

Abstract
Chronic ethanol feeding sensitizes Kupffer cells to activation by lipopolysac-charide (LPS), leading to increased production of tumor necrosis factor-α (TNF-α). Adiponectin treatment protects mice from ethanol-induced liver injury. Because adiponectin has anti-inflammatory effects on macrophages, we hypothesized that adiponectin would normalize chronic ethanol-induced sensitization of Kupffer cells to LPS-mediated signals. Serum adiponectin concentrations were decreased by 45% in rats fed an ethanol-containing diet for 4 wk compared with pair-fed rats. Adiponectin dose dependently inhibited LPS-stimulated accumulation of TNF-α mRNA and peptide in Kupffer cells from both pair- and ethanol-fed rats. Kupffer cells from ethanol-fed rats were more sensitive to both globular (gAcrp) and full-length adiponectin (flAcrp) than Kupffer cells from pair-fed controls with suppression at 10 ng/ml adiponectin after chronic ethanol feeding. Kupffer cells expressed both adiponectin receptors 1 and 2; chronic ethanol feeding did not change the expression of adiponectin receptor mRNA or protein. gAcrp suppressed LPS-stimulated ERK1/2 and p38 phosphorylation as well as IκB degradation at 100–1,000 ng/ml in Kupffer cells from both pair- and ethanol-fed rats. However, only LPS-stimulated ERK1/2 phosphorylation was sensitive to 10 ng/ml gAcrp. gAcrp also normalized LPS-stimulated DNA binding activity of early growth response-1 with greater sensitivity in Kupffer cells from rats fed chronic ethanol. In conclusion, these results demonstrate that Kupffer cells from ethanol-fed rats are more sensitive to the anti-inflammatory effects of both gAcrp and flAcrp. Suppression of LPS-stimulated ERK1/2 signaling by low concentrations of gAcrp was associated with normalization of TNF-α production by Kupffer cells after chronic ethanol exposure.
Keywords: macrophage, Toll-like receptor 4, inflammation, mitogen-activated protein kinase, early growth response-1, lipopolysaccharide, tumor necrosis factor-α
Adiponectin [a 30-kDa peptide also known as adipocyte complement-related protein (Acrp30)] is an adipokine secreted by adipose tissue (4, 27). The adiponectin concentration is decreased in the blood during obesity, insulin resistance, and type 2 diabetes, and insulin sensitivity can be restored in obese and diabetic animals by treatment with adiponectin (3, 13). Adiponectin acts on target tissues via the activation of adiponectin receptors 1 and 2 (AdipoR1 and AdipoR2) (38). The liver primarily expresses AdipoR2, whereas skeletal muscle expresses predominantly AdipoR1 (38). Treatment of mice with adiponectin increases fatty acid oxidation in the liver and glucose utilization in muscle (3).
In addition to affecting glucose and lipid metabolism, adiponectin also influences the activity of the innate immune system. Adiponectin inhibits the growth of myelomonocytic progenitor cells (43) and decreases the ability of mature macrophages to respond to activation (36, 43). Adiponectin suppresses phagocytic activity as well as lipopolysaccharide (LPS)-stimulated cytokine production in macrophages (36, 43). The response of macrophages to adiponectin may be mediated in part by complement C1q receptors (43). However, a specific interaction of adiponectin with AdipoR on the surface of macrophages is also likely (43) but not yet characterized. AdipoR1 is expressed in monocytes but decreases with differentiation, coincident with increased AdipoR2 expression (7). Adiponectin is hypothesized to dampen the early phases of macrophage inflammatory responses (43). This anti-inflammatory response to adiponectin has been implicated as a potential mechanism for the antiatherogenic properties of adiponectin (10, 23).
Chronic ethanol-induced liver injury is mediated, at least in part, by increased inflammatory activity likely involving both Kupffer cells, the resident macrophage in the liver (for a review, see Ref. 21), as well as infiltrating neutrophils and lymphocytes (2). After chronic ethanol exposure, Kupffer cells exhibit enhanced sensitivity to LPS-stimulated inflammatory cytokine production (1, 21). While the mechanisms for this increased sensitivity are not well understood, chronic ethanol exposure disrupts a number of LPS-mediated signaling events, including increased activity of the MAPK family members ERK1/2 and p38 (5, 15, 16). Increased activation of ERK1/2 after chronic ethanol is associated with increased expression of early growth response-1 (Egr-1), a transcription factor required for maximal LPS-stimulated TNF-α transcription (28), whereas increased LPS-stimulated p38 activity is associated with a stabilization of TNF-α mRNA after chronic ethanol exposure (16).
Recent studies (37, 44) have demonstrated that chronic ethanol feeding decreases circulating adiponectin concentrations in mice. Furthermore, Xu et al. (37) reported that treatment of mice with adiponectin during chronic ethanol exposure prevents the development of liver injury. The protective effect of adiponectin was associated with changes in fatty acid metabolism in the liver; adiponectin treatment during chronic ethanol feeding increased fatty acid oxidation and suppressed fatty acid synthesis (37). Hepatic TNF-α expression was also normalized by adiponectin treatment during chronic ethanol feeding (37). However, it is not clear whether decreased TNF-α was due to a direct anti-inflammatory action of adiponectin or an indirect effect of changes in lipid homeostasis leading to decreased steatosis. Because adiponectin is known to have direct anti-inflammatory effects on macrophage function in other model systems (36, 43), we hypothesized that adiponectin would suppress LPS-stimulated inflammatory cytokine production in Kupffer cells after chronic ethanol exposure by normalizing LPS-mediated signal transduction. We report that chronic ethanol feeding to rats decreased serum adiponectin concentrations. Treatment of Kupffer cells isolated from pair- and ethanol-fed rats with globular adiponectin (gAcrp) or full-length adiponectin (flAcrp) normalized LPS-stimulated TNF-α production; Kupffer cells from ethanol-fed rats were more sensitive to the suppressive effects of adiponectin. These data provide further support for a potential therapeutic role for adiponectin in treating ethanol-induced liver injury.
MATERIALS AND METHODS
Materials
Adult male Wistar rats weighing 170–180 g were purchased from Harlan Sprague Dawley (Indianapolis, IN). Lieber DeCarli ethanol diet (regular, no. 710260) was purchased from Dyets (Bethlehem, PA). Cell culture reagents were from GIBCO-BRL (Grand Island, NY). Antibodies were from the following sources: phospho-ERK1/2 (Santa Cruz Biotechnology; Santa Cruz, CA), phos-pho-p38 (Promega; Madison, WI), total ERK1/2 (Upstate Biotechnology; Lake Placid, NY), total p38 (Santa Cruz Biotechnology), transferrin receptor (Zymed; San Francisco, CA), IκB-α (Cell Signaling; Beverly, MA), and AdipoR1 (NH2 terminus) and AdipoR2 (COOH-terminus) (AlphaDiagnostic; San Antonio, TX). Anti-rabbit and anti-mouse IgG-peroxidase were purchased from Boehringer Mannheim (Indianapolis, IN). LPS from Escherichia coli serotype 026:B6 was purchased from Sigma (St. Louis, MO). Recombinant human gAcrp and flAcrp expressed in E. coli were purchased from Peprotech (Rocky Hill, NJ). Recombinant human flAcrp expressed in HEK-293 cells to produce a fully glycosylated form of flAcrp was purchased from BioVendor (Candler, NC). Endotoxin contamination of all reagents used to treat Kupffer cells was routinely monitored in the laboratory using a kinetic chromogenic test based on the Limulus amebocyte lysate assay (Kinetic-QCL, BioWhittaker; Walkersville, MD).
Chronic ethanol feeding and Kupffer cell isolation
Rats were provided with a liquid diet containing 35% of the calories from ethanol for 4 wk (1). Controls were pair fed a liquid diet that was identical to the ethanol diet except that maltose dextrins were isocalorically substituted for ethanol. Rats maintained on standard laboratory chow were also included in some experiments as additional controls. Procedures involving animals were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University. Blood was collected from the posterior vena cava before perfusion of the liver for isolation of Kupffer cells. Serum was isolated, stored at −20°C and then assayed for adiponectin by ELISA (B-Bridge; Sunnyvale, CA), alanine aminotransferase (ALT) by enzymatic assay (Sigma-Aldrich), or TNF-α by ELISA (BioLegend; San Diego, CA). In some experiments, the liver was fixed in formalin for histology. Formalin-fixed tissues were paraffin embedded, sectioned, and stained with hematoxylin-eosin.
Kupffer cells were isolated and cultured as previously described (1, 15). Isolated Kupffer cells were suspended in CMRL media with 10% FBS and penicillin-streptomycin (CMRL-FBS) at a concentration of 2 × 106 cells/ml and plated onto 96-well (0.2 × 106 cells/well for TNF-α ELISA assays or 0.03 × 106 cells/well for dihydrorhodamine fluorescence), 6-well (6 × 106 cells/well for nuclear extracts and membrane preparation), and 24-well (1.5 × 106 cells/well for ERK1/2, p38, and IκB assays) culture plates. The purity of the Kupffer cell preparations has been previously described (1) and was routinely monitored by staining with ED2, a rat macrophage marker. Two hours after cells were plated, nonadherent cells were removed by aspiration and fresh media with or without adiponectin were added. Assays were initiated after 16–18 h in culture. Treatment with gAcrp or flAcrp at concentrations up to 1,000 ng/ml had no effect on Kupffer cell number or protein content after overnight culture (data not shown).
LPS-stimulated TNF-α production
After treatment with or without adiponectin for 16–18 h, cell culture media were removed and replaced with CMRL-10% FBS with or without LPS. After 4 h stimulation with LPS, cell culture media were removed and stored at −20°C for assay of TNF-α by ELISA (R&D Systems; Minneapolis, MN). Control experiments were carried out to test whether possible endotoxin contamination of recombinant gAcrp or flAcrp was involved the observed responses of Kupffer cells to adiponectin. In these experiments, cells were treated with or without 10 μg/ml polymixin during the overnight treatment with 10 ng/ml gAcrp or flAcrp. Cells were then washed to remove polymixin-adiponectin before treatment with LPS.
Measurement of ROS
Kupffer cells were cultured for 16–18 h with or without gAcrp. Media were then changed, and cells were then stimulated with 100 ng/ml LPS for 0–15 min at 37°C in a 5% CO2 atmosphere. Media were then replaced with 100 μl of 10 μM dihydrorhodamine (DHR; Molecular Probes; Eugene, OR) diluted in CMRL with 10% FBS, and cells were incubated for 15 min at 37°C in a 5% CO2 atmosphere in the dark. Fluorescence was measured by a fluorescence plate reader (Perkin Elmer) using an excitation wavelength of 505 nm and emission detection wavelength of 530 nm.
Western blot analysis of LPS-stimulated signaling pathways
After treatment with or without gAcrp for 16–18 h, cells were treated with or without 100 ng/ml LPS for 0–30 min. Cells were then moved to ice, washed with 2 ml ice-cold PBS buffer containing 1 mM sodium orthovanadate, and then lysed in Laemmli sample buffer containing 1 mM sodium orthovanadate. Samples were then separated by SDS-PAGE and probed by Western blot analysis for phospho-ERK1/2, phospho-p38, total ERK1/2, or IκB. Membranes were then stripped and reprobed with antibodies for total p38.
Quantification of AdipoR1 and AdipoR2
After overnight cultures, Kupffer cells were homogenized in 20 mM Tris (pH 7.4), 1 mM EDTA, 255 mM sucrose, and protease inhibitors (Boehringer Mannheim Complete) in a glass on glass homogenizer. Homogenates were then centrifuged for 10 min at 200 g to remove nuclei. Supernatants were then centrifuged at 16,000 g for 15 min to isolate crude plasma membrane fractions. Crude plasma membrane proteins were separated by SDS-PAGE and probed by Western blot analysis with antibodies against AdipoR1 and AdipoR2. Skeletal muscle and livers were used for positive controls for the expression of AdipoR1 and AdipoR2. Blots were also probed with antibody to the transferrin receptor to ensure equal loading and to assess the recovery of plasma membranes in the isolation procedure.
Egr-1 DNA binding activity by EMSA
After overnight culture with or without adiponectin, Kupffer cells were treated with 100 ng/ml LPS for 60 min. Nuclear proteins were extracted as previously described (15). Nuclear extracts (15 μg) were then used to assess DNA binding activity by EMSA using an oligonucleotide probe for the sequence of the Egr-1 binding site in the TNF-α promoter (Integrated DNA Technologies; Coralville, IA), as previously described (15). The positive control was prepared from the mouse liver 60 min after an intraperitoneal injection of LPS, as previously described (19).
Real-time PCR
To measure mRNA for AdipoR1 and AdipoR2, Kupffer cells were cultured overnight in CMRL-10% FBS, and total RNA was isolated. To measure the effects of gAcrp on LPS-stimulated TNF-α mRNA accumulation, Kupffer cells were cultured with or without gAcrp for 16–18 h, media were replaced with fresh CMRL-10% FBS, and, after 2 h, cells were then treated with or without 100 ng/ml LPS for 60 min. Total RNA was isolated from Kupffer cells using the RNeasy Micro Kit (Qiagen), with on-column DNA digestion using the RNase-free DNase set (Qiagen) according to the manufacturer’s instructions. Two to three hundred nanograms of total RNA were reverse transcribed using the RETROscript kit (Ambion; Austin, TX) with randon decamers as primers. Real-time PCR amplification was performed in an iCycler (Bio-Rad; Hercules, CA) using SYBR Green PCR Core Reagents (Applied Biosystems; Warrington, UK). The relative amount of target mRNA was determined using the comparative threshold (Ct) method by normalizing target mRNA Ct values to those for GAPDH or β-actin (ΔCt). The primer sequences were as follows: AdipoR1, forward 5′-CAA ACT GGA CTA TTC AGG G-3′ and reverse 5′-CAG ACG ATG GAG AGG TAG-3′; AdipoR2, forward 5′-GCC TTT GTT CAC TTC CAT GGA-3′ and reverse 5′-ATC ACA GCG CAT CCT CTT CA-3′; GAPDH, forward 5′-ATG ATT CTA CCC ACG GCA AG-3′ and reverse 5′-CTG GAA GAT GGT GAT GGG TT-3′; TNF-α, forward 5′-GAA CAA CCC TAC GAG CAC CT-3′ and reverse 5′-GGG TAG TTT GGC TGG GAT AA-3′; and β-actin, forward 5′-CGG TCA GGT CAT CAC TAT CG-3′ and reverse 5′-TTC CAT ACC CAG GAA GGA AG-3′. All primers used for real-time PCR analysis were synthesized by Integrated DNA Technologies. Statistical analysis of real-time PCR data was performed using ΔCt values.
Statistical analysis
Because of the limited number of Kupffer cells available from each animal, data from several feeding trials are presented in this study. Values reported are means ± SE; n represents the number of different animals used per group in each assay. IC50 values were estimated by fitting a one-phase exponential decay curve using the GraphPad Prism 4 program (Graph Pad Software). Data were analyzed by Student’s t-test or a general linear models procedure followed by least-square means analysis of differences between groups (SAS; Carey, IN), blocking for trial effects if data from more than one trial were used. Data were log transformed, if needed, to obtain a normal distribution.
RESULTS
Two recent reports (37, 44) have indicated that chronic ethanol feeding to mice decreases serum adiponectin concentrations. Furthermore, treatment of mice with recombinant adiponectin prevented the development of ethanol-induced fatty liver injury (37). Because adiponectin has potent anti-inflammatory activities in other models of macrophage activation (43), we investigated whether adiponectin can ameliorate the increased sensitivity of Kupffer cells to LPS-mediated activation after chronic ethanol feeding. In a rat model of ad libitum ethanol feeding, which results in fatty liver injury (8), serum adiponectin concentrations were decreased after 4 wk of ethanol exposure compared with pair-fed controls (Fig. 1A). The 45% reduction in serum adiponectin observed in rats (Fig. 1A) was comparable to the reduction reported in mice after 4 wk of ethanol feeding (37). As previously described for this rat model, markers of ethanol-induced fatty liver injury are apparent after 4 wk of ethanol feeding, including increased serum ALT (Fig. 1B), increased TNF-α concentrations (Fig. 1C), and the appearance of micro- and macrovesicular steatosis (Fig. 1D).
Fig. 1.
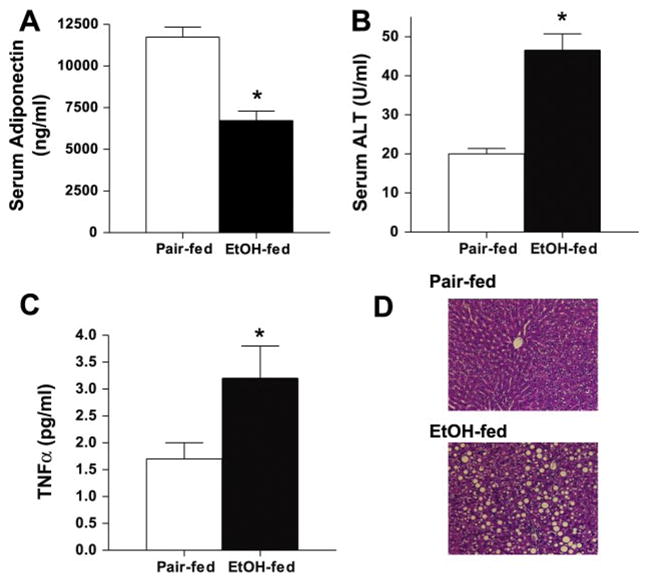
Chronic ethanol feeding decreases serum adiponectin concentration in rats. Rats were allowed free access to an ethanol (EtOH)-containing diet or pair-fed control diets for 4 wk. Serum concentrations of adiponectin (A; n = 12 for pair-fed rats and 11 for EtOH-fed rats), alanine aminotransferase (ALT; B; n = 8), and TNF-α (C; n = 8) were measured. Values represent means ± SE. *P < 0.05 compared with pair-fed rats. D: formalin-fixed liver sections were stained with hematoxylin-eosin. Images are representative of 4 animals.
Chronic ethanol feeding increases the sensitivity of isolated Kupffer cells to LPS stimulation, leading to increased production of TNF-α (Fig. 2A) (1, 15, 16, 28). Treatment of Kupffer cells with gAcrp (5–500 ng/ml) for 16 h dose dependently reduced TNF-α release by Kupffer cells stimulated by 10 ng/ml LPS from both pair- and ethanol-fed rats (Fig. 2B). Maximal inhibition, observed in cells treated with 500 ng/ml, was not affected by chronic ethanol feeding (Fig. 2B). However, Kupffer cells isolated from ethanol-fed rats were more sensitive to the inhibitory effects of gAcrp on LPS-stimulated TNF-α production; the IC50 for gAcrp-mediated inhibition was 31 ng/ml in Kupffer cells from pair-fed rats compared with 10 ng/ml in cells from ethanol-fed rats (Fig. 2B).
Fig. 2.
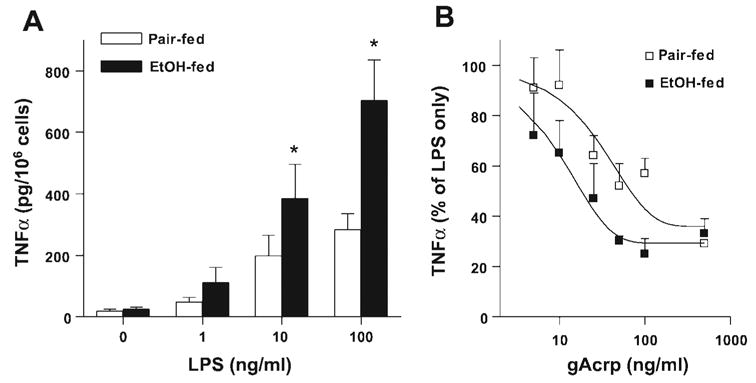
Globular adiponectin (gAcrp) suppresses LPS-stimulated TNF-α production by Kupffer cells. A: Kupffer cells from pair- and EtOH-fed rats were cultured for 16 h and then cultured in fresh CMRL-FBS for 2 h. Cells were then treated with 0–100 ng/ml LPS for 4 h. Cell culture media were removed, and TNF-α production was measured by ELISA. Values represent means ± SE; n = 7–8 except for the 1 ng/ml dose, where n = 4. *P < 0.05 compared with pair-fed rats at a given concentration of LPS. B: Kupffer cells were cultured for 16 h with or without gAcrp and then stimulated with 10 ng/ml LPS for 4 h. The TNF-α concentration was measured by ELISA. Values are expressed as percentages of TNF-α production by cells stimulated with 10 ng/ml LPS but not treated with gAcrp within each diet group. Best-fit curves for suppression of LPS-stimulated TNF-α production in Kupffer cells treated with 0–500 ng/ml gAcrp were generated as described in MATERIALS AND METHODS. Values represent means ± SE; n = 3–7 except at 500 ng/ml gAcrp, where n = 2.
Kupffer cells from ethanol-fed rats were also more sensitive to flAcrp produced in HEK-293 cells. To compare the effects of gAcrp and flAcrp, Kupffer cells were treated overnight with or without 10–1,000 ng/ml adiponectin (gAcrp or flAcrp produced in HEK-293 cells). Adiponectin was then removed, and cells were stimulated in fresh media containing 100 ng/ml LPS for 4 h. In Kupffer cells from both pair- and ethanol-fed rats, gAcrp dose dependently inhibited TNF-α production, even at this higher concentration of LPS (Fig. 3A). As in cells treated with 10 ng/ml LPS (Fig. 2B), Kupffer cells from ethanol-fed rats stimulated with 100 ng/ml LPS were more sensitive to gAcrp than Kupffer cells from pair-fed rats (Fig. 3A). flAcrp showed a similar capacity to gAcrp in suppressing LPS-stimulated TNF-α production after chronic ethanol feeding, decreasing TNF-α production at concentrations as low as 10 ng/ml (Fig. 3A). Inhibition of LPS-stimulated TNF-α production in pair-fed rats by flAcrp was observed only at 1,000 ng/ml flAcrp (Fig. 3A). flAcrp produced in E. coli, which lacks posttranslational glycosylation, did not inhibit LPS-stimulated TNF-α production in cells from either pair- or ethanol-fed rats at concentrations as high as 5,000 ng/ml (data not shown).
Fig. 3.
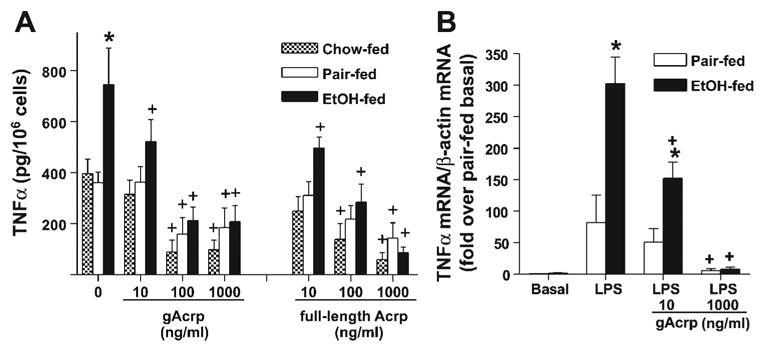
A: gAcrp and full-length adiponectin (flAcrp) suppress LPS-stimulated TNF-α production by Kupffer cells isolated from ad libitum chow-fed, pair-fed, and EtOH-fed rats. Kupffer cells were isolated and cultured with or without 10–1,000 ng/ml gAcrp or flAcrp produced in HEK-293 cells for 16 h. Cells were cultured in fresh CMRL-FBS for 2 h and then stimulated with 100 ng/ml LPS for 4 h. The TNF-α concentration in the media was measured by ELISA. TNF-α production in cells not treated with LPS was not affected by adiponectin (data not shown). Values represent means ± SE; n = 4 for chow-fed rats and 7–10 for pair- and EtOH-fed rats. *P < 0.05 compared with pair-fed rats at each concentration of adiponectin; +P < 0.05 compared with cells not treated with adiponectin in the same diet group. B: gAcrp decreased LPS-stimulated TNF-α mRNA accumulation in Kupffer cells. Kupffer cells were treated with gAcrp as described above and then stimulated with or without 100 ng/ml LPS for 60 min. RNA was then isolated, and TNF-α and β-actin mRNA were measured by real-time PCR. Values represent means ± SE; n = 3. *P < 0.05 compared with pair-fed rats at each concentration of adiponectin; +P < 0.05 compared with cells not treated with adiponectin in the same diet group.
Suppression of LPS-stimulated TNF-α by gAcrp and flAcrp produced in HEK-293 cells was also compared between Kupffer cells isolated from a group of rats allowed ad libitum access to rodent chow with Kupffer cells isolated from ethanol-and pair-fed groups. TNF-α production in response to 100 ng/ml LPS did not differ between pair-fed and ad libitum chow-fed rats (Fig. 3A). Furthermore, whereas 10 ng/ml gAcrp and flAcrp suppressed TNF-α production after chronic ethanol feeding, this dose of gAcrp had no effect on either pair-fed or ad libitum chow-fed rats (Fig. 3A); TNF-α production was only suppressed in pair-fed and ad libitum chow-fed rats at 100–1,000 ng/ml gAcrp or flAcrp. The inclusion of 10 μg/ml polymixin during treatment with gAcrp or flAcrp had no effect on the suppression of LPS-stimulated TNF-α production (data not shown), demonstrating that potential contamination of the adiponectin preparations with endotoxins did not contribute to their biological effectiveness.
The suppression of LPS-induced TNF-α production by gAcrp was paralleled by inhibition of TNF-α mRNA accumulation. LPS increased TNF-α mRNA accumulation in Kupffer cells from both pair- and ethanol-fed rats (Fig. 3B); Kupffer cells from ethanol-fed rats accumulated 3.5-fold more TNF-α mRNA than cells from pair-fed rats (Fig. 3B). Pretreatment with 10 ng/ml gAcrp suppressed TNF-α mRNA accumulation in Kupffer cells from ethanol-fed rats but had no effect on pair-fed rats at this low concentration. However, pretreatment with 1,000 ng/ml gAcrp effectively inhibited the ability of LPS to stimulate TNF-α mRNA accumulation in cells from both pair- and ethanol-fed rats (Fig. 3B).
Expression of immunoreactive AdipoR1 and AdipoR2 was assessed in crude plasma membrane fractions prepared from Kupffer cells isolated from ethanol- and pair-fed rats. Kupffer cells expressed proteins migrating to ~40 kDa on SDS-poly-acrylamide gels that were recognized by antibodies raised against murine AdipoR1 and AdipoR2 proteins (Fig. 4). The skeletal muscle and liver were used as positive controls for tissue known to express AdipoR1 and AdipoR2, respectively. Chronic ethanol feeding had no effect on the relative quantity of AdipoR1 or AdipoR2 protein (Fig. 4). The relative expression of AdipoR1 and AdipoR2 mRNA in Kupffer cells isolated from pair- and ethanol-fed rats was assessed by real-time RT-PCR. mRNA for both AdipoR1 and AdipoR2 was detectable in Kupffer cells. Relative to the liver, Kupffer cells expressed a greater quantity of AdipoR1 and lower levels of AdipoR2 (data not shown). Kupffer cells from pair- and ethanol-fed rats expressed equivalent quantities of AdipoR1 normalized to GAPDH mRNA (2.67 ± 0.7 ΔCt in pair-fed rats and 2.43 ± 0.46 in ethanol-fed rats, n = 3) and AdipoR2 (0.57 ± 0.12 ΔCt in pair-fed rats and 0.73 ± 0.19 in ethanol-fed rats, n = 3), respectively.
Fig. 4.
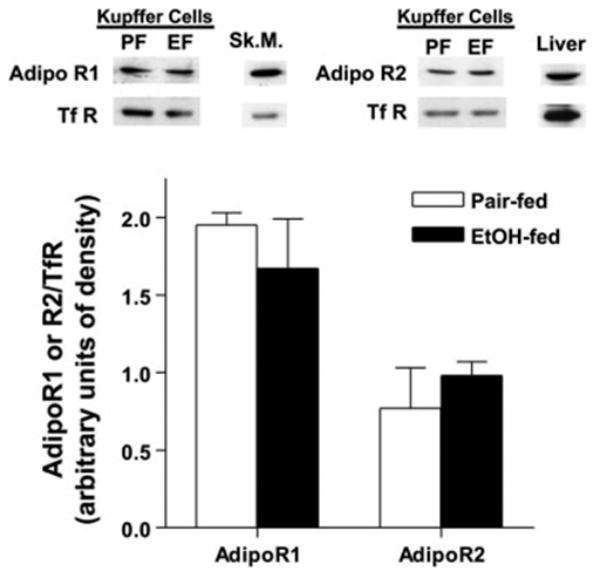
Expression of adiponectin receptors 1 and 2 (AdipoR1 and AdipoR2, respectively) by Kupffer cells from pair-fed (PF) and EtOH-fed (EF) rats. Crude plasma membrane fractions were isolated from Kupffer cells from pair-and EtOH-fed rats, and expression of immunoreactive AdipoR1 and AdipoR2 was assessed by Western blot analysis. Membranes were probed with an antibody to the transferrin receptor (TfR) as a loading control. Skeletal muscle (Sk.M.) and liver fractions were used as positive controls for AdipoR1 and AdipoR2, respectively. Values represent means ± SE; n = 3 for AdipoR1 and 5 for AdipoR2.
To understand the mechanism for the normalization of LPS-stimulated TNF-α production, we investigated the effects of gAcrp on signals activated in response to LPS. Degradation of IκB and the subsequent activation of NFκB is an important signaling pathway that contributes to LPS-stimulated TNF-α production by macrophages and monocytes (42). Whereas chronic ethanol feeding increases hepatic NF-κB activation in vivo (22), LPS-stimulated NF-κB DNA binding activity is not affected by chronic ethanol feeding as measured in the model of isolated Kupffer cells used in this study (15). Here, we monitored the degradation of IκB in response to LPS as an indirect measure of NF-κB activation to assess the effects of gAcrp on NF-κB signaling. Immunoreactive IκB was decreased by 50% in response to LPS in Kupffer cells from both ethanol- and pair-fed rats (Fig. 5A). gAcrp had no effect on IκB at baseline (i.e., before stimulation with LPS) at concentrations up to 1,000 ng/ml (data not shown). Whereas pretreatment with 10 ng/ml gAcrp had no effect on LPS-induced degradation of IκB, when cells were treated with gAcrp at 100–1,000 ng/ml, there was a gradual suppression of LPS-stimulated IκB degradation (Fig. 5A).
Fig. 5.
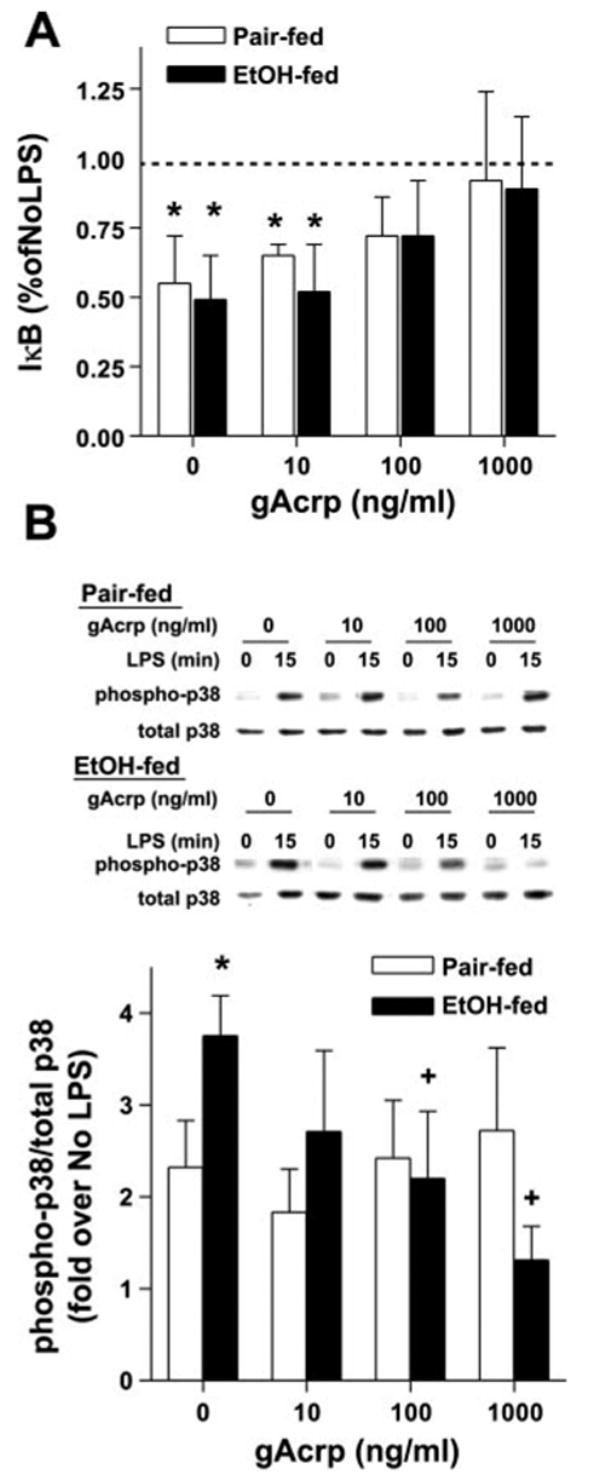
Effect of gAcrp on LPS-stimulated IκB degradation (A) and p38 MAPK phosphorylation (B) in Kupffer cells from EtOH-fed rats. Kupffer cells from pair- and EtOH-fed rats were cultured for 16 h with or without 10–1,000 ng/ml gAcrp. A: cells were then stimulated or not with 100 ng/ml LPS for 30 min to measure IκB degradation. Values represent means ± SE; n = 5. *P < 0.05 compared with cells not treated with LPS in the same diet group. B: cells were then stimulated or not with 100 ng/ml LPS for 15 min to measure phosphorylation of p38. Values represent means ± SE; n = 4. *P < 0.05 compared with pair-fed rats at each concentration of gAcrp; +P < 0.05 compared with cells not treated with gAcrp in the same diet group.
Increased LPS-stimulated TNF-α production by Kupffer cells after chronic ethanol exposure is associated with enhanced activation of two members of the MAPK family, ERK1/2 and p38 (15, 16, 28). We hypothesized that normalization of TNF-α production in response to gAcrp may be mediated by a suppression of LPS-stimulated p38 and/or ERK1/2 MAPK phosphorylation. Chronic ethanol feeding increased p38 MAPK phosphorylation in response to LPS treatment (Fig. 5B) (16). gAcrp suppressed LPS-stimulated p38 phosphorylation in Kupffer cells from ethanol-fed rats at concentrations of 100 ng/ml or greater (Fig. 5B). In contrast, gAcrp did not inhibit LPS-stimulated p38 phosphorylation in Kupffer cells from pair-fed rats, even at 1,000 ng/ml (Fig. 5B). These data are consistent with those of a previous report (36) suggesting that p38 MAPK is not involved in adiponectin-mediated suppression of TNF-α or IL-6 production in porcine macrophages.
LPS-stimulated ERK1/2 phosphorylation was also increased after chronic ethanol feeding compared with Kupffer cells from pair-fed controls (Fig. 6A) (15). Treatment of Kupffer cells with gAcrp suppressed ERK1/2 phosphorylation in both ethanol-fed and pair-fed controls (Fig. 6A). Maximal inhibition of LPS-stimulated ERK1/2 phosphorylation was observed after treatment with 1,000 ng/ml gAcrp and did not differ between Kupffer cells isolated from ethanol- and pair-fed rats. However, Kupffer cells from ethanol-fed rats were more sensitive to gAcrp-mediated suppression, with inhibition of ERK1/2 phosphorylation observed at concentrations as low as 10 ng/ml gAcrp (Fig. 6A). gAcrp treatment had no effect on the expression of total ERK1/2 (Fig. 6A).
Fig. 6.
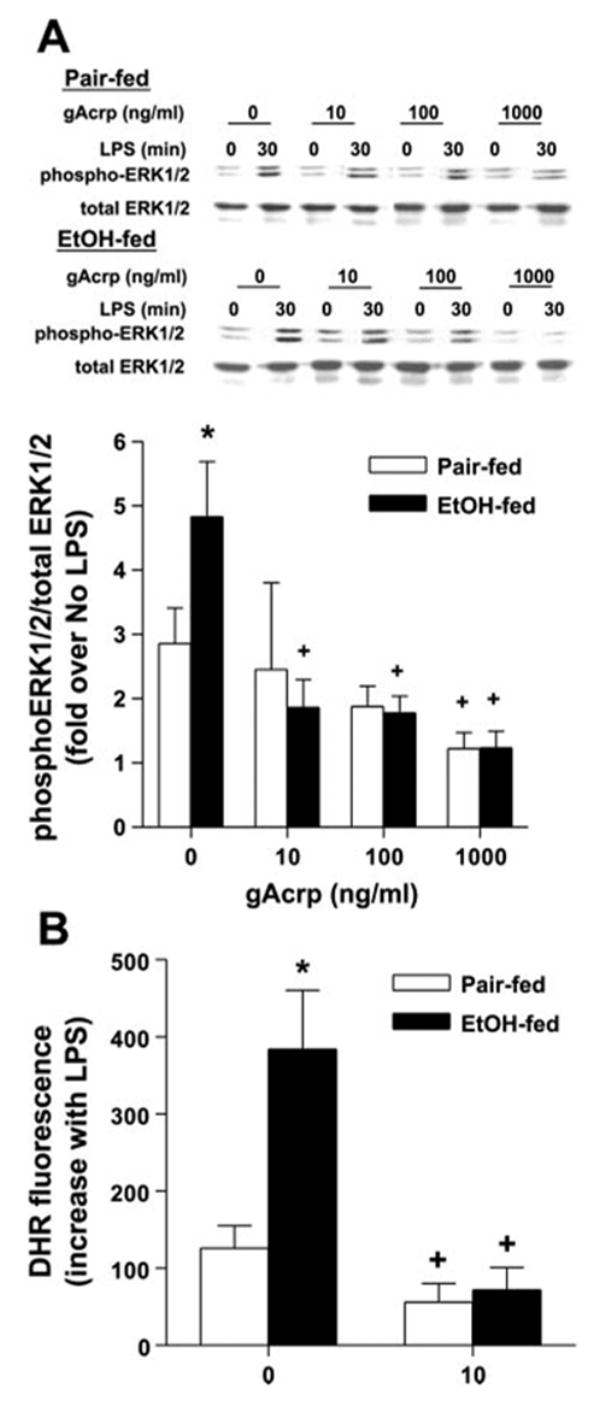
Effect of gAcrp on LPS-stimulated ERK1/2 phosphorylation and ROS production. A: Kupffer cells from pair- and EtOH-fed rats were cultured for 16 h with or without 10–1,000 ng/ml gAcrp. Cells were then stimulated or not with 100 ng/ml LPS for 30 min. Phospho-ERK1/2 and total ERK were assessed by Western blot analysis. Values represent means ± SE; n = 4. *P < 0.05 compared with pair-fed rats at each concentration of gAcrp; +P < 0.05 compared with cells not treated with gAcrp in the same diet group. B: Kupffer cells from pair- and EtOH-fed rats were cultured for 16 h with or without 10 ng/ml gAcrp and then stimulated with 100 ng/ml LPS for 15 min. Media were then replaced with 10 μM dihydrorhodamine (DHR), and cells were incubated for 15 min. Fluorescence was measured at an excitation wavelength at 505 nm and emission detection wavelength of 530 nm. Values represent means ± SE corrected for DHR fluorescence at time 0; n = 5. *P < 0.05 compared with pair-fed rats at each concentration of gAcrp; +P < 0.05 compared with cells not treated with gAcrp in the same diet group.
The data presented here for Kupffer cells and those previously reported for porcine macrophages (36) clearly demonstrate the ability of higher concentrations of adiponectin to suppress multiple LPS-stimulated signaling pathways. However, only LPS-stimulated ERK1/2 activation was suppressed at lower concentrations of gAcrp, suggesting that ERK1/2 signaling is critical to mediating the increased sensitivity of Kupffer cells from ethanol-fed rats to Acrp-mediated inhibition of TNF-α production. LPS-stimulated production of ROS has been implicated in the activation of ERK1/2 in macrophages and monocytes (12, 17). Furthermore, LPS-stimulated production of ROS also contributes to increased ERK1/2 activation in Kupffer cells after chronic ethanol feeding (5, 29). Because 10 ng/ml gAcrp effectively normalized LPS-stimulated ERK1/2 activation in Kupffer cells from rats fed chronic ethanol, we asked whether gAcrp also suppressed the ability of LPS to increase ROS production. LPS increased DHR fluorescence, a measure of ROS production, in Kupffer cells from both pair-and ethanol-fed rats, with higher fluorescence observed after chronic ethanol feeding (Fig. 6B). Overnight treatment with 10 ng/ml gAcrp suppressed ROS production in cells from both pair- and ethanol-fed rats (Fig. 6B).
We next investigated whether LPS signaling events downstream of ERK1/2 activation were also inhibited by gAcrp. One critical downstream target of ERK1/2 involved in mediating the effects of chronic ethanol on TNF-α production is the expression and DNA binding activity of Egr-1, an immediate-early gene required for maximal activation of the TNF-α promoter in response to LPS (28, 42). The functional activity of Egr-1 was assessed by EMSA using an oligonucleotide for the Egr-1 binding site in the murine TNF-α promoter. LPS-stimulated Egr-1 DNA binding activity was increased after chronic ethanol feeding (Fig. 7) (15). Overnight treatment with gAcrp suppressed LPS-stimulated Egr-1 DNA binding activity, with a more sensitive response observed in Kupffer cells isolated from ethanol-fed rats (Fig. 7B). Taken together, these data suggest that gAcrp antagonizes the ethanol-induced increase in LPS-stimulated ERK1/2 phosphorylation and, as a consequence, suppresses the activation of Egr-1 DNA binding activity, contributing to a reduction of LPS-stimulated TNF-α production.
Fig. 7.
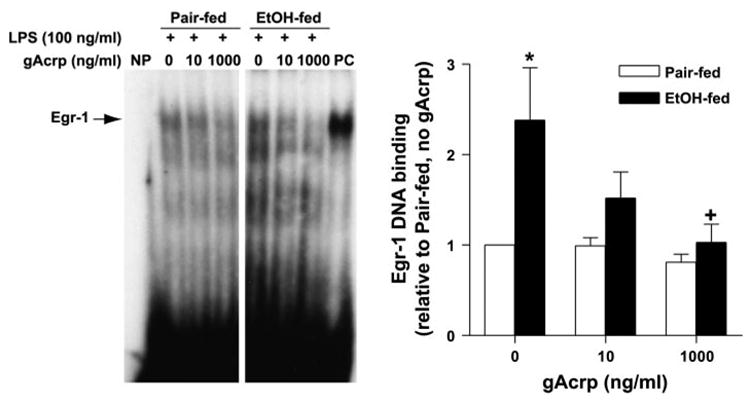
gAcrp suppresses LPS-stimulated early growth response-1 (Egr-1) DNA binding activity. Kupffer cells from pair- and EtOH-fed rats were cultured for 16 h with 0, 10, or 1,000 ng/ml gAcrp. Cells were then stimulated or not with 100 ng/ml LPS for 60 min, and nuclear proteins were extracted. DNA binding activity was then measured by EMSA. Control lanes are shown as no protein loaded (NP) and a positive control (PC) of a nuclear extract from the liver of a mouse treated with LPS via an intraperitoneal injection; n = 6. *P < 0.05 compared with pair-fed rats at each concentration of gAcrp; +P < 0.005 compared with cells not treated with gAcrp in the same diet group.
DISCUSSION
Adiponectin is an adipokine secreted by adipose tissue that regulates the function of a number of cells and tissues, including the liver, muscle, and macrophages (4). Previous work (37, 44) has shown that chronic ethanol feeding to mice decreases serum adiponectin concentrations. Importantly, treatment with adiponectin (37) or restoration of circulating adiponectin concentrations in response to high-saturated fat diets (44) prevents the development of chronic ethanol-induced liver injury in mice. Protection by adiponectin is associated with increased fatty acid oxidation and decreased fatty acid synthesis in the liver as well as a reduction in circulating TNF-α (37). Kupffer cells are an important source of TNF-α production in the liver and show increased sensitivity to activation by LPS after chronic ethanol exposure (21). Here, we report that chronic ethanol feeding to rats decreased serum adiponectin concentrations and, furthermore, that treatment of Kupffer cells isolated from ethanol-fed rats with gAcrp or flAcrp potently suppressed LPS-stimulated TNF-α production. After rats were fed chronic ethanol, their Kupffer cells were more sensitive to the inhibitory effects of gAcrp on LPS-mediated signaling. The enhancement of LPS-stimulated ERK1/2 phosphorylation and Egr-1 DNA binding activity observed after ethanol feeding were particularly sensitive to gAcrp, with 10 ng/ml gAcrp ameliorating the increased sensitivity of Kupffer cells from ethanol-fed rats to LPS. These data suggest that, in addition to the effects of adiponectin on hepatic lipid homeostasis (37), a potent anti-inflammatory effect of adiponectin on Kupffer cell responses may also contribute to the protective effect of adiponectin therapy against ethanol-induced liver injury.
The molecular mechanisms of adiponectin-mediated responses are not well understood. Adiponectin circulates in the blood in several molecular forms. flAcrp contains both the globular head region and the collagen-like domain (4). Circulating adiponectin is found primarily as trimers and hexamers as well as higher-order complexes containing 12–18 subunits (high-molecular-weight adiponectin) (25). There is a growing consensus that oligomerization of adiponectin is important for some of the biological functions of adiponectin (30, 31). In particular, higher oligomeric complexes are important in mediating the effects of adiponectin on insulin sensitivity (26, 32). Although only flAcrp oligomerizes, the globular head domain of adiponectin can form trimers (30, 32). It is not clear whether gAcrp is present in the circulation. Whereas one study (9) measured gAcrp in serum using immunoprecipitation techniques, other groups have not detected gAcrp in the circulation or found it secreted by 3T3-L1 adipocytes (34). However, a recent report (33) of proteolytic activity secreted by the monocytic THP-1 cell line that cleaves flAcrp to gAcrp suggests that localized production of gAcrp could play a role in the mediating adiponectin function, particularly in monocytes/macrophages.
Posttranslational modification of flAcrp was critical to its anti-inflammatory effects on Kupffer cells. flAcrp produced in HEK-293 cells suppressed LPS-stimulated TNF-α production by Kupffer cells (Fig. 2), whereas flAcrp generated in E. coli had no effect on Kupffer cells (data not shown). Adipocytes secrete adiponectin that is both hydroxylated and glycosylated (34). Posttranslational modifications are required for the insulin-sensitizing effects of flAcrp on hepatocytes (3, 34) and in vivo (3). A similar requirement for posttranslational modification of flAcrp can be inferred from studies (9, 41) comparing the effectiveness of gAcrp to flAcrp produced by E. coli in regulating glucose homeostasis and insulin sensitivity. In contrast to the requirement for posttranslational processing of flAcrp, gAcrp, generated in E. coli, is effective in some, but not all, cell types/model systems. For example, gAcrp enhances insulin-stimulated glucose uptake in adipocytes (35) and skeletal muscle cells (6), increases fatty acid oxidation in the heart (24), and increases fatty acid oxidation in skeletal muscle (9). gAcrp activates AMP kinase activity in myocytes but, in contrast, has no effect on AMP kinase activity in hepatocytes (32).
Two adiponectin receptors have been identified: AdipoR1 and AdipoR2 (38, 39). The studies cited above (6, 9, 24, 32, 35) demonstrating the effectiveness of gAcrp were all carried out in cells/tissues expressing AdipoR1, consistent with the higher binding affinity of gAcrp to skeletal muscle, on the order of 150 ng/ml compared with the liver (39). In contrast, flAcrp has a higher affinity to the liver than skeletal muscle, consistent with the predominant expression of AdipoR2 in hepatocytes (39). Differential distribution of AdipoR1 and AdipoR2 is thought to contribute, at least in part, to the differential efficacy of gAcrp compared with flAcrp in different tissues (11).
Several lines of investigation have suggested that gAcrp may be effective in mediating the anti-inflammatory effects of adiponectin. Restoration of serum adiponectin concentrations by overexpression of gAcrp in two mouse models of diabetes/obesity (ob/ob) and atherosclerosis (apolipoprotein E deficient) protected mice from the progression of diabetes and atherosclerosis, respectively (40). Overexpression of gAcrp was associated with decreased macrophage activation in the arterial wall (40). In isolated rat Kupffer cells from chow-, pair-, and ethanol-fed rats, both flAcrp expressed in mammalian HEK-293 cells and gAcrp produced in E. coli were effective at suppressing TNF-α production in response to LPS (Figs. 2 and 3). Human macrophages have been reported to express both AdipoR1 and AdipoR2 mRNA (7), and rat Kupffer cells expressed both AdipoR1 and AdipoR2 mRNA and protein (Fig. 4). One microgram per milliliter (1,000 ng/ml) gAcrp effectively suppressed several LPS-stimulated signaling cascades, including the degradation of IκB and phosphorylation of ERK1/2 in Kupffer cells isolated from control rats (Figs. 5 and 6). Taken together, these data suggest that gAcrp, likely acting via AdipoR1, as well as flAcrp, are important regulators of macrophage function.
One important aspect of the studies described here is the characterization of increased sensitivity of Kupffer cells isolated from ethanol-fed rats to the anti-inflammatory effects of adiponectin. Kupffer cells isolated from rats chronically exposed to ethanol are more sensitive to LPS, leading to enhanced production of TNF-α (Fig. 2) (1, 5, 28). Although TNF-α is only one of the many inflammatory mediators produced by Kupffer cells, it is a useful readout for studies investigating the mechanisms by which chronic ethanol disrupts the regulated expression of pro- and anti-inflammatory mediators. After chronic ethanol feeding, increased LPS-stimulated TNF-α production is mediated, at least in part, by increased activation of two MAPK family members, ERK1/2 and p38 (5, 16, 28). Because adiponectin normalized LPS-stimulated TNF-α production by Kupffer cells from rats fed chronic ethanol (Figs. 2 and 3), we hypothesized that adiponectin might be acting to ameliorate the chronic ethanol-induced increases in LPS-stimulated ERK1/2 and/or p38 activation. Whereas gAcrp did suppress LPS-stimulated p38 activation in Kupffer cells from ethanol-fed rats, inhibition of p38 was less sensitive to gAcrp than the effect of gAcrp on TNF-α production. In contrast, gAcrp potently suppressed LPS-stimulated ERK1/2 phosphorylation, with a sensitivity that paralleled the inhibition of TNF-α production. Similarly, gAcrp normalized LPS-stimulated Egr-1 DNA binding activity, consistent with the hypothesis that the ERK1/2-Egr-1 pathway is a primary target for the anti-inflammatory effects of gAcrp on Kupffer cells from ethanol-fed rats.
The mechanism for the enhanced sensitivity to adiponectin after chronic ethanol exposure is not clear. We first hypothesized that chronic ethanol may increase the expression of AdipoRs and/or increase the ability of adiponectin to activate specific signaling cascades. However, the expression of Adi-poR1 and AdipoR2, at both the levels of mRNA or proteins, was not different between Kupffer cells isolated from pair- and ethanol-fed rats (Fig. 5). Short-term treatment of Kupffer cells with gAcrp activated AMP kinase phosphorylation as well as p38 and ERK1/2 phosphorylation. However, chronic ethanol feeding did not enhance gAcrp-stimulated signals (data not shown). These data suggest that chronic ethanol may not enhance adiponectin-dependent signaling pathways per se. Instead, they suggest that one or more adiponectin-mediated signals may act to specifically normalize the particular defect in LPS signaling that leads to increased sensitivity of Kupffer cells to LPS after chronic ethanol feeding. Our data suggest that LPS-stimulated ERK1/2 phosphorylation is such a sensitive target of gAcrp. Although the mechanisms for increased ERK1/2 activity after chronic ethanol are not well understood, increased production of ROS after ethanol feeding is thought to contribute to enhanced LPS-stimulated ERK1/2 activation (5, 29). Here, we show that LPS-stimulated ROS production and ERK1/2 phosphorylation were potently suppressed by gAcrp, with ROS production by Kupffer cells normalized after treatment with 10 ng/ml gAcrp (Fig. 5). A similar inhibitory effect of adiponectin on oxidized LDL-stimulated ROS production and subsequent ERK1/2 activation has been reported in endothelial cells (20). In endothelial cells, it has been proposed that gAcrp inhibits the activation of NADPH oxidase (11, 20). Further experimentation will be required to determine whether adiponectin inhibits LPS-stimulated NADPH oxidase in Kupffer cells. However, such a mechanism would be consistent with the rapid activation of NADPH oxidase by LPS in macrophages (14).
Chronic ethanol feeding to mice (37, 44) and rats (Fig. 1) decreases the concentration of serum adiponectin. This decrease in adiponectin is associated with the development of liver injury in response to chronic ethanol feeding in mice (37). Adiponectin treatment protected against ethanol-induced liver injury in this mouse model of chronic ethanol exposure as well as liver injury associated with a genetic model of obesity (ob/ob) (37). This protective effect is due, at least in part, to increased fatty acid oxidation and decreased fatty acid synthesis in the liver and was associated with decreased TNF-α (37). Here, we show that adiponectin also has important anti-inflammatory effects in Kupffer cells isolated from rats fed chronic ethanol, suggesting that adiponectin therapy may also act to normalize Kupffer cell responses to LPS during chronic ethanol exposure. This hypothesis is consistent with a previous report (18) demonstrating that adiponectin also protects against LPS-induced liver injury by suppressing TNF-α expression in KK-Ay obese mice. Taken together, these data suggest that the anti-inflammatory effects of adiponectin on Kupffer cells, along with the potential to normalize fatty acid metabolism in the liver (37), likely contribute to the protective effects of adiponectin therapy in preventing chronic ethanol-induced liver injury.
Acknowledgments
We thank Gayley Woolston and Colleen Finn for the help with this project as summer students and Qifang Wang for the isolation of Kupffer cells. We also thank Dr. Abram Stavitsky for helpful discussions throughout the course of this project and Peprotech for the kind gift of globular adiponectin used in preliminary experiments.
GRANTS
This work was supported by National Insititute on Alcoholism and Alcohol Abuse Grants AA-011975 and AA-013868. We also acknowledge support from Case/UHC CFAR (NIH AI-36219).
References
- 1.Aldred A, Nagy LE. Ethanol dissociates hormone-stimulated cAMP production from inhibition of TNFα production in rat Kupffer cells. Am J Physiol Gastrointest Liver Physiol. 1999;276:G98–G106. doi: 10.1152/ajpgi.1999.276.1.G98. [DOI] [PubMed] [Google Scholar]
- 2.Bautista AP. Neutrophilic infiltration in alcoholic hepatitis. Alcohol. 2002;27:17–21. doi: 10.1016/s0741-8329(02)00206-9. [DOI] [PubMed] [Google Scholar]
- 3.Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med. 2001;7:947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- 4.Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab. 2002;13:84–89. doi: 10.1016/s1043-2760(01)00524-0. [DOI] [PubMed] [Google Scholar]
- 5.Cao Q, Mak KM, Lieber CS. Dilinoleoylphosphatidylcholine decreases LPS-induced TNFα generation in Kupffer cells of ethanol-fed rats: respective roles of MAPKs and NFκB. Biochem Biophys Res Commun. 2002;294:849–853. doi: 10.1016/S0006-291X(02)00586-7. [DOI] [PubMed] [Google Scholar]
- 6.Ceddia RB, Somwar R, Maida A, Fang X, Bikopoulos G, Sweeney G. Globular adiponectin increases GLUT4 translocation and glucose uptake but reduces glycogen synthesis in rat skeletal muscle cells. Diabetologia. 2005;48:132–139. doi: 10.1007/s00125-004-1609-y. [DOI] [PubMed] [Google Scholar]
- 7.Chinetti G, Zawadski C, Fruchart JC, Staels B. Expression of adiponectin receptors in human macrophages and regulation by agonists of the nuclear receptors PPARalpha, PPARgamma, and LXR. Biochem Biophys Res Commun. 2004;314:151–158. doi: 10.1016/j.bbrc.2003.12.058. [DOI] [PubMed] [Google Scholar]
- 8.de la Hall PM, Lieber CS, DeCarli LM, French SW, Lindros KO, Jarvelainen H, Bode C, Parlesak A, Bode JC. Models of alcoholic liver disease in rodents: a critical evaluation. Alcohol Clin Exp Res. 2001;25:254S–261S. doi: 10.1097/00000374-200105051-00041. [DOI] [PubMed] [Google Scholar]
- 9.Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, Bihain BE, Lodish HF. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci USA. 2001;98:2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furukawa K, Hori M, Ouchi N, Kihara S, Funahashi T, Matsuzawa Y, Miyazaki A, Nakayama H, Horiuchi S. Adiponectin down-regulates acyl-coenzyme A:cholesterol acyltransferase-1 in cultured human monocyte-derived macrophages. Biochem Biophys Res Commun. 2004;317:831–836. doi: 10.1016/j.bbrc.2004.03.123. [DOI] [PubMed] [Google Scholar]
- 11.Goldstein BJ, Scalia R. Adiponectin: a novel adipokine linking adipocytes and vascular function. J Clin Endocrinol Metab. 2004;89:2563–2568. doi: 10.1210/jc.2004-0518. [DOI] [PubMed] [Google Scholar]
- 12.Hsu HY, Wen MH. Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J Biol Chem. 2002;277:22131–22139. doi: 10.1074/jbc.M111883200. [DOI] [PubMed] [Google Scholar]
- 13.Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem. 1996;271:10697–10703. doi: 10.1074/jbc.271.18.10697. [DOI] [PubMed] [Google Scholar]
- 14.Iles KE, Forman HJ. Macrophage signaling and respiratory burst. Immunol Res. 2002;26:95–105. doi: 10.1385/IR:26:1-3:095. [DOI] [PubMed] [Google Scholar]
- 15.Kishore R, Hill JR, McMullen MR, Frenkel J, Nagy LE. ERK1/2 and Egr-1 contribute to increased TNFα production in rat Kupffer cells after chronic ethanol feeding. Am J Physiol Gastrointest Liver Physiol. 2002;282:G6–G15. doi: 10.1152/ajpgi.00328.2001. [DOI] [PubMed] [Google Scholar]
- 16.Kishore R, McMullen MR, Nagy LE. Stabilization of TNFα mRNA by chronic ethanol: role of A+U rich elements and p38 mitogen activated protein kinase signaling pathway. J Biol Chem. 2001;276:41930–41937. doi: 10.1074/jbc.M107181200. [DOI] [PubMed] [Google Scholar]
- 17.Landmann R, Scherer F, Schumann R, Link S, Sansano S, Zimmerli W. LPS directly induces oxygen radical production in human monocytes via LPS binding protein and CD14. J Leukoc Biol. 1995;57:440–449. doi: 10.1002/jlb.57.3.440. [DOI] [PubMed] [Google Scholar]
- 18.Masaki T, Chiba S, Tatsukawa H, Yasuda T, Noguchi H, Seike M, Yoshimatsu H. Adiponectin protects LPS-induced liver injury through modulation of TNF-alpha in KK-Ay obese mice. Hepatology. 2004;40:177–184. doi: 10.1002/hep.20282. [DOI] [PubMed] [Google Scholar]
- 19.McMullen MR, Pritchard MT, Wang Q, Millward CA, Croniger CM, Nagy LE. Early growth response-1 transcription factor is essential for ethanol-induced fatty liver injury in mice. Gastroenterology. 2005;128:2066–2076. doi: 10.1053/j.gastro.2005.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motoshima H, Wu X, Mahadev K, Goldstein BJ. Adiponectin suppresses proliferation and superoxide generation and enhances eNOS activity in endothelial cells treated with oxidized LDL. Biochem Biophys Res Commun. 2004;315:264–271. doi: 10.1016/j.bbrc.2004.01.049. [DOI] [PubMed] [Google Scholar]
- 21.Nagy LE. New insights into the role of the innate immune response in the development of alcoholic liver disease. Exp Biol Med. 2003;228:882–890. doi: 10.1177/153537020322800803. [DOI] [PubMed] [Google Scholar]
- 22.Nanji AA, Jokelainen K, Rahemtulla A, Miao L, Fogt F, Matsumoto H, Tahan SR, Su GL. Activation of nuclear factor kappa B and cytokine imbalance in experimental alcoholic liver disease in the rat. Hepatology. 1999;30:934–943. doi: 10.1002/hep.510300402. [DOI] [PubMed] [Google Scholar]
- 23.Okamoto Y, Kihara S, Ouchi N, Nishida M, Arita Y, Kumada M, Ohashi K, Sakai N, Shimomura I, Kobayashi H, Terasaka N, Inaba T, Funahashi T, Matsuzawa Y. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2002;106:2767–2770. doi: 10.1161/01.cir.0000042707.50032.19. [DOI] [PubMed] [Google Scholar]
- 24.Onay-Besikci A, Altarejos JY, Lopaschuk GD. gAd-globular head domain of adiponectin increases fatty acid oxidation in newborn rabbit hearts. J Biol Chem. 2004;279:44320–44326. doi: 10.1074/jbc.M400347200. [DOI] [PubMed] [Google Scholar]
- 25.Pajvani UB, Du X, Combs TP, Berg AH, Rajala MW, Schulthess T, Engel J, Brownlee M, Scherer PE. Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications fpr metabolic regulation and bioactivity. J Biol Chem. 2003;278:9073–9085. doi: 10.1074/jbc.M207198200. [DOI] [PubMed] [Google Scholar]
- 26.Pajvani UB, Hawkins M, Combs TP, Rajala MW, Doebber T, Berger JP, Wagner JA, Wu M, Knopps A, Xiang AH, Utzschneider KM, Kahn SE, Olefsky JM, Buchanan TA, Scherer PE. Complex distribution, not absolute amount of adiponectin, correlates with thiazo-lidinedione-mediated improvement in insulin sensitivity. J Biol Chem. 2004;279:12152–12162. doi: 10.1074/jbc.M311113200. [DOI] [PubMed] [Google Scholar]
- 27.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 28.Shi L, Kishore R, McMullen M, Nagy LE. Chronic ethanol increases LPS-stimulated Egr-1 expression in RAW 264.7 macrophages: contribution to enhanced TNFα production. J Biol Chem. 2002;277:14777–14785. doi: 10.1074/jbc.M108967200. [DOI] [PubMed] [Google Scholar]
- 29.Thakur V, Pritchard MT, McMullen MR, Wang Q, Nagy LE. Chronic ethanol feeding increased activation of NADPH oxidase by LPS in rat Kupffer cells. J Leuk Biol. 2006 doi: 10.1189/jlb.1005613. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsao TS, Murrey HE, Hug C, Lee DH, Lodish HF. Oligomerization state-dependent activation of NF-kappa B signaling pathway by adipocyte complement-related protein of 30 kDa (Acrp30) J Biol Chem. 2002;277:29359–29362. doi: 10.1074/jbc.C200312200. [DOI] [PubMed] [Google Scholar]
- 31.Tsao TS, Tomas E, Murrey HE, Hug C, Lee DH, Ruderman NB, Heuser JE, Lodish HF. Role of disulfide bonds in Acrp30/adiponectin structure and signaling specificity. Different oligomers activate different signal transduction pathways. J Biol Chem. 2003;278:50810–5087. doi: 10.1074/jbc.M309469200. [DOI] [PubMed] [Google Scholar]
- 32.Waki H, Yamauchi T, Kamon J, Ito Y, Uchida S, Kita S, Hara K, Hada Y, Vasseur F, Froguel P, Kimura S, Nagai R, Kadowaki T. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J Biol Chem. 2003;278:40352–40363. doi: 10.1074/jbc.M300365200. [DOI] [PubMed] [Google Scholar]
- 33.Waki H, Yamauchi T, Kamon J, Kita S, Ito Y, Hada Y, Uchida S, Tsuchida A, Takekawa S, Kadowaki T. Generation of globular fragment of adiponectin by leukocyte elastase secreted by monocytic cell line THP-1. Endocrinology. 2005;146:790–796. doi: 10.1210/en.2004-1096. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Xu A, Knight C, Xu LY, Cooper GJ. Hydroxylation and glycosylation of the four conserved lysine residues in the collagenous domain of adiponectin. Potential role in the modulation of its insulin-sensitizing activity. J Biol Chem. 2002;277:19521–19529. doi: 10.1074/jbc.M200601200. [DOI] [PubMed] [Google Scholar]
- 35.Wu X, Motoshima H, Mahadev K, Stalker TJ, Scalia R, Goldstein BJ. Involvement of AMP-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes. 2003;52:1355–1363. doi: 10.2337/diabetes.52.6.1355. [DOI] [PubMed] [Google Scholar]
- 36.Wulster-Radcliffe MC, Ajuwon KM, Wang J, Christian JA, Spurlock ME. Adiponectin differentially regulates cytokines in porcine macrophages. Biochem Biophys Res Commun. 2004;316:924–929. doi: 10.1016/j.bbrc.2004.02.130. [DOI] [PubMed] [Google Scholar]
- 37.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 39.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 40.Yamauchi T, Kamon J, Waki H, Imai Y, Shimozawa N, Hioki K, Uchida S, Ito Y, Takakuwa K, Matsui J, Takata M, Eto K, Terauchi Y, Komeda K, Tsunoda M, Murakami K, Ohnishi Y, Naitoh T, Yamamura K, Ueyama Y, Froguel P, Kimura S, Nagai R, Kadowaki T. Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J Biol Chem. 2003;278:2461–2468. doi: 10.1074/jbc.M209033200. [DOI] [PubMed] [Google Scholar]
- 41.Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 42.Yao J, Mackman N, Edgington TS, Fan ST. Lipopolysaccharide induction of the tumor necrosis factor α promoter in human monocytic cells. Regulation by egr-1, c-jun and NFkB transcription factors. J Biol Chem. 1997;272:17795–17801. doi: 10.1074/jbc.272.28.17795. [DOI] [PubMed] [Google Scholar]
- 43.Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N, Kihara S, Funahashi T, Tenner AJ, Tomiyama Y, Matsuzawa Y. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood. 2000;96:1723–1732. [PubMed] [Google Scholar]
- 44.You M, Considine RV, Leone TC, Kelly DP, Crabb DW. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology. 2005;42:568–577. doi: 10.1002/hep.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]