

Abstract
Several studies have shown that PKA-mediated phosphorylation of IP3R1 at serines S1588 and S1755 enhances the receptor's ability to mobilize Ca2+. In contrast, much less is known about whether Ca2+ mobilization via IP3R2 and IP3R3 is regulated by PKA. We report here that IP3R2 is only very weakly phosphorylated in response to PKA activation and is probably not a physiological substrate for this kinase. IP3R3, however, is known to be phosphorylated by PKA at three sites (S916, S934 and S1832) and, thus, we examined how phosphorylation of these sites affects Ca2+ mobilization in DT40-3KO cells stably expressing either exogenous wild-type or mutant IP3R3s; an antibody raised against phospho-serine 934 of IP3R3 was used to demonstrate that the exogenous IP3R3s are strongly phosphorylated in response to PKA activation. Surprisingly, our data show that IP3R3-mediated Ca2+ mobilization is unaffected by phosphorylation of S916, S934 and S1832. In contrast, phosphorylation of exogenous IP3R1 (monitored with an antibody against phospho-serine 1755) enhances Ca2+ mobilization, indicating that DT40-3KO cells have the capacity to respond to phosphorylation of IP3Rs. Overall, these data suggest that modification of Ca2+ flux may not be the primary effect of IP3R3 phosphorylation by PKA.
Keywords: Inositol 1,4,5-trisphosphate receptor; cAMP-dependent protein kinase; phosphorylation; calcium mobilization; DT40
1. Introduction
Inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) are a family of proteins which form Ca2+ channels in endoplasmic reticulum (ER) membranes and govern Ca2+ release from this organelle by opening in response to IP3 and Ca2+ binding [1-3]. In mammals, type I, II and III IP3 receptors (IP3R1, IP3R2, and IP3R3) are highly homologous and have the same overall structure; with an N-terminal “ligand-binding domain”, a C-terminal “channel domain”, and an intervening “coupling domain” containing putative binding sites for several regulatory factors [1-3]. Despite these similarities, however, expression of IP3R types varies between tissues [4, 5] and subtle differences in their functional properties, such as their regulation by IP3, Ca2+ and ATP, are apparent [3, 6-8]. Functional differences between the three receptor types may also arise from their differential phosphorylation by cAMP-dependent protein kinase (PKA) [9-11] but, with the exception of IP3R1, there is no clear consensus regarding the functional consequences of these phosphorylation events.
PKA phosphorylates IP3R1 at two sites (S1588 and S1755) [12-14] and studies on IP3R1 expressed in IP3R-null DT40-3KO cells (hereafter referred to as 3KO cells) indicate that this phosphorylation enhances IP3R1-mediated Ca2+ release [15, 16], a finding consistent with biophysical studies on purified IP3R1 reconstituted into lipid bilayers [17]. In contrast, PKA-mediated phosphorylation of IP3R2 has not been examined in detail, and while IP3R3 is known to be phosphorylated at three sites (S916 , and S934 and S1832) [18], the effects of phosphorylation at these sites has not been examined. Attempts to ascribe functional effects to PKA-mediated IP3R2 and IP3R3 phosphorylation, however, have been made by activating PKA in cells that express relatively high levels of each receptor type. Enhancement of Ca2+ signaling in parotid acinar cells, for example, has been attributed to IP3R2 phosphorylation [19]. In contrast, inhibition of Ca2+ signaling has been attributed to phosphorylation of IP3R3 in pancreatic acinar cells [20, 21]. While these findings suggest that IP3R2 and IP3R3 might be differentially affected by PKA, the presence of multiple IP3R types and other PKA-regulated proteins in these systems makes it impossible to unequivocally attribute effects on Ca2+ signaling to the phosphorylation of one particular IP3R type.
It should be possible to overcome these difficulties using 3KO cells [22], as exogenous wild-type and mutant IP3Rs can be expressed and analyzed without interference from endogenous receptors [15, 16]. Thus, our initial goal was to define the effects of PKA activation on IP3R2 and IP3R3-mediated Ca2+ mobilization in 3KO cells. However, as our preliminary studies showed that IP3R2 is phosphorylated only very weakly by PKA, we focused on IP3R3. We report here that, surprisingly, phosphorylation of S916, S934 and S1832 has no effect on IP3R3-medicated Ca2+ mobilizatior in transfected 3KO cells. In contrast, phosphorylation of IP3R1 enhances Ca2+ signaling, demonstrating that the 3KO cell-based system has the capacity to respond to IP3R phosphorylation.
2. Materials and methods
2.1 Materials
CT1, CT2 and CT3, affinity purified rabbit antisera specific to the C-termini of IP3R1, IP3R2 and IP3R3, respectively, have been described previously [4]. Anti-IgM (clone M-4), a mouse monoclonal antibody directed against chicken IgM, was from Southern Biotechnology Associates. Trypsin was from Sigma (bovine pancreas; #T4665). TL3, a mouse monoclonal antibody directed against amino acids 22–230 of IP3R3, was from BD Biosciences. DT40 and 3KO cells [22] were kindly provided by Dr. D. Yule (University of Rochester, NY, U.S.A) with the permission of Dr. T. Kurosaki (Kansai Medical University, Japan).
2.2 IP3R cDNAs
Mouse SI+/SII+ IP3R1 (a kind gift from Dr, K. Mikoshiba, The University of Tokyo, Japan), rat IP3R2 (a kind gift from Dr. G. Mignery, Loyola University, Chicago, IL, U.S.A.) and rat IP3R3 (a kind gift from Dr. G. Bell, University of Chicago, Chicago, IL, U.S.A.) cDNAs have been described elsewhere [14, 23-25]. To facilitate transfection of 3KO cells, the coding region of wild-type rat IP3R3 was excised from its original vector backbone [24] with NotI and was ligated into the multiple cloning region of correspondingly cut pcDNA3 (Invitrogen). The mutation of PKA phosphorylation sites was then performed as described [18].
2.3 Generation of phospho-IP3R antibodies
Anti-pS934 and anti-pS1755 were raised in rabbits as described [4] against the synthetic peptides C-V-L-R-K-Q-phosphoS-V-F and C-S-G-R-R-E-phosphoS-L-T-S, which correspond to amino acids 929-936 of rat IP3R3 [24] and 1750-1758 of mouse IP3R1 [25], respectively, with an additional cysteine at the N-termini to facilitate coupling reactions. Crude antisera were affinity-purified on peptide-coupled SulfoLink columns (Pierce). Briefly, columns were incubated with antisera overnight at 4 °C and washed with 1 M guanidine HCl, followed by 50 mM Tris, pH 7.4. Specifically bound antibodies were eluted from the columns with 4.5 M MgCl2 in 50 mM Tris-HCl, pH 7.4, containing 0.1% bovine serum albumin. The eluate was then dialyzed against 155 mM NaCl in 50 mM Tris-HCl, pH 7.4, for 2 hr at room temperature and then overnight at 4 °C against the same buffer supplemented with 10% glycerol and 0.005% NaN3.
2.4 Cell culture
DT40 and 3KO cell lines were cultured at 40°C, under 5% CO2 in L-glutamine-containing RPMI 1640 (Mediatech Inc) supplemented with 1% chicken serum, 10% fetal bovine serum and antibiotics (100 units penicillin and 100 μg streptomycin/ml). Human embryonic kidney (HEK) 293 and African green monkey COS kidney cells were cultured as described [4]. The concentration of DMSO, used as a vehicle for forskolin and 3-isobutyl-1-methylxanthine (IBMX), was less than 1% in all experiments.
2.5 Transfection of IP3R cDNAs
Stable transfection of 3KO cells was achieved by electroporation at 550 V and 25 μF (4-mm gap cuvette) using a BioRad GenePulser XCell. Briefly, 10 million cells were incubated for 10 min on ice in 800 μl of phosphate buffered saline (PBS) containing 25 μg of IP3R cDNA linearized in the non-coding region. The mixture was then electroporated, incubated for 10 min on ice, and incubated overnight in 25 ml culture medium. Finally, the cells were centrifuged, re-suspended in 80 ml culture medium supplemented with 2 mg/ml G418, seeded into 96-well plates, and grown for 7-10 days prior to selection of G418-resistant clones for expansion. Transient transfection of HEK and COS cells was performed as described [14].
2.6 IP3R phosphorylation in intact cells
Phosphorylation of IP3Rs was analyzed using [32P]Pi-labeled HEK cells, as described [14], or through immunoblotting of electrophoresed cell lysates or immunoprecipitated IP3Rs (the ∼200-300 kDa regions of gels are shown). Cell lysates were prepared via solubilization with ice-cold, phosphatase-inhibitor-supplemented lysis buffer [14] and were either probed directly, or phosphorylated IP3Rs were immunoprecipitated by overnight incubation at 4°C with Protein A–Sepharose CL-4B beads and anti-pS1755 or anti-pS934. Immune complexes were then electrophoresed and probed with IP3R1-specific CT1 or IP3R3-specific TL3. To ensure that the conditions for measurement of IP3R phosphorylation were the same as those used in Ca2+ mobilization experiments (see section 2.8), DT40 or transfected 3KO cells were incubated with 3 μM Fura-2AM, washed, and resuspended in Krebs-HEPES buffer (KHB) [26] prior to stimulation.
2.7 Immunofluorescence microscopy
COS cells were seeded on poly-l-lysine-coated glass coverslips (500,000 cells/ 9.6 cm2), transfected with 1 μg IP3R3SSS cDNA, stimulated, fixed with 1% paraformaldehyde in PBS for 1 hr at room temperature, washed with PBS, incubated at −20°C for 10 min with methanol, rinsed for 10 sec with ice-cold acetone, and re-hydrated in PBS for 30 min at room temperature. Fixed cells were then incubated for 45 min in blocking buffer (10% goat serum, 0.1% BSA in PBS) before overnight incubation at 4°C in blocking buffer supplemented with primary antibody (TL3 or anti-pS934). Following three washes in PBS, the cells were incubated for 45 min at room temperature in blocking buffer supplemented with secondary antibody (FITC- or TRITC-conjugated goat anti-rabbit IgG, respectively). Finally, the fixed cells were washed three times, stained with 100 ng/ml 4'-6-diamidino-2-phenylindole (DAPI) for 5 min, washed three more times, and coverslips were mounted using Vectashield reagent (Vector Labs Inc). Images were acquired using a Zeiss Axioplan 2 microscope equipped with a 63x oil immersion objective (Plan Apochromat from Zeiss).
2.8 Ca2+ mobilization
Five million cells were harvested by centrifugation (2000 x g for 10 min), re-suspended in 1 ml culture medium, and incubated with 3 μM Fura-2AM for 20 min at 37°C. Following three washes with KHB, the cells were incubated for 10 min at 37°C in 1 ml KHB to allow for de-esterfication of Fura-2AM, were washed once more, resuspended in 2 ml KHB, and excited at 340 and 380 nm and emission intensity at 509 nm was recorded with an LS-50B luminescence spectrometer (Perkin-Elmer). Cytoplasmic Ca2+ concentration ([Ca2+]i) was calculated as described [27] utilizing 0.1% Triton X-100 and 7 mM EGTA to obtain Rmax and Rmin, respectively. All combined data are presented as mean ± S.E.M from ≥ 3 independent experiments except as noted. Values of “[Ca2+]i increase” were calculated by subtracting basal [Ca2+]i values from [Ca2+]i values recorded ∼60 s after stimulation with anti-IgM or ∼15 s after trypsin, and are expressed as a percentage of maximum; the average response (n ≥ 3) to a maximal dose of stimulus in the absence of forskolin plus IBMX. Statistical significance was then assessed using the unpaired Student's t test; asterisks indicate when p < 0.05.
3 Results
3.1 PKA-mediated phosphorylation of IP3Rs in intact cells
As our overall goal was to define the effects of PKA-mediated phosphorylation of IP3Rs on Ca2+ signaling in intact cells, we first sought to define which of the receptors are phosphorylated. We, and others, have already shown that IP3R1 and IP3R3 are phosphorylated by PKA in intact cells; at Ser1588 and Ser1755 in IP3R1 [12-14], and Ser916, Ser934 and Ser1832 in IP3R3 [18]. The extent to which IP3R2 is phosphorylated, however, is not clear. To determine this, we examined exogenous IP3R2 expressed in [32P]Pi-labeled HEK cells stimulated with 10 μM forskolin, a condition that results in maximal and equivalent phosphorylation of exogenous IP3R1 and IP3R3 [14, 18]. Fig 1A demonstrates that IP3R2 (lanes 1-2) was only very weakly phosphorylated under conditions that allowed for substantial phosphorylation of IP3R3 (lanes 3-4) and IP3R1 [14, 18]. Fig 1B shows that there was ∼90% less radioactivity associated with exogenous IP3R2 than IP3R3. Thus, IP3R2 is a very poor substrate for PKA and we did not examine it further. Rather, we focused on elucidating the functional effects of PKA-dependent IP3R3 phosphorylation.
Figure 1. Phosphorylation of IP3Rs in intact cells.

(A) [32P]Pi-labeled HEK cells transfected with cDNAs encoding wild-type IP3R2 (lanes 1-2), or IP3R3 (lanes 3-4) were exposed to 10 μM forskolin for 5 min as indicated, and IP3Rs were immunoprecipitated with CT2 or CT3, electrophoresed and assessed for Coomassie blue staining capacity (upper panel) and associated radioactivity (lower panel). Negligible staining and radioactivity was obtained from control (vector-transfected) cells (not shown) [14, 18]. (B) Histogram of the data presented in A. Exogenous IP3R phosphorylation was calculated by subtracting the negligible radioactivity obtained from control cells from that in lanes 1-4 and is expressed as a percentage of the radioactivity associated with IP3R3 in the presence of forskolin (lane 4). Data shown are representative of three independent experiments.
3.2 Validation of anti-pS934
To facilitate the study of IP3R3 phosphorylation, we developed an antibody against phospho-S934 of IP3R3, the predominant site of PKA-mediated phosphorylation [18]. The specificity of this antibody, termed anti-pS934, was demonstrated in immunoblots of cell lysates prepared from HEK cells expressing exogenous wild-type and mutant IP3R3s (Fig 2A), a system in which we have previously characterized IP3R3 phosphorylation using [32P]Pi [18]. Anti-pS934 exhibited minor immunoreactivity towards wild-type IP3R3 (IP3R3SSS; lane 3) and IP3R3 mutated at S916 and S1832 (IP3R3ASA; lane 9) in the absence of stimulus, and this was increased substantially under conditions of maximal IP3R phosphorylation (lanes 4 and 10). Anti-pS934 did not, however, exhibit any immunoreactivity towards IP3R3 mutated at S934 (IP3R3SAS; lanes 7-8), IP3R3 mutated at S916, S934, and S1832 (IP3R3AAA; lanes 5-6), or wild-type IP3R1 (IP3R1SS; lanes 11-12) in the absence or presence of forskolin, demonstrating that it is entirely specific towards IP3R3 phosphorylated at S934. This was confirmed by immunofluorescence microscopy of transfected COS cells. Exogenous IP3R3SSS was recognized by TL3 in ∼50% of the cells, as indicated by punctate staining throughout the cytoplasm (panels v and vi), and anti-pS934 produced a similar signal in IP3R3SSS-expressing cells after the addition of forskolin (panel viii). Thus, anti-pS934 is specific and is effective in both immunoblots and immunofluorescence (Fig 2), and subsequent studies (Fig 3) show it also to be effective in immunoprecipitations.
Figure 2. Validation of anti-pS934.

(A) HEK cells transfected with empty vector (lanes 1-2), or cDNA encoding wild-type IP3R3 (IP3R3SSS; lanes 3-4), IP3R3 mutated at S916, S934, S916, and S1832 (IP3R3AAA; lanes 5-6), mutated at S934 (IP3R3SAS; lanes 7-8), mutated at S916 and S1832 (IP3R3ASA; lanes 9-10) or wild-type IP3R1 (IP3R1SS; lanes 11-12) were incubated with or without 10 μM forskolin for 5 min as indicated, and lysates were prepared and probed with anti-pS934 (upper panel), TL3 (lower panel; lanes 1-10), or CT1 (lower panel; lanes 11-12). (B) COS cells transfected with cDNA encoding IP3R3SSS were incubated without (upper panels) or with (lower panels) 10 μM forskolin for 10 min, fixed, and stained with DAPI to visualize nuclei (panels iii and iv), TL3 to visualize IP3R3 (panels v and vi), and anti-pS934 to visualize IP3R3 phosphorylated at S934 (panels vii and viii).
Figure 3. Phosphorylation of IP3R3s expressed in 3KO cells.

(A) DT40 cells (lanes 1-2), 3KO cells (lanes 3-4), or IP3R3-expressing 3KO cells (lanes 5-12) were exposed to 1 μM forskolin plus 200 μM IBMX for 10 min as indicated, lysates were prepared, and IP3R3s phosphorylated at S934 were immunoprecipitated with anti-pS934. Total IP3R3 in lysates prior to immunoprecipitation (upper panel) or phosphorylated IP3R3 in immunoprecipitates (lower panel) were assessed in immunoblots with TL3. (B) 3KO-SSS cells were exposed to 0-100 μM forskolin in the absence (upper panel) or presence (lower panel) of 200 μM IBMX for 10 min, and IP3R3 phosphorylation was assessed as in A. (C) IP3R3SSS-expressing HEK cells were exposed to 0.01-3.0 μM forskolin for 10 min and IP3R3 phosphorylation was assessed using anti-pS934 in immunoblots of cell lysates. (D) Traces of typical Ca2+ responses to forskolin (added at t = 0.5 min) in 3KO cells (dashed line) and 3KO-SSS cells (solid lines). (E) 3KO-SSS cells were incubated with or without 1μM forskolin plus 200 μM IBMX for 10 min as indicated and levels of IP3R3 in cell lysates prior to (pre) and following (post) immunoprecipitation of phosphorylated IP3R3 with anti-pS934 were assessed in immunoblots with TL3. The lack of difference in signal intensity in control lysates pre and post immunoprecipitation (lanes 1 and 2) shows that IP3R3SSS is not significantly phosphorylated, whereas the difference in signal intensity in stimulated cells (lanes 3 and 4; a reduction of ∼50%) shows that at least 50% of IP3R3SSS is phosphorylated.
3.3 Phosphorylation of IP3R3s expressed in 3KO cells
To study the effects of phosphorylation on IP3R3-mediated Ca2+ signaling, we chose to use transfected 3KO cells, since they provide a system to examine Ca2+ signaling mediated exclusively via exogenous IP3Rs, free from the interference of endogenous IP3Rs [15, 16]. As expected, endogenous IP3R3 was present in DT40 cells (Fig 3A, upper panel, lanes 1-2), but was absent from 3KO cells (lanes 3-4). Stable expression of exogenous IP3R3SSS (3KO-SSS, lanes 5-6), IP3R3SAS (3KO-SAS, lanes 7-8), IP3R3ASA (3KO-ASA, lanes 9-10), and IP3R3AAA(3KO-AAA, lanes 11-12) was achieved at levels comparable to, or above, endogenous IP3R3. To examine whether these exogenous receptors could be phosphorylated, cell lysates were incubated with anti-pS934 and immunoprecipitated IP3R3s were visualized with TL3 (Fig 3A, lower panel). This showed that IP3R3SSS (lane 6) and IP3R3ASA (lane 10) were phosphorylated at S934 upon PKA activation and that, as expected, IP3R3SAS (lane 8) or IP3R3AAA (lane 12) were not. Surprisingly, we did not detect phosphorylation of endogenous IP3R3 (lanes 1-2), most likely because anti-pS934 does not recognize phosphorylated chicken IP3R3, due to minor sequence differences in the antibody epitope.
We next set out to define the basic characteristics of IP3R3 phosphorylation in 3KO-SSS cells. Fig 3B (upper panel) shows, surprisingly, that forskolin was not a potent stimulus and that very little phosphorylation of IP3R3 was detected at doses of forskolin (10-30 μM) considered supramaximal in many mammalian cell lines; e.g. maximal phosphorylation of exogenous IP3R3 in HEK cells (Fig 3C) or endogenous IP3R3 in Rat-1 fibroblasts (data not shown) occurs at 1 μM forskolin. However, inclusion of IBMX (200 μM), an inhibitor of cAMP phosphodiesterases, greatly enhanced IP3R3 phosphorylation in 3KO-SSS cells (Fig 3B, lower panel) and reduced the concentration of forskolin required for maximal IP3R3 phosphorylation to ∼10 μM.
The use of IBMX to enhance IP3R3 phosphorylation proved to be essential for the studies on Ca2+ signaling in 3KO-derived cells, as we found that forskolin alone caused Ca2+ mobilization in 3KO-SSS cells at concentrations of 10 μM and above (Fig 3D). The cause of this mobilization remains unknown, but appears to reflect IP3R-independent Ca2+ release from intracellular stores, as it still occurred in 3KO cells (Fig 3D), and in the absence of extracellular Ca2+ (data not shown). Thus, to study the effects of IP3R3 phosphorylation on Ca2+ mobilization, we chose to use 1 μM forskolin plus 200 μM IBMX, as this condition causes near maximal IP3R3 phosphorylation (Fig 3B), but did not itself cause Ca2+ mobilization (Figs 4-5). Further, we could demonstrate that at least 50% of exogenous IP3R3SSS was phosphorylated by forskolin plus IBMX under these conditions (Fig 3E).
Figure 4. Effect of PKA activation on IP3R3-mediated Ca2+ mobilization in response to BCR stimulation.
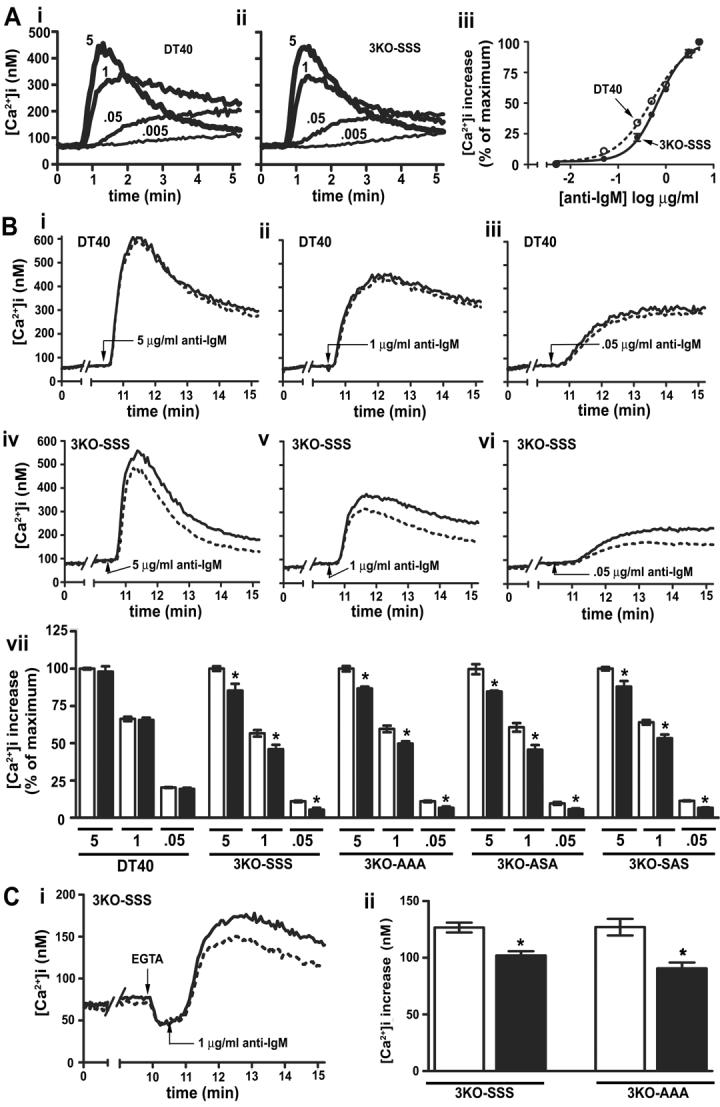
(A) Traces of typical Ca2+ responses to 5, 1, 0.05 and 0.005 μg/ml anti-IgM (added at t = 0.5 min) in DT40 (i) or 3KO-SSS (ii) cells. Dose-response curves (iii) for DT40 and 3KO-SSS cells are expressed as a percentage of the [Ca2+]i increase in response to 5 μg/ml anti-IgM. Error bars may be occluded by symbols. (B) Traces of typical Ca2+ responses in DT40 (i-iii) or 3KO-SSS (iv-vi) cells exposed to 5 (i and iv), 1 (ii and v), or 0.05 (iii and vi) μg/ml anti-IgM (added at t = 10.5 min) in the absence (solid line) or presence (broken line) of 1 μM forskolin plus 200 μM IBMX (added at t = 0.5 min). Histogram (vii) of [Ca2+]i increases in DT40 or 3KO cell lines in response to 5, 1, or 0.05 μg/ml anti-IgM in the absence (clear bars) or presence (solid bars) of forskolin plus IBMX. (C) Typical traces of Ca2+ responses in 3KO-SSS cells (i) exposed to 5mM EGTA (added at t = 10 min) and 1 μg/ml anti-IgM (added at t = 10.5 min) in the absence (solid line) or presence (broken line) of 1μM forskolin plus 200 μM IBMX (added at t = 0.5 min). Histogram (ii) of [Ca2+]i increases in 3KO-SSS and 3KO-AAA cells in the absence (clear bars) or presence (solid bars) of forskolin plus IBMX.
Figure 5. Effect of PKA activation on IP3R1-mediated Ca2+ mobilization in response to BCR stimulation.

(A) HEK cells transfected with empty vector (lanes 1-2), or cDNA encoding wild-type IP3R1 (IP3R3SS; lanes 3-4), IP3R1 mutated at S1755 (IP3R3SA; lanes 5-6), mutated at S1588 (IP3R3AS; lanes 7-8), mutated at S1588 and S1755 (IP3R1AA; lanes 9-10), or wild-type IP3R3 (IP3R3SSSS ; lanes 11-12) were exposed to 10 μM forskolin for 5 min as indicated, lysates were prepared, and IP3R1 phosphorylated at S1755 were immunoprecipitated with anti-pS1755 and visualized in immunoblots with CT1. (B) DT40 (lanes 1-2), or IP3R1-expressing 3KO (lanes 3-6) cells were exposed to 1 μM forskolin plus 200 μM IBMX for 10 min as indicated, cell lysates were prepared, and IP3R1 phosphorylated at S1755 was immunoprecipitated with anti-pS1755 and visualized in immunoblots with CT1 (lower panel). IP3R1 levels in lysates prior to immunoprecipitation were assessed with CT1 (upper panel). (C) 3KO-SS cells were incubated with 1μM forskolin plus 200 μM IBMX for 10 min and levels of IP3R1 in cell lysates prior to (pre) and following (post) immunoprecipitation of phosphorylated IP3R1 with anti-pS1755 were assessed in immunoblots with CT1. The ∼50% reduction in signal intensity post immunoprecipitation indicates that at least 50% of IP3R1 is phosphorylated. (D) Traces of typical Ca2+ responses to 5 μg/ml anti-IgM (added at t = 10.5) in the absence (solid line) or presence (broken line) of 1 μM forskolin plus 200 μM IBMX (added at t = 0.5 min) in 3KO-SS cells. (E) Histogram of [Ca2+]i increases in 3KO-SS and 3KO-AA cells in response to 5 μg/ml or 1 μg /ml anti-IgM in the absence (clear bars) or presence (solid bars) of forskolin plus IBMX.
3.4 Effects of IP3R3 phosphorylation on BCR-mediated Ca2+ mobilization
Stimulation of DT40 cells with anti-IgM results in endogenous B cell receptor (BCR) activation, PLCγ-mediated generation of IP3, and IP3R-mediated Ca2+ mobilization [22, 28]. Fig 4A shows typical Ca2+ responses to various doses of anti-IgM in DT40 (i) and 3KO-SSS (ii) cells, and dose-response curves (iii). Clearly, exogenous IP3R3SSS (ii) couples BCR activation to Ca2+ mobilization essentially identically to endogenous IP3Rs (i), with the exception that 3KO-SSS cells are slightly less sensitive to anti-IgM than DT40 cells (EC50 = 0.70 ± 0.06 and 0.50 ± 0.04 μg/ml, respectively). This minor difference may be explained by the fact that, in addition to IP3R3, DT40 cells express IP3R1 and IP3R2 [22], both of which have a higher affinity for IP3 than IP3R3 [6, 7, 22]. 3KO-SSS, 3KO-AAA, 3KO-SAS and 3KO-ASA cells exhibited virtually identical maximum responses to anti-IgM (544 ± 40, 570 ± 52, 533 ± 37 and 522 ± 48 nM Ca2+, respectively) and anti-IgM dose-response curves (EC50 = 0.70 ± 0.04, 0.72 ± 0.08, 0.79 ± 0.10, and 0.72 ± 0.06 μg/ml), demonstrating that basal phosphorylation of S916, S934 and S1832 does not affect IP3R3-mediated Ca2+ mobilization.
To determine the effects of increased IP3R3 phosphorylation on anti-IgM - induced Ca2+ mobilization, we stimulated cells with a high (5 μg/ml), medium (1 μg/ml) and low (0.05 μg/ml) dose of anti-IgM, 10 min after addition of either vehicle or 1 μM forskolin plus 200 μM IBMX, a condition which phosphorylates at least 50% of IP3R3s in transfected 3KO cells (Fig 3E) and causes phosphorylation of endogenous IP3Rs in DT40 cells (Fig 5B). Fig 4B shows that treatment with forskolin plus IBMX had no effect on anti-IgM induced Ca2+ mobilization in DT40 cells (i-iii), but slightly inhibited Ca2+ mobilization in 3KO-SSS cells (iv-vi). However, this inhibition was unrelated to IP3R3 phosphorylation at S916, S934 or S1832, as it occurred to a similar extent in all IP3R3 mutant-expressing cell lines (vii). It also occurred in 3KO-SSS and 3KO-AAA cells incubated with extracellular EGTA (Fig 4C), indicating that it is due to reduced IP3R3-mediated Ca2+ release from intracellular stores, rather than reduced Ca2+ entry. Surprisingly, therefore, the data in Fig 4 demonstrate that phosphorylation of IP3R3 at S916, S934 and S1832 has no effect on Ca2+ mobilization in response to BCR activation, but also reveals a slight inhibitory effect of forskolin plus IBMX that is mediated by an, as yet, unidentified mechanism. This inhibition appears to be a consequence of cAMP elevation and/or PKA activation, as it was not seen when cells were incubated with either 1 μM forskolin or 200 μM IBMX alone (data not shown).
3.5 IP3R1 phosphorylation enhances BCR-mediated Ca2+ mobilization
Given the lack of effect of forskolin plus IBMX on Ca2+ signaling in DT40 cells and 3KO-SSS cells (Fig 4), we sought evidence that IP3R phosphorylation can have measurable effects on Ca2+ signaling in our experimental system. In this regard, it has been shown previously that PKA-mediated phosphorylation of IP3R1 at S1588 and/or S1755 enhances Ca2+ release in individual IP3R1-expressing 3KO cells [15, 16]. Thus, PKA activation should also enhance IP3R1-mediated Ca2+ release in our experiments with cell suspensions.
To measure IP3R1 phosphorylation, we developed an antibody against phospho- S1755. This antibody, termed anti-pS1755, did not recognize phosphorylated IP3R1 in immunoblots or via immunofluorescence microscopy (not shown), but was effective in immunoprecipitations and was specific towards phospho-S1755 of IP3R1 (Fig 5A). Thus, we could demonstrate forskolin plus IBMX-induced phosphorylation of endogenous IP3R1 in DT40 cells (Fig 5B, lane 2, lower panel), and of exogenous IP3R1SS in 3KO-SS cells (lane 4, lower panel), but not in 3KO-AA cells (lane 6 lower panel). Further, using the same approach utilized for IP3R3 (Fig 3E), we could show that at least 50% of the receptors in 3KO-SS cells are phosphorylated in response to forskolin plus IBMX (Fig 5C).
Fig 5D shows that treatment with forskolin plus IBMX enhanced the Ca2+ response to 5 μg/ml anti-IgM by ∼30 % in 3KO-SS cells. This effect was also seen at 1 μg/ml anti-IgM, but was not evident in 3KO-AA cells (Fig 5E), indicating that it is a direct consequence of IP3R1 phosphorylation at S1755 and/or S1588. Thus, IP3R phosphorylation at defined sites is capable of modulating Ca2+ signals in our experimental system.
3.6 Effects of IP3R phosphorylation on trypsin-mediated Ca2+ mobilization
To determine whether activation of Ca2+ mobilization via a different signaling pathway might uncover effects of IP3R3 phosphorylation, we examined Ca2+ signaling in response to trypsin, which activates the PAR2 G protein-coupled receptor in DT40 cells [29] and which elevates intracellular Ca2+ in our experimental system (Fig 6A). However, as with anti-IgM, trypsin-induced Ca2+ mobilization was not affected by forskolin plus IBMX treatment in DT40 cells, and was inhibited slightly by this treatment in both 3KO-SSS and 3KO-AAA cells (Fig 6B). Thus, again, this inhibitory effect is not due to phosphorylation of S916, S934, or S1832. Enhancement of trypsin-induced responses by forskolin plus IBMX treatment in 3KO-SS cells, but not in 3KO-AA cells, again confirms that Ca2+ mobilization can be regulated by IP3R phosphorylation (Fig 6B).
Figure 6. Effect of PKA activation on trypsin-induced Ca2+ mobilization.

(A) Trace showing a typical Ca2+ response to a maximal dose of trypsin (1000 U/ml) in DT40 cells. (B) Histogram of [Ca2+]i increases in DT40 and 3KO cell lines in response to 1000 U/ml or 50 U/ml trypsin in the absence (clear bars) or presence (solid bars) of 1 μM forskolin plus 200 μM IBMX.
4. Discussion
The major findings presented herein are (i) that IP3R2 is a very poor substrate for PKA and (ii) that phosphorylation of S916, S934 and S1832 has no effect on the ability of IP3R3 to mobilize Ca2+.
The inability of maximally activated PKA to substantially phosphorylate exogenous IP3R2 expressed in HEK cells (Fig 1) is consistent with findings that endogenous IP3R2 in AR42J cells is only very weakly phosphorylated by PKA [9], and that PKA catalytic subunit does not affect IP3-mediated Ca2+ release in permeabilized cells that predominantly express IP3R2 [11]. Taken together, these data indicate that IP3R2 is not a physiological substrate for PKA. Therefore, the enhanced Ca2+ signaling that has been attributed to PKA-mediated phosphorylation of IP3R2 in parotid acinar cells [19] is most likely the result of phosphorylation of other proteins, possibly other IP3R types. Indeed, the evidence for IP3R2 phosphorylation in parotid acinar cells was that IP3R2 was immunoprecipitated from forskolin-stimulated cells by an anti-phosphoserine/threonine antibody [19]; however, as parotid cells also express IP3R1 and IP3R3 [30], and IP3Rs heterotetramerize [4, 5], it is entirely plausible that non-phosphorylated IP3R2 was immunoprecipitated because of its interaction with phosphorylated IP3R1 and/or IP3R3.
We have also found that phosphorylation of IP3R3 at S916, S934 and S1832, the only sites in IP3R3 known to be modified by PKA [18], does not affect the receptor's ability to mobilize Ca2+. This conclusion was validated by the positive control that Ca2+ mobilization via IP3R1SS, but not IP3R1AA, was enhanced by PKA activation; thus, 3KO cells contain the requisite proteins to allow for Ca2+ signaling via exogenous IP3Rs to be modified by their phosphorylation. Phosphorylation of IP3R3 at S916, S934 and S1832, therefore, may play a role other than regulation of Ca2+ channel activity, such as providing docking sites for ancillary proteins. Interestingly, similar findings have recently been reported for Akt kinase-mediated phosphorylation of IP3R1; this kinase phosphorylates IP3R1 at S2681, but this modification does not affect the receptor's ability to mobilize Ca2+ [31].
Surprisingly, we also found that forskolin plus IBMX treatment inhibited Ca2+ release by ∼10% for each of the exogenous IP3R3s, irrespective of whether S916, S934 or S1832 were mutated or not. Conceivably, this inhibition is a result of phosphorylation of a previously uncharacterized site in IP3R3 that is revealed when the receptor is expressed in 3KO cells, although this possibility is unlikely, as S916, S934 and S1832 comprise the full complement of sites phosphorylated by PKA in intact HEK cells and in vitro [18]. Alternatively, it could be the effect of an, as yet, unidentified PKA substrate that specifically associates with and inhibits IP3R3. Either event may go some way to explaining the PKA-mediated inhibition of Ca2+ signaling that has been attributed to IP3R3 phosphorylation in pancreatic acinar cells [20, 21].
The development of anti-pS934 and anti-pS1755 allowed us, for the first time, to characterize the phosphorylation of exogenous IP3Rs expressed in 3KO cells and endogenous IP3Rs in DT40 cells. Interestingly, our data revealed that forskolin is a very weak stimulus in these cells and that co-incubation with IBMX is required for efficient IP3R phosphorylation (Fig 3B), perhaps indicating that DT40-derived cell lines contain high levels of cAMP phosphodiesterase activity. Clearly, this should be taken into account when examining PKA-mediated events in DT40-derived cells. Further, and somewhat surprisingly, while endogenous IP3Rs are phosphorylated by PKA in DT40 cells (Fig 5B), this does not affect Ca2+ signaling (Fig 4). As IP3R1, IP3R2 and IP3R3 are all present in DT40 cells [22], this could reflect the combined effects of enhanced IP3R1 activity and inhibited IP3R3 activity. Alternatively, as DT40 cell IP3R complexes are likely to be heterotetrameric [5], and as nothing is currently known about how phosphorylation affects IP3R heterotetramers, it is possible that the IP3R complexes in DT40 cells are unaffected by phosphorylation, even though they contain IP3R1 and/or IP3R3.
In summary, we demonstrate the differential phosphorylation of IP3R types by PKA, with IP3R2 being only very weakly phosphorylated compared to IP3R1 and IP3R3. Further, phosphorylation of IP3R3 at the sites known to be targeted by PKA (S916, S934 and S1832) does not affect its ability to mobilize Ca2+, suggesting that modification of Ca2+ flux may not be the primary effect of IP3R3 phosphorylation.
Acknowledgements
This work was supported by grants from the American Heart Association (0256225T) and the National Institute of Health (DK49194). The authors wish to thank Yuan Wang for technical support, Dr. Karen Vikstrom for assistance with immunofluorescence microscopy, and Margaret Pearce, Danielle Sliter, and Drs. Greg Mignery and David Yule for many helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Matthew D. Soulsby and Richard J.H. Wojcikiewicz, the authors of the submitted manuscript “Calcium mobilization via type III inositol 1,4,5-trisphophate receptors is not altered by PKA mediated phosphorylation of serines 916, 934 and 1832”, state that there are no conflicts of interest that could inappropriately influence the work presented in our manuscript.
5. References
- 1.Taylor CW, da Fonseca PCA, Morris EP. IP3 receptors: the search for structure. Trends in Biochemical Sciences. 2004;29:210–219. doi: 10.1016/j.tibs.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Patterson RL, Boehning D, Snyder SH. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annual Review of Biochemistry. 2004;73:437–465. doi: 10.1146/annurev.biochem.73.071403.161303. [DOI] [PubMed] [Google Scholar]
- 3.Bezprozvanny I. The inositol 1,4,5-trisphosphate receptor's. Cell Calcium. 2005;38:261–272. doi: 10.1016/j.ceca.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 4.Wojcikiewicz RJH. Type I, II, and III inositol 1,4,5-trisphosphate receptors are unequally susceptible to down-regulation and are expressed in markedly different proportions in different cell types. J. Biol. Chem. 1995;270:11678–11683. doi: 10.1074/jbc.270.19.11678. [DOI] [PubMed] [Google Scholar]
- 5.Taylor CW, Genazzani AA, Morris SA. Expression of inositol trisphosphate receptors. Cell Calcium. 1999;26:237–251. doi: 10.1054/ceca.1999.0090. [DOI] [PubMed] [Google Scholar]
- 6.Wojcikiewicz RJH, Luo SG. Differences Among Type I, II, and III Inositol-1,4,5-Trisphosphate Receptors in Ligand-Binding Affinity Influence the Sensitivity of Calcium Stores to Inositol-1,4,5-Trisphosphate. Mol Pharmacol. 1998;53:656–662. doi: 10.1124/mol.53.4.656. [DOI] [PubMed] [Google Scholar]
- 7.Tu H, Wang Z, Nosyreva E, et al. Functional Characterization of Mammalian Inositol 1,4,5-Trisphosphate Receptor Isoforms. Biophys. J. 2005;88:1046–1055. doi: 10.1529/biophysj.104.049593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maes K, Missiaen L, Parys JB, et al. Mapping of the ATP-binding Sites on Inositol 1,4,5-Trisphosphate Receptor Type 1 and Type 3 Homotetramers by Controlled Proteolysis and Photoaffinity Labeling. J. Biol. Chem. 2001;276:3492–3497. doi: 10.1074/jbc.M006082200. [DOI] [PubMed] [Google Scholar]
- 9.Wojcikiewicz RJH, Luo SG. Phosphorylation of Inositol 1,4,5-Trisphosphate Receptors by cAMP-dependent Protein Kinase. Type I, II, and III receptors are differentially susceptible to phosphorylation and are phosphorylated in intact cellls. J. Biol. Chem. 1998;273:5670–5677. doi: 10.1074/jbc.273.10.5670. [DOI] [PubMed] [Google Scholar]
- 10.Tu H, Tang T-S, Wang Z, et al. Association of Type 1 Inositol 1,4,5-Trisphosphate Receptor with AKAP9 (Yotiao) and Protein Kinase A. J. Biol. Chem. 2004;279:19375–19382. doi: 10.1074/jbc.M313476200. [DOI] [PubMed] [Google Scholar]
- 11.Dyer JL, Mobasheri H, Lea EJA, et al. Differential effect of PKA on the Ca2+ release kinetics of the type I and III InsP3 receptors. Biochemical and Biophysical Research Communications. 2003;302:121–126. doi: 10.1016/s0006-291x(03)00120-7. [DOI] [PubMed] [Google Scholar]
- 12.Ferris CD, Cameron AM, Bredt DS, et al. Inositol 1,4,5-trisphosphate receptor is phosphorylated by cyclic AMP-dependent protein kinase at serines 1755 and 1589. Biochemical and Biophysical Research Communications. 1991;175:192–198. doi: 10.1016/s0006-291x(05)81219-7. [DOI] [PubMed] [Google Scholar]
- 13.Haug LS, Jensen V, Hvalby O, et al. Phosphorylation of the Inositol 1,4,5-Trisphosphate Receptor by Cyclic Nucleotide-dependent Kinases in Vitro and in Rat Cerebellar Slices in Situ. J. Biol. Chem. 1999;274:7467–7473. doi: 10.1074/jbc.274.11.7467. [DOI] [PubMed] [Google Scholar]
- 14.Soulsby MD, Alzayady K, Xu Q, et al. The contribution of serine residues 1588 and 1755 to phosphorylation of the type I inositol 1,4,5-trisphosphate receptor by PKA and PKG. FEBS Letters. 2004;557:181–184. doi: 10.1016/s0014-5793(03)01487-x. [DOI] [PubMed] [Google Scholar]
- 15.Wagner LE, II, Li W-H, Joseph SK, et al. Functional Consequences of Phosphomimetic Mutations at Key cAMP-dependent Protein Kinase Phosphorylation Sites in the Type 1 Inositol 1,4,5-Trisphosphate Receptor. J. Biol. Chem. 2004;279:46242–46252. doi: 10.1074/jbc.M405849200. [DOI] [PubMed] [Google Scholar]
- 16.Wagner LE, II, Li W-H, Yule DI. Phosphorylation of Type-1 Inositol 1,4,5-Trisphosphate Receptors by Cyclic Nucleotide-dependent Protein Kinases: A mutational analysis of the functionally important sites in the S2+ and S2− splice variants. J. Biol. Chem. 2003;278:45811–45817. doi: 10.1074/jbc.M306270200. [DOI] [PubMed] [Google Scholar]
- 17.Tang T, Tu H, Wang Z, et al. Modulation of Type I inositol (1,4,5)-trisphosphate Receptor Function by Protein Kinase A and Protein Phosphatase 1 alpha. J. Neurosci. 2003;23:403–415. doi: 10.1523/JNEUROSCI.23-02-00403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soulsby MD, Wojcikiewicz RJH. The type III inositol 1,4,5-trisphosphate receptor is phosphorylated by cAMP-dependent protein kinase at three sites. Biochemical Journal. 2005;392:493–497. doi: 10.1042/BJ20051325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruce JIE, Shuttleworth TJ, Giovannucci DR, et al. Phosphorylation of Inositol 1,4,5-Trisphosphate Receptors in Parotid Acinar Cells. A mechanism for the synergistic effects of cAMP on Ca2+ signaling. J. Biol. Chem. 2002;277:1340–1348. doi: 10.1074/jbc.M106609200. [DOI] [PubMed] [Google Scholar]
- 20.Giovannucci DR, Groblewski GE, Sneyd J, et al. Targeted Phosphorylation of Inositol 1,4,5-Trisphosphate Receptors Selectively Inhibits Localized Ca2+ Release and Shapes Oscillatory Ca2+ Signals. J. Biol. Chem. 2000;275:33704–33711. doi: 10.1074/jbc.M004278200. [DOI] [PubMed] [Google Scholar]
- 21.Straub SV, Giovannucci DR, Bruce JIE, et al. A Role for Phosphorylation of Inositol 1,4,5-Trisphosphate Receptors in Defining Calcium Signals Induced by Peptide Agonists in Pancreatic Acinar Cells. J. Biol. Chem. 2002;277:31949–31956. doi: 10.1074/jbc.M204318200. [DOI] [PubMed] [Google Scholar]
- 22.Sugawara H, Kurosaki M, Takata M, et al. Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. The EMBO Journal. 1997;16:3078–3088. doi: 10.1093/emboj/16.11.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sudhof TC, Newton CL, Archer BT, III, et al. Structure of a novel InsP3 receptor. The EMBO Journal. 1991;10:3199–3206. doi: 10.1002/j.1460-2075.1991.tb04882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blondel O, Takeda J, Janssen H, et al. Sequence and functional characterization of a third inositol trisphosphate receptor subtype, IP3R-3, expressed in pancreatic islets, kidney, gastrointestinal tract, and other tissues. J. Biol. Chem. 1993;268:11356–11363. [PubMed] [Google Scholar]
- 25.Furuichi T, Yoshikawa S, Miyawaki A, et al. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature. 1989;342:32–38. doi: 10.1038/342032a0. [DOI] [PubMed] [Google Scholar]
- 26.Soulsby MD, Wojcikiewicz RJH. 2-Aminoethoxydiphenyl borate inhibits inositol 1,4,5-trisphosphate receptor function, ubiquitination and downregulation, but acts with variable characteristics in different cell types. Cell Calcium. 2002;32:175–181. doi: 10.1016/s0143416002001525. [DOI] [PubMed] [Google Scholar]
- 27.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 28.Kubista H, Hawkins T, Moss SE. Characterisation of calcium signalling in DT40 chicken B-cells. Biochimica et Biophysica Acta - Molecular Cell Research. 1998;1448:299–310. doi: 10.1016/s0167-4889(98)00132-3. [DOI] [PubMed] [Google Scholar]
- 29.Morita T, Tanimura A, Nezu A, Kurosaki T, Tojyo Y. Functional analysis of the green fluorescent protein-tagged inositol 1,4,5-trisphosphate receptor type 3 in Ca2+ release and entry in DT40 B lymphocytes. Biochemical Journal. 2004;382:793–801. doi: 10.1042/BJ20031970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Wen J, Bidasee KR, et al. Ryanodine and inositol trisphosphate receptors are differentially distributed and expressed in rat parotid gland. Biochem. J. 1999;340:519–527. [PMC free article] [PubMed] [Google Scholar]
- 31.Khan MT, Wagner L, II, Yule DI, Bhanumathy C, Joseph SK. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 2006;281:3731–3737. doi: 10.1074/jbc.M509262200. [DOI] [PubMed] [Google Scholar]