suspension, which was plated in a 24 well plate at 1 ml/well, or one half TG/well. TG cell cultures were tested daily for the presence of infectious virus as described for explant cultures. The mean time to detection of reactivated virus was performed as described for TG explants.
Images of ex vivo cultures were captured as described above. Magnification is indicated in the legends for the figures.
Immunofluorescence of TG cell cultures
At 30 to 35 d p.i., TG from mice infected with KOS-CMVGFP were excised and plated on collagen-coated 12-mm coverslips with 3 x 104 uninfected TG feeder cells per well of a 24 well plate, in the presence of 200 μM acyclovir. The cultures were incubated at 37°C with 5% CO2 for two or four d p.p. All subsequent steps were carried out at room temperature. The cultures were washed twice with PBS, fixed with 4% paraformaldehyde in PBS (pH 7.4) for 15 minutes, washed with PBS for 5 minutes, then blocked and permeabilized in block solution (PBS; 5% goat serum, 0.1% Triton X-100) for 1 h TuJ1 monoclonal antibody (Covance, Berkeley, CA) was applied in block solution at a dilution of 1:5000 for 1 h. The cultures were washed with PBS three times for 5 minutes. Rhodamine Red-X-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA) was diluted 1:300 in 1.5% goat serum in PBS and incubated for 1 hr in a dark, humidified chamber. The cultures were washed in PBS three times for 5 minutes, stained with DAPI, rinsed with distilled water and mounted on glass slides with ProLong Antifade (Molecular Probes, Eugene, Oregon).
Statistics
The statistical tests used in this study are those recommended by Richardson and Overbaugh (Richardson and Overbaugh, 2005). Data are presented as the mean ± standard error of the mean (SEM). Two-way ANOVA was used to determine the significance of differences among experimental groups at multiple time points. Linear regression was used to evaluate the reliability of the standard curves for competitive PCR. The significance of differences in mean time to reactivation was determined by two-tailed t test. The significance of differences in reactivation efficiencies were determined by Fisher’s exact test.
Acknowledgments
We acknowledge the generosity of Holly Hans and James Alwine for providing plasmid pUPAS. We thank Tim Foster for communication of unpublished results. We also thank Jonathan Min, William Fay vonZagorski and A. Scott McWilliams for excellent technical assistance. We are indebted to past and present members of the Schaffer laboratory for helpful discussions and ideas.
This work was supported by NIAID research grant R01 AI28537 and NINDS Program Project grant P01 NS35138 from the Public Health Service. J.W.B. was supported, in part, by National Research Service Award F32 AI10557.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balliet JW, Min JC, Cabatingan MS, Schaffer PA. Site-directed mutagenesis of large DNA palindromes: construction and in vitro characterization of herpes simplex virus type 1 mutants containing point mutations that eliminate the oriL or oriS initiation function. J Virol. 2005;79(20):12783–97. doi: 10.1128/JVI.79.20.12783-12797.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balliet JW, Schaffer PA. Point mutations in herpes simplex virus type 1 oriL, but not in oriS, reduce pathogenesis during acute infection of mice and impair reactivation from latency. J Virol. 2006;80(1):440–50. doi: 10.1128/JVI.80.1.440-450.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer CB, Roth MG. A single amino acid change in the cytoplasmic domain alters the polarized delivery of influenza virus hemagglutinin. J Cell Biol. 1991;114(3):413–21. doi: 10.1083/jcb.114.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzik DJ, Fox BA, DeLuca NA, Person S. Nucleotide sequence specifying the glycoprotein gene, gB, of herpes simplex virus type 1. Virology. 1984;133(2):301–14. doi: 10.1016/0042-6822(84)90397-0. [DOI] [PubMed] [Google Scholar]
- Cai W, Astor TL, Liptak LM, Cho C, Coen DM, Schaffer PA. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J Virol. 1993;67(12):7501–7512. doi: 10.1128/jvi.67.12.7501-7512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carswell S, Alwine JC. Efficiency of utilization of the simian virus 40 late polyadenylation site: effects of upstream sequences. Mol Cell Biol. 1989;9(10):4248–58. doi: 10.1128/mcb.9.10.4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173(1 Spec):33–8. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Ecob-Prince MS, Hassan K, Denheen MT, Preston CM. Expression of beta-galactosidase in neurons of dorsal root ganglia which are latently infected with herpes simplex virus type 1. J Gen Virol. 1995;76 (Pt 6):1527–32. doi: 10.1099/0022-1317-76-6-1527. [DOI] [PubMed] [Google Scholar]
- Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc Natl Acad Sci U S A. 2002;99(2):978–83. doi: 10.1073/pnas.022301899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Caceres A. Expression of the class III beta-tubulin isotype in developing neurons in culture. J Neurosci Res. 1992;32(4):516–29. doi: 10.1002/jnr.490320407. [DOI] [PubMed] [Google Scholar]
- Foster TP, Chouljenko VN, Kousoulas KG. Functional characterization of the HveA homolog specified by African green monkey kidney cells with a herpes simplex virus expressing the green fluorescence protein. Virology. 1999;258(2):365–74. doi: 10.1006/viro.1999.9743. [DOI] [PubMed] [Google Scholar]
- Gebhardt BM, Halford WP. Evidence that spontaneous reactivation of herpes virus does not occur in mice. Virol J. 2005;2:67. doi: 10.1186/1743-422X-2-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodpasture EW. Herpetic infection with special reference to involvement of the nervous system. Journal Of Clinical Endocrinology. 1929;8(2):223–243. doi: 10.1097/00005792-199303000-00006. [DOI] [PubMed] [Google Scholar]
- Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36(1):59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- Halford WP, Balliet JW, Gebhardt BM. Re-evaluating natural resistance to herpes simplex virus type 1. J Virol. 2004;78(18):10086–95. doi: 10.1128/JVI.78.18.10086-10095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Gebhardt BM, Carr DJ. Mechanisms of herpes simplex virus type 1 reactivation. J Virol. 1996;70(8):5051–60. doi: 10.1128/jvi.70.8.5051-5060.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Schaffer PA. Optimized viral dose and transient immunosuppression enable herpes simplex virus ICP0-null mutants to establish wild-type levels of latency in vivo. J Virol. 2000;74(13):5957–67. doi: 10.1128/jvi.74.13.5957-5967.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Schaffer PA. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J Virol. 2001;75(7):3240–9. doi: 10.1128/JVI.75.7.3240-3249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hans H, Alwine JC. Functionally significant secondary structure of the simian virus 40 late polyadenylation signal. Mol Cell Biol. 2000;20(8):2926–32. doi: 10.1128/mcb.20.8.2926-2932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland LE, Sandri-Goldin RM, Goldin AL, Glorioso JC, Levine M. Transcriptional and genetic analyses of the herpes simplex virus type 1 genome: Coordinates 0.29 to 0.45. J Virol. 1984;49(3):947–959. doi: 10.1128/jvi.49.3.947-959.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann RH, Brown C, Efstathiou S. A murine RNA polymerase I promoter inserted into the herpes simplex virus type 1 genome is functional during lytic, but not latent, infection. J Gen Virol. 1996;77 (Pt 10):2575–82. doi: 10.1099/0022-1317-77-10-2575. [DOI] [PubMed] [Google Scholar]
- LaVail JH, Johnson WE, Spencer LC. Immunohistochemical identification of trigeminal ganglion neurons that innervate the mouse cornea: relevance to intercellular spread of herpes simplex virus. J Comp Neurol. 1993;327(1):133–40. doi: 10.1002/cne.903270111. [DOI] [PubMed] [Google Scholar]
- Liu FY, Roizman B. The promoter, transcriptional unit, and coding sequence of herpes simplex virus 1 family 35 proteins are contained within and in frame with the UL26 open reading frame. J Virol. 1991;65(1):206–12. doi: 10.1128/jvi.65.1.206-212.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker GD, Bardill JP, Prior JL, Pica CM, Piwnica-Worms D, Leib DA. Noninvasive bioluminescence imaging of herpes simplex virus type 1 infection and therapy in living mice. J Virol. 2002;76(23):12149–61. doi: 10.1128/JVI.76.23.12149-12161.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggioncalda J, Mehta A, Su YH, Fraser NW, Block TM. Correlation between herpes simplex virus type 1 rate of reactivation from latent infection and the number of infected neurons in trigeminal ganglia. Virology. 1996;225(1):72–81. doi: 10.1006/viro.1996.0576. [DOI] [PubMed] [Google Scholar]
- Margolis TP, Dawson CR, Lavail JH. Herpes simplex viral infection of the mouse trigeminal ganglion. Ophthalmol Visual Sci. 1992;33:259–267. [PubMed] [Google Scholar]
- Margolis TP, Sedarati F, Dobson AT, Feldman LT, Stevens JG. Pathways of viral gene expression during acute neuronal infection with HSV-1. Virol. 1992;189:150–160. doi: 10.1016/0042-6822(92)90690-q. [DOI] [PubMed] [Google Scholar]
- Mehta A, Maggioncalda J, Bagasra O, Thikkavarapu S, Saikumari P, Valyi-Nagy T, Fraser NW, Block TM. In situ DNA PCR and RNA hybridization detection of herpes simplex virus sequences in trigeminal ganglia of latently infected mice. Virol. 1995;206:633–640. doi: 10.1016/s0042-6822(95)80080-8. [DOI] [PubMed] [Google Scholar]
- Palmer JA, Branston RH, Lilley CE, Robinson MJ, Groutsi F, Smith J, Latchman DS, Coffin RS. Development and optimization of herpes simplex virus vectors for multiple long-term gene delivery to the peripheral nervous system. J Virol. 2000;74(12):5604–18. doi: 10.1128/jvi.74.12.5604-5618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys J. 1997;73(5):2782–90. doi: 10.1016/S0006-3495(97)78307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson BA, Overbaugh J. Basic statistical considerations in virological experiments. J Virol. 2005;79(2):669–76. doi: 10.1128/JVI.79.2.669-676.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks WR, Greene CC, Aschman DP, Schaffer PA. Herpes simplex virus type 1 ICP27 is an essential regulatory protein. J Virol. 1985;55(3):796–805. doi: 10.1128/jvi.55.3.796-805.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM. Comprehensive quantification of herpes simplex virus latency at the single-cell level. J Virol. 1997;71(7):5423–31. doi: 10.1128/jvi.71.7.5423-5431.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schek N, Cooke C, Alwine JC. Definition of the upstream efficiency element of the simian virus 40 late polyadenylation signal by using in vitro analyses. Mol Cell Biol. 1992;12(12):5386–93. doi: 10.1128/mcb.12.12.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimeld C, Efstathiou S, Hill T. Tracking the spread of a lacZ-tagged herpes simplex virus type 1 between the eye and the nervous system of the mouse: comparison of primary and recurrent infection. J Virol. 2001;75(11):5252–62. doi: 10.1128/JVI.75.11.5252-5262.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KO. Relationship between the envelope and the infectivity of herpes simplex virus. Proc Soc Exp Biol Med. 1964;115:814–816. doi: 10.3181/00379727-115-29045. [DOI] [PubMed] [Google Scholar]
- Soboleski MR, Oaks J, Halford WP. Green fluorescent protein is a quantitative reporter of gene expression in individual eukaryotic cells. Faseb J. 2005;19(3):440–2. doi: 10.1096/fj.04-3180fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JG. Herpes simplex virus latency analyzed by in situ hybridization. Curr Top Microbiol Immunol. 1989;143:1–8. doi: 10.1007/978-3-642-74425-9_1. [DOI] [PubMed] [Google Scholar]
- Summers BC, Margolis TP, Leib DA. Herpes simplex virus type 1 corneal infection results in periocular disease by zosteriform spread. J Virol. 2001;75 (11):5069–75. doi: 10.1128/JVI.75.11.5069-5075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SP, Yeomans DC, Bender MA, Lu Y, Goins WF, Glorioso JC. Antihyperalgesic effects of infection with a preproenkephalin-encoding herpes virus. Proc Natl Acad Sci U S A. 1999;96(6):3211–6. doi: 10.1073/pnas.96.6.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Voytek CC, Margolis TP. Immunohistochemical analysis of primary sensory neurons latently infected with herpes simplex virus type 1. J Virol. 2000;74(1):209–17. doi: 10.1128/jvi.74.1.209-217.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasmura Y, Kawakita Y. Research into SV 40 by tissue culture. Nippon Rinsho. 1963;21:1201–1205. [Google Scholar]