

Abstract
Transglutaminase 2 (TG2) is a multi-domain, multi-functional enzyme that post-translationally modifies proteins by catalyzing the formation of intermolecular isopeptide bonds between glutamine and lysine side-chains. It plays a role in diverse biological functions, including extracellular matrix formation, integrin mediated signaling, and signal transduction involving 7-transmembrane receptors. While some of the roles of TG2 under normal physiological conditions remain obscure, the protein is believed to participate in the pathogenesis of several unrelated diseases including celiac sprue, neurodegenerative diseases, and certain types of cancer. A variety of small molecule and peptidomimetic inhibitors of the TG2 active site have been identified. Here we summarize the biochemistry, biology, pharmacology and medicinal chemistry of human TG2.
Keywords: transglutaminase, inhibitors, chemical structures, celiac sprue, neurodegeneration, cancer
1. Introduction
Transglutaminases were first discovered in the 1950s as enzymes found in mammalian liver homogenates capable of catalyzing calcium-dependent covalent bonds between small molecule amines and certain proteins with the corresponding release of ammonia (Borsook et al., 1949; Clarke et al., 1959). Since that time, nine members have been added to the transglutaminase family of enzymes, including transglutaminases 1–7, factor XIIIA, and the enzymatically inactive erythrocyte band 4.2 (for reviews see Griffin et al., 2002; Lorand & Graham, 2003). Of these nine family members, transglutaminase 2 (tissue transglutaminase, transglutaminase C, tTG, TG2, Ga h) is among the most well studied and biologically characterized presumably because it is present in most bodily tissues (Thomazy & Fesus, 1989) and has been implicated to play a role in a number of diseases, such as celiac sprue (Molberg et al., 2000), Alzheimer’s disease, Huntington’s disease (Hoffner & Djian, 2005), and certain types of cancer (Mangala & Mehta, 2005).
In an effort to treat patients that have these debilitating and often fatal diseases, small molecule and peptidomimetic inhibitors capable of blocking TG2 enzymatic activity have been designed and biochemically characterized. In this review, we will briefly summarize what is known about the biology of TG2, compare the different classes of TG2 inhibitors that have been reported, and discuss the application of these inhibitors to biological systems.
2. Functions of transglutaminase 2
Transglutaminase 2 is an ~ 80 kDa protein with enzymatic, G-protein, and non-enzymatic biological functions (for reviews see Zemskov, 2006; ; Lorand & Graham, 2003; Griffin et al., 2002; Chen & Mehta, 1999; Aeschlimann & Paulsson, 1994). As an enzyme, TG2 is able to catalyze the calcium-dependent (Ka ~ 1 mM) (Piper et al., 2002) covalent modification of protein-bound glutamine side chains through transamidation or deamidation reactions. The first step in both types of modifications is the acylation of the active site cysteine (Cys277) of TG2 by a protein-bound glutamine residue, resulting in the liberation of ammonia and the formation of a thioester intermediate between TG2 and the glutamine bearing protein substrate (see Figure 1). In TG2 catalyzed transamidation, the thioester intermediate is attacked by a nucleophilic primary amine, either a small molecule amine such as putrescine or the e-amino group of protein-bound lysine residues. This results in the formation of relatively stable isopeptide bonds. In TG2 catalyzed deamidation, water acts as the nucleophile that attacks the thioester intermediate resulting in the conversion of the glutamine residue into a glutamate residue (Lorand & Graham, 2003; Case & Stein, 2003). The transamidation reaction is kinetically favored over deamidation at pH > 7, but the deamidation reaction becomes kinetically competitive as the pH is lowered below 7 or as the concentration of amine substrates is lowered below their Km values (Fleckenstein et al., 2002). Despite a certain level of substrate specificity, the abundance of glutamine and lysine residues found on the surface of many proteins renders them potential targets of TG2 modification (Esposito & Caputo, 2005). Therefore, the enzymatic activity of TG2 is tightly regulated both by cofactors and spatial localization in order to prevent excessive protein aggregation through e-(?-glutamyl) lysine crosslink formation and to prevent potential autoimmune activation through protein deamidation.
Figure 1.

Transglutaminase 2 catalytic mechanism. In the presence of calcium, the TG2 active site cysteine (Cys277) thiol attacks the ?-glutaminyl side chain of a protein- or peptide-bound glutamine residue forming a thioester intermediate with the release of ammonia. In transamidation, a primary amine nucleophile attacks the thioester carbonyl displacing the TG2 thiol and resulting in an isopeptide crosslink between the glutamine side chain and the primary amine. In deamidation, water acts as the thiol-displacing nucleophile resulting in the net conversion of glutamine to glutamate. The presence of GTP or GDP inhibits transglutaminase activity.
GTP and GDP are both able to bind to TG2 and inhibit enzymatic activity (Kdapp(GTP?S) = 320 nM, K dapp(GDP) = 1.5 μM, IC50(GTP) = 9 μM) (Datta et al., 2006; Lai et al., 1998). TG2 localized to the cell cytosol, where the GTP concentration is approximately 100 μM and the free calcium level is around 100 nM, is enzymatically latent under normal physiological conditions (Smethurst & Griffin, 1996; Zhang et al., 1998). This cytosolic fraction of TG2 instead fulfills a second function, namely it acts as a G-protein that transduces signals received from the a1B and a 1D adrenergic receptors (Chen et al., 1996) through activation of phospholipase Cd1 (Hwang et al., 1995; Baek et al., 2001). Calcium ionophores, such as calcimycin A23187, capable of transporting free calcium across cell membranes by diffusion, can increase intracellular calcium levels high enough to activate intracellular TG2 (Fok et al., 2006; Zhang et al., 1998, Siefring et al., 1978). This system may mimic a cell that has lost calcium homeostasis due to tissue damage, apoptosis, or necrosis (Afford & Randhawa, 2000; Nicholas et al., 2003). It has been suggested that activated cytosolic TG2 may crosslink intracellular proteins to prevent them from spilling out of a dying cell and causing inflammation (Fesus et al., 1987).
A fraction of cellular TG2 is exported to the cell surface even though the protein contains no export leader sequence or post-translational modifications despite the presence of potential N-linked glycosylation sites, suggesting a non-ER/Golgi route of externalization (Lorand & Graham, 2003; Chen & Mehta, 1999). Cell surface TG2 associates with integrins expressing the beta 1, 3, or 5 subunits and has been shown to enhance cell adhesion, cell spreading, and cell migration on the extracellular matrix protein (ECM) fibronectin (Akimov et al., 2000; Akimov & Belkin, 2001a; Zemskov et al., 2006). Although the protein-protein surface interactions between TG2 and integrins have not been mapped out yet, the high affinity (Kd =7.1 nM) (Akimov & Belkin, 2001b) interaction between TG2 and fibronectin has been localized to a hairpin loop in the N-terminal domain of TG2 consisting of amino acids Trp88 – Thr106 (Hang et al., 2005) and the 42-kDa gelatin binding domain of fibronectin (Radek et al., 1993). TG2 has been shown to act as a cell surface adhesion co-receptor forming a ternary complex with cell surface integrins and ECM fibronectin capable of relaying signals from outside of the cell to the intracellular environment activating RhoA/ROCK (Janiak et al, 2006) and focal adhesion kinase (Akimov et al., 2000) cell signaling cascades. Studies in which the active site cysteine (Cys277) has been mutated to serine (C277S) to inactivate enzymatic crosslinking activity have shown that the cell adhesion, spreading, migration, and signaling functions of cell surface TG2 are independent of crosslinking activity (Akimov et al., 2000; Janiak et al., 2006; Balklava et al., 2002). It is currently unclear whether the crosslinking activity of cell surface TG2 is active or held latent due to its interaction with integrins despite the high calcium, low GTP content of the extracellular environment.
Histological staining of mammalian organs for TG2 has revealed the ubiquitous expression pattern of this protein throughout the body. While some common cell types, such as endothelial cells, macrophages, certain fibroblasts, and smooth muscle cells, show consistently high expression of the protein (Thomazy & Fesus, 1989), a fraction of extracellular TG2 colocalizes with fibronectin in the ECM (Korponay-Szabo et al., 2000; Korponay-Szabo et al., 2003). This TG2 fraction appears distinct from the extracellular TG2 that is directly associated with the cell surface (Maiuri et al., 2005) and is able to crosslink small molecule amines to tissue proteins in situ when the tissue section is incubated in a buffer containing millimolar levels of calcium (Maiuri et al., 2005, Esposito et al., 2003). However, it has not been shown whether this population of ECM-bound TG2 is constitutively active under normal physiological conditions in vivo or whether its crosslinking activity is induced as a result of the incubation conditions.
Many other biological functions have been attributed to TG2. An abbreviated list of these functions includes wound healing (Haroon et al., 1999; Upchurch et al., 1991), macrophage phagocytosis (Szondy et al., 2003), TGF-β activation (Rose et al., 2006), NF-?B activation (Lee et al., 2004), protein kinase activity (Mishra & Murphy, 2004), association with calreticulin (Feng et al., 1999), and association with G-protein coupled receptor GPR56 (Xu et al., 2006). Despite the plethora of biological functions ascribed to TG2, the majority of these functions have been shown to be independent of the enzymatic transamidation activity of the protein, an important point when one considers designing inhibitors of the enzyme. It is also worth noting that TG2 knockout mice have no reproductive or developmental defects (Nanda et al., 2001; De Laurenzi & Melino, 2001), although some abnormalities at the cellular level, such as decreased fibroblast adhesion and macrophage phagocytosis, have been noted. TG2 knockout mice develop lupus-like symptoms including hyperactive B-cell proliferation and anti-nuclear antibody production at about one year of age (Szondy et al., 2003), they respond to chemical wounding more severely than wild-type mice (Sarang et al., 2005; Nardacci et al., 2003), and they have a defect in the rate of mitochondrial ATP synthesis following strenuous exercise (Szondy et al., 2006) suggesting that TG2 has important, non-redundant physiological functions. Before discussing the pathological states TG2 is believed to play a role in, we first review the conformational states of the enzyme and how they relate to its biological functions.
3. Conformational states of transglutaminase 2
While the C277S TG2 mutant has been widely used to determine the relevance of the enzymatic transamidation activity of TG2 for a given biological function, one key biochemical property of TG2 often overlooked is its structure. TG2 can assume multiple conformations. The binding of GTP or irreversible inhibitors to TG2 causes significant shifts in electrophoretic mobility of the protein under native conditions (Murthy et al., 1999; D. Pinkas, unpublished observation). Further, proteolysis studies have shown that TG2 is efficiently proteolyzed by calpain and trypsin in the presence of calcium while GTP protects the protein from proteolysis (Begg et al., 2006; Zhang et al., 1998). Finally, certain anti-TG2 antibodies have a high affinity for one population of TG2 while other antibodies bind preferentially to a distinct population of the enzyme (Maiuri et al., 2005; Monsonego et al., 1998; Fesus & Laki, 1977). Although it is clear that multiple conformations of TG2 exist, very little is known about the biological relevance of each conformation.
Recently, two distinct conformations of human TG2 have been characterized via x-ray crystallography, one with GDP bound (Liu et al., 2002) and the other with an active site covalent inhibitor bound to it (D. Pinkas, unpublished observation). Transglutaminase 2 consists of four distinct domains: 1) an N-terminal β-sandwich domain that contains the fibronectin binding site, 2) the catalytic core domain composed of interspersed α-helices and β-sheets containing the substrate binding pocket and catalytic triad 3) a β-barrel domain with a binding pocket for GTP and interaction sites with the a1B adrenergic receptor and 4) a C-terminal β-barrel that includes the phospholipase Cd1 interaction site. In the GDP bound crystal structure, the two C-terminal β-barrels overlap a significant surface area of the catalytic core domain effectively blocking substrate access to the active site. On the other hand, in the structure with the irreversible inhibitor bound, the two C-terminal β-barrels are extended away from the catalytic core and twisted 180 degrees giving the protein a rod-like shape (D. Pinkas, unpublished observation). The active site is easily accessible to substrates in this conformation. A second interesting feature of the inhibitor bound crystal structure is the disulfide bond formed between Cys370 and Cys371 (D. Pinkas, unpublished observation). In the GDP bound crystal structure, the peptide bond between these two cysteine residues is in the normal trans configuration. However, this bond is twisted into a cis conformation in the inhibitor bound crystal structure and is presumably stabilized by the formation of the disulfide bond. Future studies should aim to clarify the biological significance of each TG2 conformation.
4. Transglutaminase 2 in disease states
It is the role TG2 plays in diseases that makes it a potential therapeutic target. Transglutaminase 2 has been implicated in the pathogenesis of a number of diseases, such as celiac sprue (Molberg et al., 2000), neurodegenerative disorders (Hoffner & Djian, 2005), diabetes (Bernassola et al., 2002), liver cirrhosis and fibrosis (Issa et al., 2004;Mirza et al., 1997), renal scarring (Johnson et al., 2003), and certain types of cancer (Mangala & Mehta, 2005). Importantly, it is the enzymatic function of TG2 that is thought to contribute to the pathology or etiology of most of the aforementioned diseases. Therefore, inhibition of the TG2 active site offers a potential strategy to therapeutically treat these diseases. Before examining the design of such active site inhibitors, a brief review of the role TG2 plays in celiac sprue, neurodegenerative disorders, and cancer will help illustrate the potential benefits these inhibitors may provide.
4.1. Transglutaminase 2 in celiac sprue
Celiac sprue is a T cell mediated inflammatory disorder of the small intestine caused by a class of proteins called prolamins found in wheat, barley, and rye (Sollid, 2000). The high proline and glutamine content of these proteins makes them resistant to natural gastric, pancreatic, and intestinal proteases and peptidases during digestion (Shan et al., 2002; Piper et al., 2004). As such, peptide fragments longer than nine amino acids remain undigested well into the small intestine and gain access to the intestinal lamina propria where they ignite a T cell mediated immune response leading to inflammation and destruction of intestinal architecture (Sollid, 2000).
Transglutaminase 2 is central to both the cell-mediated and humoral immune responses elicited in the small intestine of celiac sprue patients. Intestinal TG2 deamidates specific glutamine residues in the prolamin peptides to glutamate residues, thereby increasing their affinity for the disease associated HLA-DQ2/8 proteins, which in turn mediate the T cell response (Quarsten et al., 1999; Xia et al., 2006). Although prolamins intrinsically have a high glutamine content (around 30–35%), only a few of these glutamine residues are recognized by human TG2 (Piper et al., 2002; Quarsten et al., 1999). The excellent correlation between TG2 substrate specificity, DQ2 binding affinity, and T cell stimulatory potential of TG2 treated prolamins strongly suggests peptide deamidation is caused by TG2 and plays a significant role in the severity of the disease (Molberg et al., 1998; Qiao et al., 2005). Further, celiac patients generate an antibody autoimmune response against TG2 (Dieterich et al., 1997). Anti-TG2 antibodies are found in both the small intestine, where they have been shown to colocalize with extracellular TG2 (Korponay-Szabo et al., 2004), and in the blood, where they are currently being used as a diagnostic tool for the disease (Sardy et al., 1999).
4.2. Transglutaminase 2 in neurodegeneration
There are several neurodegenerative diseases characterized by the presence of protein aggregates in the degenerative region of the brain. For example, huntingtin protein (htt) containing expanded polyglutamine repeats in its N-terminal domain (wild-type htt contains less than 35 consecutive glutamines while disease-related htt typically has over 40 consecutive glutamines (The Huntington’s Disease Collaborative Research Group, 1993)) forms insoluble aggregates in the striatum and cortex of Huntington’s disease patients, and the frequency of aggregates correlates well with the severity of the disease (Hoffner & Djian, 2005). Alzheimer’s disease is typified by the presence of extracellular senile plaques composed of aggregated amyloid β-protein and intracellular neurofibrillary tangles consisting of a highly phosphorylated form of the protein tau. Finally, a hallmark of Parkinson’s disease is the presence of a -synuclein aggregates called Lewy bodies in the cytoplasm of affected neurons. All of the aforementioned proteins are good substrates of TG2 in vitro and, for polyglutamine proteins and a -synuclein, in transfected cells as well (de Cristofaro et al., 1999; Junn et al., 2003). Furthermore, the afflicted region of the brain contains higher levels of TG2 protein than non-affected regions of the brain in the same patients (Ruan & Johnson, 2007). The correlation between the TG2 substrate specificity for disease-relevant aggregated proteins and increased TG2 expression levels suggest a role for enzymatically active TG2 in each disease.
On the other hand, it should be noted that recent evidence indicates that aggregate formation in the mouse model of Huntington’s disease (R6/2) is independent of TG2 activity since crossing the R6/2 mouse with the TG2 knockout mouse led to an increase in the number of protein aggregates. However, TG2 knockout R6/2 mice showed improved motor function and survival rates compared with controls indicating a role for TG2 in this disease model (Mastroberardino et al., 2002; Bailey & Johnson, 2005). Thus, it is currently unclear whether enzymatically active TG2 plays a role in neurodegenerative aggregate formation and, if it does, whether this activity is pathogenic or the consequence of the pre-existing disease.
4.3. Transglutaminase 2 in cancer
A number of experimental observations support the hypothesis that TG2 plays a role in the development of certain types of cancer. First, multiple studies have shown that TG2 protein is upregulated in cancerous tissue relative to healthy tissue in cancers such as glioblastomas (Yuan et al., 2006), malignant melanomas (Fok et al., 2006), and pancreatic ductal adenocarcinomas (Verma et al., 2006) to name a few. Second, a positive correlation between the chemotherapeutic resistance and metastatic potential of certain cancers with TG2 expression levels has been demonstrated (Mehta et al., 2004; Han & Park, 1999). Third, in certain cell types TG2 has been shown to exert anti-apoptotic effects on cells while siRNA down-regulation of TG2 protein expression levels or treatment with TG2 inhibitors sensitizes these cells to apoptosis (Yuan et al., 2005; Mangala et al., 2006; Antonyak et al., 2004). On the other hand, there are also reports of the down-regulation of TG2 expression in certain types of cancer (Birckbichler et al., 2000; Jones et al., 2006). Xu et al. recently identified GPR56 as a protein down-regulated in highly metastatic cancer cells and showed that TG2 was a binding partner for GPR56 suggesting that TG2 can act as a tumor suppressing protein through its interaction with GPR56 (Xu et al., 2006). Therefore, the relevance of TG2 to cancer biology may depend upon the cancer cell type, the type of cancer, the location of the cancer, and possibly the stage of the cancer.
Even though there is strong evidence suggesting a role for TG2 in cancer, a unifying molecular mechanism that explains why TG2 promotes oncogenesis in certain cell or tissue types has not been discovered, although a few mechanisms have been proposed. Recent data suggest that intracellular TG2 is able to crosslink the inhibitory subunit alpha of NF-?B (I?Ba) and, thus, constitutively activate the transcription factor NF-?B which, in turn, promotes expression of anti-apoptotic proteins such as Bcl-xL and BFL1 (Lee et al., 2004; Mann et al., 2006). Other data suggest that TG2 expression activates the tyrosine kinase focal adhesion kinase (FAK) through a mechanism independent of its enzymatic activity. Activated FAK subsequently activates anti-apoptotic pathways, such as the PI3K/Akt pathway, which may lead to uncontrolled cell growth if constitutively activated (Verma et al., 2006). Until the underlying molecular connection between TG2 expression and cancer is ascertained, it may be difficult to predict whether certain cancers are dependent upon the enzymatic activity of TG2 or not and, therefore, whether TG2 inhibition has therapeutic potential in such situations.
5. Chemistry of transglutaminase 2 inhibitors
Transglutaminase 2 inhibitors can be divided into three classes based upon their mechanism of inhibition. These three classes are 1) competitive amine inhibitors 2) reversible inhibitors and 3) irreversible inhibitors. The structures and chemical parameters of inhibitors that fall into these three classes will be reviewed.
5.1. Competitive amine inhibitors
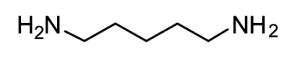
Competitive amine inhibitors are probably the most widely used TG2 inhibitors because they are commercially available, chemically stable, and relatively non-toxic in living systems (Karpuj et al., 2002; Verderio et al., 1998). The structure of this class of inhibitors is typically characterized by a primary amine bound to an aliphatic unbranched carbon chain of around 4–5 saturated carbon atoms, although shorter amines such as hydroxylamine and methylamine are also TG2 substrates. Some of the most commonly used competitive amine inhibitors, including putrescine, monodansylcadaverine, and 5-(biotinamido)pentylamine, are shown in Table 1. For a more complete list of amine substrates and their chemical parameters, see Lorand & Conrad, 1984.
Table 1.
Chemical structures of common competitive amine inhibitors of TG2
| Structure | Cmpd | Common Name |
|---|---|---|
|
1 | Putrescine |
|
|
2 | Cystamine |

|
3 | Spermidine |

|
4 | Histamine |

|
5 | Monodansyl cadaverine |

|
6 | Biotin cadaverine/5-(biotinamido) pentylamine |

|
7 | Fluorescein cadaverine |
Competitive amine inhibitors inhibit TG2 activity by competing with natural amine substrates, such as protein-bound lysine residues, in the transamidation reaction. Thus, TG2 is still enzymatically active and transamidation continues to occur in the presence of competitive amine inhibitors. However, the resulting isopeptide crosslink is mainly formed between the natural glutamine substrate and the competitive amine inhibitor rather than between the natural glutamine substrate and natural amine substrate. The chemical parameter used to assess the inhibition potency of this class of inhibitors is the specificity constant kcat/KM where kcat is the turnover rate and KM is the Michaelis constant.
Cystamine is a unique inhibitor of TG2 because it may have multiple inhibition mechanisms. As shown in Table 1, cystamine is a diamine capable of competitive amine inhibition. Indeed, the reduced form of cystamine, called cysteamine or 2-mercaptoethylamine (MEA), has been shown to be a competitive amine inhibitor of TG2 (Jeitner et al., 2005). However, cystamine has additionally been shown to inactivate TG2 in a time-dependent manner suggesting an irreversible inhibition mechanism (Lorand & Conrad, 1984). This time-dependent inhibition was initially hypothesized to occur due to the formation of mixed disulfides between the TG2 active site cysteine and cysteamine (Lorand & Conrad, 1984; Lorand, 1998) although it is also feasible that cystamine oxidizes the aforementioned disulfide bond in TG2 thereby inactivating the enzyme. Cystamine has been shown to inhibit the thiol-dependent protease caspase 3 and cause increased production of glutathione inside of cells (Lesort et al., 2003). Thus, cystamine, like all TG2 inhibitors, should not be considered a specific TG2 inhibitor when applied in a biological setting.
5.2. Reversible inhibitors
Reversible TG2 inhibitors prevent enzyme activity by blocking substrate access to the active site without covalently modifying the enzyme. The kinetic parameter used to assess the potency of reversible inhibitors is KI, the inhibitor dissociation constant. Transglutaminase 2 cofactors, such as GTP and GDP, are examples of allosteric, reversible inhibitors of the enzyme (Lai et al., 1998). Also, the divalent metal ion Zn2+ is able to reversibly inhibit TG2 activity by competing with Ca2+ for metal binding sites in the protein (Lorand & Conrad, 1984; Aeschlimann & Paulsson, 1994). There are a few GTP analogs, such as GTP?S and GMP-PCP, which have been shown to inhibit transglutaminase activity reversibly (Lai et al., 1998).
In the course of screening a library of 110,000 compounds for TG2 inhibitors, Case and coworkers recently identified a new class of allosteric, reversible inhibitors with thieno[2,3-d]pyrimidin-4-one acylhydrazide backbones (See Table 2) (Case & Stein, 2007; Duval et al., 2005). These inhibitors exhibit slow-binding kinetics, and kinetic analysis suggests that they bind to TG2 exclusive of GTP possibly by competing for the same binding site. While compound 12 was nontoxic to cultured cells at concentrations up to 100 μM (Case & Stein, 2007), no extensive biological characterization of these compounds has been reported.
Table 2.
Chemical Structures of Reversible TG2 Inhibitors
| Structure | Cmpd | R1 | R2 | Common Name |
|---|---|---|---|---|

|
8 | O | O | GTP |
| 9 | CH2 | O | GMP-PCP | |
| 10 | O | S | GTPγS | |

|
11 | GDP | ||

|
12 | LDN-27219 | ||

|
13a |
5.3. Irreversible inhibitors
Irreversible TG2 inhibitors (suicide inhibitors) prevent enzyme activity by covalently modifying the enzyme thereby preventing substrate binding. Most irreversible TG2 inhibitors are designed to target the active site cysteine using chemical functional groups that are reactive in the presence of a nucleophilic atom but form relatively stable chemical bonds after reacting. The structures of different classes of irreversible inhibitors are shown in Table 3.
Table 3.
Chemical Structures and Inhibition Parameters for Irreversible TG2 Inhibitors
| Structure | Cmpd | R1 | n | ki (min−1) | KI (μM) | ki/KI (mM−1min−1) |
|---|---|---|---|---|---|---|
|
14 | 0.90a | 0.075a | 12000a, 96b, 7200c | ||

|
15 | (S-)-CH2-p-OH-phenyl | 0.86d | 420d | 2.0d | |
| 16 | (S-/R-)-CH2-3-(5-F-indolyl) | 0.071e | 19e | 3.6e | ||

|
17 | (S-)-CH2-p-OH-phenyl | 0.21e | 41e | 5.1e | |
| 18 | (S-/R-)-CH2-3-(5-F-indolyl)7 | 0.19e | 18e | 10e | ||
| 19 | (S-)-CH2-3-(5-F-indolyl) | 0.19e | 4.0e | 45e | ||

|
20 | (S-)-CH2-p-OH-phenyl | 0.14e | 30e | 4.6e | |
| 21 | (S-)-CH2-3-(5-F-indolyl) | 0.072e | 1.3e | 57e | ||

|
22a |
|
1 | 1.34f | 2.75f | 490f |
| 22b | 2 | 0.59f | 0.48f | 1240f | ||
| 22c | 3 | 0.54f | 0.28f | 1950f | ||
| 23a |
|
1 | 0.60f | 1.10f | 550f | |
| 23b | 2 | 0.81f | 1.23f | 660f | ||
| 23c | 3 | 0.63f | 0.56f | 1120f | ||
| 24a |
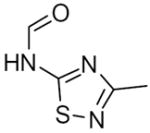
|
1 | No Inhibg | |||
| 24b | 2 | 4.7g | 14g | 330 g | ||

|
25 | 0.20h | 0.070h | 2900h | ||

|
26 | 1140c | ||||

|
27 | 1.4h, 318c | ||||

|
28 | 1560c |
(de Macedo et al., 2000; guinea pig liver TG2),
(Folk et al., 1968; guinea pig liver TG2),
(Freund et al., 1994; human erythrocyte TG),
(Choi et al., 2005; recombinant human TG2),
(Watts et al., 2006; recombinant human TG2),
(Marrano et al., 2001a; guinea pig liver TG2),
(Marrano et al., 2001b; guinea pig liver TG2),
(Hausch et al., 2003; recombinant human TG2)
The chemical parameter used to assess the potency of irreversible inhibitors is ki/KI, where ki is the kinetic parameter describing the reaction rate for formation of the inhibitor-enzyme covalent bond and KI is the dissociation constant of the inhibitor-enzyme complex (Gray & Duggleby, 1989). Because there are a number of different methods for calculating the inhibition constants (Hausch et al., 2003; Gray & Duggleby, 1989; de Macedo et al., 2000) and many different continuous TG2 activity assays which track substrate deamidation (Piper et al., 2002), transamidation (de Macedo et al., 2000), or even esterolysis (Leblanc et al., 2001), it is not reasonable to compare inhibition constants between different studies. Differences in assay temperature, pH, buffer composition, species of TG2 used, and transglutaminase isoenzyme used further preclude an accurate comparison of irreversible inhibitor potencies determined in separate studies.
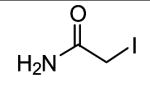
Iodoacetamide is among the structurally simplest irreversible inhibitors of TG2 (Folk & Cole, 1966; de Macedo et al., 2000). The iodide moiety is a good leaving group allowing the TG2 active site thiol to form a relatively stable thioether bond with the inhibitor. Although highly reactive, the small structure of iodoacetamide prevents it from having much interaction with the enzyme making it a fairly non-specific inhibitor of active site cysteines (Beynon & Bond, 1989). Furthermore, depending upon the reaction conditions, iodoacetamide labels TG2 in a greater than 1:1 molar ratio suggesting reactions with TG2 nucleophilic residues other than the active site cysteine (Folk & Cole, 1966).
One of the most well-studied classes of TG2 irreversible inhibitors, from both a chemical and biological perspective, are 3-halo-4,5-dihydroisoxazoles. The structure of these inhibitors is based on the natural product acivicin (Castelhano et al., 1988), which is a glutamine isostere able to inhibit several enzymes bearing active site cysteines (Tso et al., 1980). Structurally these compounds are typified by a 3-bromo-4,5-dihydroisoxazole warhead group bound to a natural or unnatural amino acid via a peptide bond typically followed by a hydrophobic, aromatic moiety, which increases binding specificity to the TG2 active site (see Table 3) (Choi et al., 2005, Watts et al., 2006). Purification of the R and S enantiomers around the C-5 carbon in the dihydroisoxazole ring demonstrated the asymmetric nature of the interaction between dihydroisoxazoles and the TG2 active site since only the S enantiomer was found to irreversibly inhibit the enzyme (Castelhano et al., 1988, Killackey et al., 1989, Watts et al., 2006). The originally proposed mechanism for dihydroisoxazole inhibition involving the TG2 active site thiol displacing the dihydroisoxazole halide atom and forming a stable imino thioether (Castelhano et al, 1988) was recently confirmed via mass spectrometry (Watts et al., 2006). While this class of inhibitors shows good bioavailability and low toxicity in mice (Choi et al., 2005), their main drawback is their low solubility (typically under 500 μM, M. Siegel, unpublished observation) in buffers at physiological pH.
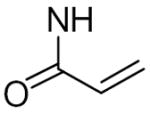
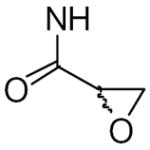
Keillor and coworkers have synthesized and characterized a family of irreversible TG2 inhibitors with different electrophilic functional groups using the TG2 substrate carbobenzyloxy-L-glutaminylglycine (Cbz-gln-gly) as the inhibitor backbone and substituting the reactive moieties in the gln position. Among the reactive functional groups they have examined are a,β-unsaturated amides (de Macedo et al., 2002), epoxides (de Macedo et al., 2002), 1,2,4-thiadiazoles (Marrano et al., 2001), maleimides (Halim et al., 2006), and chloroacetamides (Pardin et al., 2006) (see Table 3). This group also made a series of Cbz-phe-gln derivatives although these inhibitors were not as potent as their Cbz-gln-gly analogs (Pardin et al., 2006). The Cbz-gln(epoxide) and Cbz-gln(a,β-unsaturated)-gly inhibitors are the two most potent inhibitors reported in these studies.
Gluten peptides have been shown to be high-affinity substrates of TG2 (Bruce et al., 1985; Piper et al., 2002). This high-affinity interaction was exploited to design a set of peptidomimetic irreversible inhibitors using a gluten peptide sequence as the inhibitor backbone (Hausch et al., 2003) (Table 3). The glutamine targeted by TG2 in these gluten peptides was substituted with the inhibitory glutamine isosteres acivicin or 6-diazo-5-oxo-norleucine (DON). While the acivicin analog irreversibly inhibited TG2, the DON peptide was shown to be five orders of magnitude more potent than the acivicin analog with a KI in the nM range.
Although shown to be an effective class of factor XIIIa inhibitors, 2-[(2-oxopropyl)thio]imidazolium derivatives have also been used as TG2 inhibitors (Freund et al., 1994). Despite a limited level of structure-activity development of these compounds, they have been shown to have fairly good inhibition potency (Freund et al., 1994, Hausch et al., 2003) as well as effectiveness in a number of different biological settings (Hausch et al., 2003; Maiuri et al., 2005; Barsigian et al., 2001). Nevertheless, the potential side-effects of inhibiting factor XIIIa make the currently available 2-[(2-oxopropyl)thio]imidazolium inhibitors inappropriate for treatment of TG2 mediated diseases.
6. Transglutaminase 2 inhibition in disease states
Application of TG2 inhibitors to biological systems has yielded promising results in a number of different disease models suggesting the potential use of these inhibitors for therapeutic treatment of human diseases. In this section, we will review the effects TG2 inhibitors have shown in biological models of the previously discussed disease states, celiac sprue, neurodegeneration, and cancer.
6.1. Transglutaminase 2 inhibition in celiac sprue
Because there is no mouse model for celiac sprue, evaluation of potential therapeutics to treat the disease is typically performed on simplified biological systems. There have been two notable ex vivo demonstrations that TG2 inhibition has the potential to benefit patients with celiac sprue. In the first study by Molberg and coworkers (Molberg et al., 2001), the authors showed that culturing celiac patient small intestinal biopsies with either TG2 treated (deamidated) or non-TG2 treated (non-deamidated) gluten digests both typically resulted in the generation of patient T-cell lines that preferentially recognized deamidated gluten peptides rather than non-deamidated gluten peptides. Further, by blocking the activity of endogenous TG2 in the celiac biopsies with cystamine, the authors were able to show that more than half of the resultant T cell lines had reduced proliferative responses to deamidated gluten digests compared to non-cystamine treated controls and that these cell lines still did not respond well to the non-deamidated digests. Taken together, these two results imply that the gluten responsive T cell populations in celiac intestinal biopsies are naturally biased towards recognizing deamidated gluten peptides as opposed to non-deamidated peptides, that endogenous TG2 activity in these biopsies can result in gluten peptide deamidation in situ, and that treatment of celiac biopsies with TG2 inhibitors can reduce the proliferative response of gluten-reactive T cells.
In another study by Maiuri and coworkers (Maiuri et al., 2005), the authors showed that the 2-[(2-oxopropyl)thio]imidazolium inhibitor L682777 (28 in Table 3, a.k.a. R283) was able to prevent the in situ crosslinking of gluten peptides to endogenous proteins in thin tissue sections taken from both celiac sprue patients and controls. More importantly, the authors showed that incubation of intact celiac small intestinal biopsies with L682777 prevented T cell activation induced by the non-deamidated form of an immunodominant gluten peptide. In contrast, L682777 was ineffective at controlling T cell activation when the biopsies were incubated with the deamidated version of the same peptide. These results suggest that irreversible inhibition of endogenous TG2 in celiac patient biopsies can prevent gluten peptide deamidation and, therefore, reduce T cell activation.
6.2. Transglutaminase 2 inhibition in neurodegenration
Transglutaminase 2 inhibitors have been shown to exert therapeutic effects in multiple biological models of neurodegenerative diseases. In a cell culture model of Parkinson’s disease, Junn and coworkers showed that by transfecting COS-7 cells with a -synuclein and TG2 simultaneously, covalent a -synuclein aggregates, reminiscent of Lewy bodies in Parkinson’s disease, form and are dependent upon enzymatically active TG2 since the C277S TG2 mutant failed to induce aggregate formation. Further, treatment of these co-transfected cells with cystamine significantly reduced the quantity of a -synuclein aggregates as well as the percentage of cells containing the aggregates (Junn et al., 2003). There have been two other reports in which proteins with normal length and expanded polyglutamine repeat proteins, representative of expanded CAG diseases such as Huntington’s disease, have been transfected into cell lines and shown to form aggregates (Igarashi et al., 1998; de Cristofaro et al., 1999). Treatment of these cell lines with the TG2 competitive amine inhibitor monodansylcadaverine led to a decrease in nuclear fragmentation while treatment with cystamine lead to both a decrease in nuclear fragmentation and a decrease in protein aggregate formation.
Cystamine has a beneficial therapeutic effect in vivo when dosed in mouse models of Huntington’s disease. Huntington R6/2 mice dosed with cystamine showed improved motor function, less severe weight loss, and increased survival compared to non-treated controls (Karpuj et al., 2002; Dedeoglu et al., 2002). Importantly, ex vivo TG2 activity in brain homogenates was lower after dosing with cystamine at least 60 minutes after injection (Karpuj et al., 2002). In a different mouse model of Huntington’s disease, the YAC128 strain, cystamine was able to decrease the level of striatal atrophy but unable to improve animal weight or motor function indicating a beneficial effect of cystamine at the cellular and tissue level but not in disease symptoms (Van Raamsdonk et al., 2002).
Despite the activity of cystamine in mouse models of Huntington’s disease, it is not clear how much of the therapeutic effect is due to TG2 enzymatic inhibition. Cystamine has been shown to cause an increase in cysteine concentration in the brain of cystamine treated mice (Fox et al., 2004). It also induces a change in the transcript levels of a number of genes in treated mice (Karpuj et al., 2002), including a heat shock protein HSJ1b that promotes the release of the neuroprotective protein brain-derived neurotrophic factor (BDNF) (Borrell-Pages et al., 2006). Further, cystamine inhibits caspase-3 and increases glutathione levels in a neuroblastoma cell line, proving that cystamine interacts with proteins other than TG2 (Lesort et al., 2003).
Probably the most convincing evidence that the beneficial therapeutic effect of cystamine on Huntington mice is independent of TG2 inhibition was provided by Bailey and Johnson. They crossed the R6/2 Huntington mouse with the TG2 knockout mouse to create a strain susceptible to neurodegeneration in the absence of TG2. When the R6/2 TG2−/− mice were treated with cystamine, the improved motor function and increased lifespan were not statistically different from the improvement seen in R6/2 TG2+/+ mice treated with cystamine (Bailey & Johnson, 2006). Additionally, R6/1 and R6/2 TG2−/− mice had increased levels of neuronal protein aggregates compared to R6/1 and R6/2 TG2+/+ mice suggesting a mechanism of protein aggregation independent of TG2 transamidation activity in these models (Mastroberardino et al., 2002; Bailey & Johnson, 2005). However, it is noteworthy that R6/2 TG2−/− mice showed a delay in the onset of motor dysfunction and improved survival compared to R6/2 TG2+/+ mice implying a role for TG2 in the pathogenesis of neurodegeneration in the R6/2 model (Mastroberardino et al., 2002; Bailey and Johnson, 2005). Whether this role is dependent upon the enzymatic activity of TG2, and, thus, treatable with TG2 inhibitors, or is related to a non-enzymatic function of TG2 has yet to be determined.
6.3. Transglutaminase 2 inhibition in cancer
Cultured cancer cells have provided a convenient system for studying the molecular details linking TG2 enzymatic activity and inhibition to cell growth and chemoresistance. Antonyak and coworkers showed that the upregulation of TG2 caused by retinoic acid or epidermal growth factor (EGF) treatment in breast cancer cells protected the cells from doxorubicin-induced apoptosis. However, treatment of these cells with monodansylcadaverine reversed this anti-apoptotic effect implying that TG2 activity contributes to chemoresistance in human breast cancer cells (Antonyak et al., 2004). In an effort to explain the anti-apoptotic property of TG2 activity on cancer cells, Mann and coworkers found a correlation between TG2 activity and the activation of the pro-survival transcription factor NF-?B. Treatment of cultured cells with the calcium ionophore A23187 increased cytosolic calcium levels high enough to enzymatically activate endogenous TG2. This activated TG2 crosslinked the inhibitory subunit alpha of NF-?B (I?Ba), thereby inactivating I?Ba and constitutively activating NF-?B. Addition of the TG2 competitive amine inhibitors monodansylcadaverine and 5-(biotinamido)pentylamine greatly reduced NF-?B activation by preventing TG2 mediated I?Ba polymerization (Mann et al., 2006). Interestingly, in a separate report, treatment of cancer cells expressing high levels of TG2 with A23187 was shown to induce apoptosis that could be reversed using 5-(biotinamido)pentylamine (Fok et al., 2006). Therefore, enzymatically active TG2 appears to have both pro-apoptotic and anti-apoptotic effects on cancer cells depending upon the assay.
Irreversible TG2 inhibitors have also shown therapeutic value for treatment of glioblastomas both in vitro and in vivo. Yuan and coworkers showed that both monodansylcadaverine and a dihydroisoxazole inhibitor, compound 15 (KCC009) in Table 3, were able to decrease phosphorylation of pro-survival protein Akt and upregulate pro-apoptotic protein Bim resulting in enhanced cell apoptosis when the inhibitors were incubated with a cultured mouse glioblastoma cell line (Yuan et al., 2005). Co-administration of these inhibitors with the chemotherapeutic N, N′-bis(2-chloroethyl)-N-nitrosourea (BCNU) decreased the size of subcutaneously implanted glioblastoma tumors in mice by 50% compared to controls treated with BCNU only (Yuan et al., 2005; Choi et al., 2005). In an orthotopic mouse model of glioblastoma, the size of intracranial tumors was monitored over time by implanting luciferase transfected glioblastoma cells and tracking the tumor growth using the bioluminescence signal resulting from luciferin injection. While compound 15 and BCNU alone had little effect on tumor size relative to vehicle treated mice, the combination therapy of compound 15 and BCNU dramatically decreased the tumor size. Further, the combination enzyme therapy extended the lifespan of mice compared with vehicle and monotherapy treated animals proving the therapeutic utility of dihydroisoxazoles in a mouse model of glioblastoma (Yuan et al., 2006).
7. Conclusions and future directions
Transglutaminase 2 is an enzyme with diverse biological functions postulated to participate in the pathology of a number of diseases. While extensive research has investigated the role of TG2 in fatal neurodegenerative diseases and cancer, the molecular mechanism by which TG2 contributes to the etiology of these diseases has not been discovered. From a pharmacological perspective, application of TG2 inhibitors to mouse models of these diseases has shown therapeutic potential. Cystamine delays the onset of the neurological symptoms associated with Huntington’s disease when applied to the R6/2 Huntington’s mouse model, and dihydroisoxazoles, when used in tandem with BCNU, are able to decrease tumor size and extend survival in a mouse model of glioblastoma. However, without knowing the molecular details of TG2’s contribution to these diseases, it is difficult to conclude whether the improvement in symptoms seen in these models is due to TG2 inhibition alone, due to off-target inhibition of other disease relevant proteins, or both. The application of cystamine to the R6/2 TG2−/− mouse provides a clever paradigm for experiments exploring the contribution of TG2 inhibition to disease amelioration in mouse models.
In contrast to neurodegerative diseases and cancer, the contribution of TG2 activity to celiac sprue pathogenesis has been fairly well established. The deamidation of specific glutamine residues targeted by TG2 in gluten peptides has been shown to enhance peptide binding to the disease-associated proteins HLA-DQ2/8, which are the only known genetic risk factors, as well as significantly increase the activation of disease relevant T cells compared to non-TG2 treated peptides. However, unlike Huntington’s disease and cancer, there is no mouse model for celiac sprue to explore the therapeutic effects of inhibiting intestinal TG2. Therefore, all experiments studying the pharmacology of TG2 inhibitors in celiac sprue have been on ex vivo systems. Because celiac sprue can be treated with a strict gluten exclusion diet, TG2 inhibitors will have to be shown efficacious and safe with virtually no side effects before they can be justified as a therapy for this ailment.
Although many biological functions have been attributed to transglutaminase 2, the precise in vivo function of this protein remains a mystery. A significant fraction of these functions have arisen from tissue culture studies in which TG2 is either transfected into the cell, upregulated using cell differentiation factors such as retinoic acid, or artificially activated by inducing large intracellular calcium fluxes. However, the overexpression and activation of TG2 in certain cell types may never occur in vivo resulting in potential experimental artifacts due to excess TG2 protein interacting with an unnatural and incompatible cellular environment. Instead of forcing TG2 expression, an effort must be made to study TG2 in its natural environment both in cell culture and, especially, in vivo. The use of specific, potent irreversible TG2 inhibitors may help to clarify the biological function of enzymatically active TG2 by blocking only the fraction of the enzyme that is active under physiological conditions. However, the two caveats in interpreting resultant biological phenotypes caused by TG2 inhibitors are the potential for off-target inhibitor binding and the potential effect of trapping TG2 into an unnatural conformation relative to its cellular localization.
Transglutaminase 2 conformation may be a critical factor in the success or failure of enzymatic inhibitors of TG2. It is not possible to predict the biological consequence of trapping TG2 in certain conformations, such as that observed in the inhibitor bound crystal structure, due to a lack of data correlating TG2 conformation to biological function. This unpredictability is further enhanced by the lack of in vivo data concerning the enzymatic activity status of the different populations of TG2. It may be that only a small fraction of TG2 is active in a disease state and that inhibition of this population has little biological consequence. However, it is just as easy to imagine that the conformational constraints placed upon TG2 when inhibited prevent it from performing one or more of its natural biological functions causing potential side effects. Careful biochemical studies correlating TG2 conformation to biological function as well as the design of TG2 inhibitors that allow for conformational flexibility may be vital to the success of pharmacological therapy of TG2 mediated diseases.
Acknowledgments
The authors would like to thank Dan Pinkas for sharing unpublished observations. M. Siegel is funded by an ARCS Foundation fellowship
Abbreviations
- ECM
extracellular matrix
- GDP
guanosine diphosphate
- GPR56
G protein-coupled receptor 56
- GTP
guanosine triphosphate
- HLA
human leukocyte antigen
- NF-?B
nuclear factor ?B
- ROCK
Rho-associated coiled-coil containing serine/threonine protein kinase
- TG2
transglutaminase 2
- TGF-β
transforming growth factor β
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aeschlimann D, Paulsson M. Transglutaminases: Protein Cross-Linking Enzymes in Tissues and Body Fluids. Thromb Haemost. 1994;71(4):402–415. [PubMed] [Google Scholar]
- Afford S, Randhawa S. Apoptosis. Mol Pathol. 2000;53(2):55–63. doi: 10.1136/mp.53.2.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue Transglutaminase is an Integrin Binding Adhesion Coreceptor for Fibronectin. J Cell Biol. 2000;148(4):825–838. doi: 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimov SS, Belkin AM. Cell surface tissue transglutaminase is involved in adhesion and migration of monocytic cells on fibronectin. Blood. 2001a;98(5):1567–1576. doi: 10.1182/blood.v98.5.1567. [DOI] [PubMed] [Google Scholar]
- Akimov SS, Belkin AM. Cell-surface transglutaminase promotes fibronectin assembly via interaction with the gelatin-binding domain of fibronectin: a role in TGFβ-dependent matrix deposition. J Cell Sci. 2001b;114(16):2989–3000. doi: 10.1242/jcs.114.16.2989. [DOI] [PubMed] [Google Scholar]
- Antonyak MA, Miller AM, Jansen JM, Boehm JE, Balkman CE, Wakshlag JJ, et al. Augmentation of tissue transglutaminase expression and activation by epidermal growth factor inhibit doxorubicin-induced apoptosis in human breast cancer cells. J Biol Chem. 2004;279(40):41461–41467. doi: 10.1074/jbc.M404976200. [DOI] [PubMed] [Google Scholar]
- Baek KJ, Kang SK, Damron DS, Im MJ. Phospholipase Cd1 is a Guanine Nucleotide Exchanging Factor for Transglutaminase II (G a h) and Promotes a1B-adrenoreceptor-mediated GTP Binding and Intracellular Calcium Release. J Biol Chem. 2001;276(8):5591–5597. doi: 10.1074/jbc.M008252200. [DOI] [PubMed] [Google Scholar]
- Bailey CD, Johnson GV. Tissue transglutaminase contributes to disease progression in the R6/2 Huntington’s disease mouse model via aggregate-independent mechanisms. J Neurochem. 2005;88:1253–1260. doi: 10.1111/j.1471-4159.2004.02839.x. [DOI] [PubMed] [Google Scholar]
- Bailey CD, Johnson GV. The protective effects of cystamine in the R6/2 Huntington’s disease mouse involve mechanisms other than the inhibition of tissue transglutaminase. Neurobiol aging. 2006;27(6):871–879. doi: 10.1016/j.neurobiolaging.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Balklava Z, Verderio E, Collighan R, Gross S, Adams J, Griffin M. Analysis of Tissue Transglutaminase Function in the Migration of Swiss 3T3 Fibroblasts. J Biol Chem. 2002;277(19):16567–16575. doi: 10.1074/jbc.M109836200. [DOI] [PubMed] [Google Scholar]
- Barsigian C, Stern AM, Martinez J. Tissue (type II) transglutaminase covalently incorporates itself, fibrinogen, or fibronectin into high molecular weight complexes on the extracellular surface of isolated hepatocytes. Use of 2-[(2-oxopropyl)thio] imidazolium derivatives as cellular transglutaminase inactivators. J Biol Chem. 1991;266(33):22501–22509. [PubMed] [Google Scholar]
- Begg GE, Holman SR, Stokes PH, Matthews JM, Graham RM, Iismaa SE. Mutation of a critical arginine in the GTP-binding site of transglutaminase 2 disinhibits intracellular cross-linking activity. J Biol Chem. 2006;281(18):12603–12609. doi: 10.1074/jbc.M600146200. [DOI] [PubMed] [Google Scholar]
- Bernassola F, Federici M, Corazzari M, Terrinoni A, Hribal ML, De Laurenzi V, et al. Role of transglutaminase 2 in glucose tolerance: knockout mice studies and a putative mutation in a MODY patient. FASEB J. 2002;16(11):1371–1378. doi: 10.1096/fj.01-0689com. [DOI] [PubMed] [Google Scholar]
- Beynon RJ, Bond JS. Proteolytic Enzymes: A Practical Approach. IRL Press; Oxford, England: 1989. p. 245. [Google Scholar]
- Birckbichler PJ, Bonner RB, Hurst RE, Bane BL, Pitha JV, Hemstreet GP., 3rd Loss of tissue transglutaminase as a biomarker for prostate adenocarcinoma. Cancer. 2000;89(2):412–423. doi: 10.1002/1097-0142(20000715)89:2<412::aid-cncr29>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Borrell-Pages M, Canals JM, Cordelieres FP, Parker JA, Pineda JR, Grange G, et al. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J Clin Invest. 2006;116(5):1410–1424. doi: 10.1172/JCI27607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsook H, Deasy CL, Haagen-Smit AJ, Keighley G, Lowy PH. The Incorporation of Labeled Lysine into the Proteins of Guinea Pig Liver Homogenate. J Biol Chem. 1949;179:689–704. [PubMed] [Google Scholar]
- Bruce SE, Bjarnason I, Peters TJ. Human jejunal transglutaminase: demonstration of activity, enzyme kinetics and substrate specificity with special relation to gliadin and coeliac disease. Clin Sci. 1985;68(5):573–579. doi: 10.1042/cs0680573. [DOI] [PubMed] [Google Scholar]
- Case A, Stein RL. Kinetic Analysis of the Action of Tissue Transglutaminase on Peptide and Protein Substrates. Biochemistry. 2003;42:9466–9481. doi: 10.1021/bi030084z. [DOI] [PubMed] [Google Scholar]
- Case A, Stein RL. Kinetic analysis of the interaction of tissue transglutaminase with a nonpeptidic slow-binding inhibitor. Biochemistry. 2007;46(4):1106–1115. doi: 10.1021/bi061787u. [DOI] [PubMed] [Google Scholar]
- Castelhano AL, Billedeau R, Pliura DH, Bonaventura BJ, Krantz A. Synthesis, Chemistry, and Absolute Configuration of Novel Transglutaminase Inhibitors Containing a 3-Halo-4,5-dihydroisoxazole. Bioorg Chem. 1988;16(3):335–340. [Google Scholar]
- Chen JSK, Mehta K. Tissue transglutaminase: an enzyme with a split personality. Int J Biochem Cell Biol. 1999;31(8):817–836. doi: 10.1016/s1357-2725(99)00045-x. [DOI] [PubMed] [Google Scholar]
- Chen S, Lin F, Iismaa S, Lee KN, Birckbichler PJ, Graham RM. a1-Adrenergic Receptor Signaling via Gh Is Subtype Specific and Independent of Its Transglutaminase Activity. J Biol Chem. 1996;271(50):32385–32391. doi: 10.1074/jbc.271.50.32385. [DOI] [PubMed] [Google Scholar]
- Choi K, Siegel M, Piper JL, Yuan L, Cho E, Strnad P, et al. Chemistry and biology of dihydroisoxazole derivatives: selective inhibitors of human transglutaminase 2. Chem Biol. 2005;12(4):469–475. doi: 10.1016/j.chembiol.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Clarke DD, Mycek MJ, Neidle A, Waelsch H. The Incorporation of Amines into Protein. Arch Biochem Biophys. 1959;79:338–354. doi: 10.1016/0003-9861(59)90613-7. [DOI] [PubMed] [Google Scholar]
- Datta S, Antonyak MA, Cerione RA. Importance of Ca(2+)-dependent transamidation activity in the protection afforded by tissue transglutaminase against doxorubicin-induced apoptosis. Biochemistry. 2006;45(44):13163–13174. doi: 10.1021/bi0606795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cristofaro T, Affaitati A, Cariello L, Avvedimento EV, Varrone S. The length of polyglutamine tract, its level of expression, the rate of degradation, and the transglutaminase activity influence the formation of intracellular aggregates. Biochem Biophys Res Commun. 1999;260(1):150–158. doi: 10.1006/bbrc.1999.0851. [DOI] [PubMed] [Google Scholar]
- De Laurenzi V, Melino G. Gene disruption of tissue transglutaminase. Mol Cell Biol. 2001;21(1):148–155. doi: 10.1128/MCB.21.1.148-155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Macedo P, Marrano C, Keillor JW. A direct continuous spectrophotometric assay for transglutaminase activity. Anal Biochem. 2000;285(1):16–20. doi: 10.1006/abio.2000.4713. [DOI] [PubMed] [Google Scholar]
- de Macedo P, Marrano C, Keillor JW. Synthesis of dipeptide-bound epoxides and alpha,beta-unsaturated amides as potential irreversible transglutaminase inhibitors. Bioorg Med Chem. 2002;10(2):355–360. doi: 10.1016/s0968-0896(01)00292-9. [DOI] [PubMed] [Google Scholar]
- Dedeoglu A, Kubilus JK, Jeitner TM, Matson SA, Bogdanov M, Kowall NW, et al. Therapeutic effects of cystamine in a murine model of Huntington’s disease. J Neurosci. 2002;22(20):8942–8950. doi: 10.1523/JNEUROSCI.22-20-08942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, Schuppan D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3(7):797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- Duval E, Case A, Stein RL, Cuny GD. Structure-activity relationship study of novel tissue transglutaminase inhibitors. Bioorg Med Chem Lett. 2005;15(7):1885–1889. doi: 10.1016/j.bmcl.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Esposito C, Caputo I. Mammalian Transglutaminases: Identification of substrates as a key to physiological function physiopathological relevance. FEBS J. 2005;272(3):615–631. doi: 10.1111/j.1742-4658.2004.04476.x. [DOI] [PubMed] [Google Scholar]
- Esposito C, Paparo F, Caputo I, Porta R, Salvati VM, Mazzarella G, et al. Expression and enzymatic activity of small intestinal tissue transglutaminase in celiac disease. Am J Gastroenterol. 2003;98(8):1813–1820. doi: 10.1111/j.1572-0241.2003.07582.x. [DOI] [PubMed] [Google Scholar]
- Feng JF, Readon M, Yadav SP, Im MJ. Calreticulin down-regulates both GTP binding and transglutaminase activities of transglutaminase II. Biochemistry. 1999;38(33):10743–10749. doi: 10.1021/bi9905009. [DOI] [PubMed] [Google Scholar]
- Fesus L, Laki K. Two antigenic sites of tissue transglutaminase. Biochemistry. 1977;16(18):4061–4066. doi: 10.1021/bi00637a019. [DOI] [PubMed] [Google Scholar]
- Fesus L, Thomazy V, Falus A. Induction and activation of tissue transglutaminase during programmed cell death. FEBS Lett. 1987;224(1):104–108. doi: 10.1016/0014-5793(87)80430-1. [DOI] [PubMed] [Google Scholar]
- Fleckenstein B, Molberg O, Qiao SW, Schmid DG, von der Mulbe F, Elgstoen K, et al. Gliadin T cell epitope selection by tissue transglutaminase in celiac disease. Role of enzyme specificity and pH influence on the transamidation versus deamidation processes. J Biol Chem. 2002;277(37):34109–34116. doi: 10.1074/jbc.M204521200. [DOI] [PubMed] [Google Scholar]
- Fok JY, Ekmekcioglu S, Mehta K. Implications of tissue transglutaminase expression in malignant melanoma. Mol Cancer Ther. 1996;5(6):1493–1503. doi: 10.1158/1535-7163.MCT-06-0083. [DOI] [PubMed] [Google Scholar]
- Folk JE, Cole PW. Identification of a functional cysteine essential for the activity of guinea pig liver transglutaminase. J Chem Biol. 1966;241(13):3238–3240. [PubMed] [Google Scholar]
- Folk JE, Cole PW, Mullooly JP. Mechanim of action of guinea pig liver transglutaminase. V. The hydrolysis reaction. J Biol Chem. 1968;243(2):418–427. [PubMed] [Google Scholar]
- Fox JH, Barber DS, Singh B, Zucker B, Swindell MK, Norflus F, et al. Cystamine increases L-cysteine levels in Huntington’s disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. J Neurochem. 2004;91(2):413–422. doi: 10.1111/j.1471-4159.2004.02726.x. [DOI] [PubMed] [Google Scholar]
- Freund KF, Doshi KP, Gaul SL, Claremon DA, Remy DC, Baldwin JJ, et al. Transglutaminase inhibition by 2-[(2-oxopropyl)thio]imidazolium derivatives: mechanism of factor XIIIa inactivation. Biochemistry. 1994;33(33):10109–10119. doi: 10.1021/bi00199a039. [DOI] [PubMed] [Google Scholar]
- Gray PJ, Duggleby RG. Analysis of Kinetic Data for Irreversible Enzyme Inhibition. Biochem J. 1989;257(2):419–424. doi: 10.1042/bj2570419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin M, Casadio R, Bergamini CM. Transglutaminases: Nature’s biological glues. Biochem J. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim D, Caron K, Keillor JW. Synthesis and evaluation of peptidic maleimides as transglutaminase inhibitors. Bioorg Med Chem Lett. 2006 doi: 10.1016/j.bmcl.2006.10.061. [DOI] [PubMed] [Google Scholar]
- Han JA, Park SC. Reduction of transglutaminase 2 expression is associated with an induction of drug sensitivity in the PC-14 human lung cancer cell line. J Cancer Res Clin Oncol. 1999;125(2):89–95. doi: 10.1007/s004320050247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang J, Zemskov EA, Lorand L, Belkin AM. Identifiction of a Novel Recognition Sequence for Fibronectin within the NH2-terminal β-Sandwich Domain of Tissue Transglutaminase. J Biol Chem. 2005;280(25):23675–23683. doi: 10.1074/jbc.M503323200. [DOI] [PubMed] [Google Scholar]
- Haroon ZA, Hettasch JM, Lai TS, Dewhirst MW, Greenberg CS. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999;13(13):1787–1795. doi: 10.1096/fasebj.13.13.1787. [DOI] [PubMed] [Google Scholar]
- Hausch F, Halttunen T, Maki M, Khosla C. Design, synthesis, and evaluation of gluten peptide analogs as selective inhibitors of human tissue transglutaminase. Chem Biol. 2003;10(3):225–231. doi: 10.1016/s1074-5521(03)00045-0. [DOI] [PubMed] [Google Scholar]
- Hoffner G, Djian P. Transglutaminase and diseases of the central nervous system. Front Biosci. 2005;10:3078–3092. doi: 10.2741/1764. [DOI] [PubMed] [Google Scholar]
- Hwang KC, Gray CD, Sivasubramaninan N, Im MJ. Interaction Site of GTP Binding Gh (Transglutaminase II) with Phospholipase C. J Biol Chem. 1995;270(45):27058–27062. doi: 10.1074/jbc.270.45.27058. [DOI] [PubMed] [Google Scholar]
- Igarashi S, Koide R, Shimohata T, Yamada M, Hayashi Y, Takano H, et al. Suppression of aggregate formation and apoptosis by transglutaminase inhibitors in cells expressing truncated DRPLA protein with an expanded polyglutamine stretch. Nat Gen. 1998;18(2):111–117. doi: 10.1038/ng0298-111. [DOI] [PubMed] [Google Scholar]
- Issa R, Zhou X, Constandinou CM, Fallowfield J, Millward-Sadler H, Gaca MD, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126(7):1795–1808. doi: 10.1053/j.gastro.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Janiak A, Zemskov EA, Belkin AM. Cell Surface Transglutaminase Promotes RhoA Activation via Integrin Clustering and Suppression of the Src-p190RhoGAP Signaling Pathway. Mol Biol Cell. 2006;17(4):1606–1619. doi: 10.1091/mbc.E05-06-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeitner TM, Delikatny EJ, Ahlqvist J, Capper H, Cooper AJ. Mechanism for the inhibition of transglutaminase 2 by cystamine. Biochem Pharmacol. 2005;69(6):961–970. doi: 10.1016/j.bcp.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Johnson TS, El-Koraie AF, Skill NJ, Baddour NM, El Nahas AM, Njloma M, et al. Tissue transglutaminase and the progression of human renal scarring. J Am Soc Nephrol. 2003;14(8):2052–2062. doi: 10.1097/01.asn.0000079614.63463.dd. [DOI] [PubMed] [Google Scholar]
- Jones RA, Kotsakis P, Johnson TS, Chau DY, Ali S, Melino G, et al. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2006;13(9):1442–1453. doi: 10.1038/sj.cdd.4401816. [DOI] [PubMed] [Google Scholar]
- Junn E, Ronchetti RD, Quezado MM, Kim SY, Mouradian MM. Tissue transglutaminase-induced aggregation of alpha-synuclein: Implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci. 2003;100(4):2047–2052. doi: 10.1073/pnas.0438021100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, et al. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med. 2002;8(2):143–149. doi: 10.1038/nm0202-143. [DOI] [PubMed] [Google Scholar]
- Killackey JJ, Bonaventura BJ, Castelhano AL, Billedeau RJ, Farmer W, DeYoung L, et al. A new class of mechanism-based inhibitors of transglutaminase enzymes inhibits the formation of cross-linked envelopes by human malignant keratinocytes. Mol Pharmacol. 1989;35(5):701–706. [PubMed] [Google Scholar]
- Korponay-Szabo IR, Halttunen T, Szalai Z, Laurila K, Kiraly R, Kovacs JB, et al. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac autoantibodies. Gut. 2004;53(5):641–648. doi: 10.1136/gut.2003.024836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korponay-Szabo IR, Laurila K, Szondy Z, Halttunen T, Szalai Z, Dahlbom I, et al. Missing endomysial and reticulin binding of coeliac antibodies in transglutaminase knockout tissues. Gut. 2003;52(2):199–204. doi: 10.1136/gut.52.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korponay-Szabo IR, Sulkanen S, Halttunen T, Maurano F, Rossi M, Mazzarella G, et al. Tissue transglutaminase is the target in both rodent and primate tissues for celiac disease-specific autoantibodies. J Pediatr Gastroenterol Nutr. 2000;31(5):520–527. doi: 10.1097/00005176-200011000-00013. [DOI] [PubMed] [Google Scholar]
- Lai TS, Slaughter TF, Peoples KA, Hettasch JM, Greenberg CS. Regulation of Human Tissue Transglutaminase Function by Magnesium-Nucleotide Complexes. J Biol Chem. 1998;273(3):1776–1781. doi: 10.1074/jbc.273.3.1776. [DOI] [PubMed] [Google Scholar]
- Leblanc A, Gravel C, Labelle J, Keillor JW. Kinetic studies of guinea pig liver transglutaminase reveal a general-base-catalyzed deacylation mechanism. Biochemistry. 2001;40(28):8335–8342. doi: 10.1021/bi0024097. [DOI] [PubMed] [Google Scholar]
- Lee J, Kim YS, Choi DH, Bang MS, Han TR, Joh TH, et al. Transglutaminase 2 induces nuclear factor-kappaB activation via a novel pathway in BV-2 microglia. J Biol Chem. 2004;279(51):53725–53735. doi: 10.1074/jbc.M407627200. [DOI] [PubMed] [Google Scholar]
- Lesort M, Lee M, Tucholski J, Johnson GV. Cystamine inhibits caspase activity. Implications for the treatment of polyglutamine disorders. J Biol Chem. 2003;278(6):3825–3830. doi: 10.1074/jbc.M205812200. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Liu S, Cerione RA, Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci. 2002;99(5):2743–2747. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorand L. DRPLA aggregation and transglutaminase, revisited. Nat Genet. 1998;20(3):231. doi: 10.1038/3033. [DOI] [PubMed] [Google Scholar]
- Lorand L, Graham RM. Transglutaminases: Crosslinking Enzymes with Pleiotropic Functions. Nature Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- Lorand L, Conrad SM. Transglutaminases. Mol Cell Biochem. 1984;58(1–2):9–35. doi: 10.1007/BF00240602. [DOI] [PubMed] [Google Scholar]
- Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Rispo A, et al. Unexpected Role of Surface Transglutaminase Type II in Celiac Disease. Gastroenterology. 2005;129(5):1400–1413. doi: 10.1053/j.gastro.2005.07.054. [DOI] [PubMed] [Google Scholar]
- Mangala LS, Mehta K. Tissue transglutaminase (TG2) in cancer biology. Prog Exp Tumor Res. 2005;38:125–138. doi: 10.1159/000084237. [DOI] [PubMed] [Google Scholar]
- Mangala LS, Fok JY, Zorrilla-Calancha IR, Verma A, Mehta K. Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene. 2006 doi: 10.1038/sj.onc.1210035. [DOI] [PubMed] [Google Scholar]
- Mann AP, Verma A, Sethi G, Manavathi B, Wang H, Fok JY, et al. Overexpression of Tissue Transglutaminase Leads to Constitutive Activation of Nuclear Factor-{kappa}B in Cancer Cells: Delineation of a Novel Pathway. Cancer Res. 2006;66(17):8788–8795. doi: 10.1158/0008-5472.CAN-06-1457. [DOI] [PubMed] [Google Scholar]
- Marrano C, de Macedo P, Keillor JW. Evaluation of novel dipeptide-bound alpha,beta-unsaturated amides and epoxides as irreversible inhibitors of guinea pig liver transglutaminase. Bioorg Med Chem. 2001a;9(7):1923–1928. doi: 10.1016/s0968-0896(01)00101-8. [DOI] [PubMed] [Google Scholar]
- Marrano C, de Macedo P, Gagnon P, Lapierre D, Gravel C, Keillor JW. Synthesis and evaluation of novel dipeptide-bound 1,2,4-thiadiazoles as irreversible inhibitors of guinea pig liver transglutaminase. Bioorg Med Chem. 2001b;9(12):3231–3241. doi: 10.1016/s0968-0896(01)00228-0. [DOI] [PubMed] [Google Scholar]
- Mastroberardino PG, Iannicola C, Nardacci R, Bernassola F, De Laurenzi V, Melino, et al. ‘Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington’s disease. Cell Death Differ. 2002;9:873–880. doi: 10.1038/sj.cdd.4401093. [DOI] [PubMed] [Google Scholar]
- Mehta K, Fok J, Miller FR, Koul D, Sahin AA. Prognostic significance of tissue transglutaminase in drug resistant and metastatic breast cancer. Clin Cancer Res. 2004;10(23):8068–8076. doi: 10.1158/1078-0432.CCR-04-1107. [DOI] [PubMed] [Google Scholar]
- Melino G, Annicchiarico-Petruzzelli M, Piredda L, Candi E, Gentile V, Davies PJ, et al. Tissue transglutaminase and apoptosis: sense and antisense transfection studies with human neuroblastoma cells. 1994;14(10):6584–6596. doi: 10.1128/mcb.14.10.6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milakovic T, Tucholski J, McCoy E, Johnson GV. Intracellular localization and activity state of tissue transglutaminase differentially impacts cell death. J Biol Chem. 2004;279(10):8715–8722. doi: 10.1074/jbc.M308479200. [DOI] [PubMed] [Google Scholar]
- Mirza A, Liu SL, Frizell E, Zhu J, Maddukuri S, Martinez J, et al. A role for tissue transglutaminase in hepatic injury and fibrogenesis, and its regulation by NF-kappaB. Am J Physiol. 1997;272(2 Pt 1):G281–288. doi: 10.1152/ajpgi.1997.272.2.G281. [DOI] [PubMed] [Google Scholar]
- Mishra S, Murphy LJ. Tissue transglutaminase has intrinsic kinase activity: identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. J Biol Chem. 2004;279(23):23863–23868. doi: 10.1074/jbc.M311919200. [DOI] [PubMed] [Google Scholar]
- Molberg O, McAdam S, Lundin KE, Kristiansen C, Arentz-Hansen H, Kett K, et al. T cells from celiac disease lesions recognize gliadin epitopes deamidated in situ by endogenous tissue transglutaminase. Eur J Immunol. 2001;31(5):1317–1323. doi: 10.1002/1521-4141(200105)31:5<1317::AID-IMMU1317>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Molberg O, Mcadam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4(6):713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- Molberg O, McAdam SN, Sollid LM. Role of tissue transglutaminase in celiac disease. J Pediatr Gastroenterol Nutr. 2000;30(3):232–240. doi: 10.1097/00005176-200003000-00005. [DOI] [PubMed] [Google Scholar]
- Monsonego A, Friedmann I, Shani Y, Eisenstein M, Schwartz M. GTP-dependent conformational changes associated with the functional switch between Galpha and cross-linking activities in brain-derived tissue transglutaminase. J Mol Biol. 1998;282(4):713–720. doi: 10.1006/jmbi.1998.2052. [DOI] [PubMed] [Google Scholar]
- Murthy SN, Lomasney JW, Mak EC, Lorand L. Interactions of G(h)/transglutaminase with phospholipase Cdelta1 and with GTP. Proc Natl Acad Sci. 1999;96(21):11815–11819. doi: 10.1073/pnas.96.21.11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem. 276(23):20673–20678. doi: 10.1074/jbc.M010846200. [DOI] [PubMed] [Google Scholar]
- Nardacci R, Lo Iacono O, Ciccosanti F, Falasca L, Addesso M, Amendola A, et al. Transglutaminase type II plays a protective role in hepatic injury. Am J Pathol. 2003;162(4):1293–1303. doi: 10.1016/S0002-9440(10)63925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas B, Smethurst P, Verderio E, Jones R, Griffin M. Cross-linking of cellular proteins by tissue transglutaminase during necrotic cell death: a mechanism for maintaining tissue integrity. Biochem J. 2003;371:413–422. doi: 10.1042/BJ20021949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliverio S, Amendola A, Rodolfo C, Spinedi A, Piacentini M. Inhibition of “tissue” transglutaminase increases cell survival by preventing apoptosis. J Biol Chem. 1999;274(48):34123–34128. doi: 10.1074/jbc.274.48.34123. [DOI] [PubMed] [Google Scholar]
- Pardin C, Gillet SM, Keillor JW. Synthesis and evaluation of peptidic irreversible inhibitors of tissue transglutaminase. Bioorg Med Chem. 2006;14(24):8379–8385. doi: 10.1016/j.bmc.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Piper JL, Gray GM, Khosla C. High Selectivity of Human Tissue Transglutaminase for Immunoactive Gliadin Peptides: Implications for Celiac Sprue. Biochemistry. 2002;41:386–393. doi: 10.1021/bi011715x. [DOI] [PubMed] [Google Scholar]
- Piper JL, Gray GM, Khosla C. Effect of prolyl endopeptidase on digestive-resistant gliadin peptides in vivo. J Pharmacol Exp Ther. 2004;311(1):213–219. doi: 10.1124/jpet.104.068429. [DOI] [PubMed] [Google Scholar]
- Qiao SW, Bergseng E, Molberg O, Jung G, Fleckenstein B, Sollid LM. Refining the rules of gliadin T cell epitope binding to the disease-associated DQ2 molecule in celiac disease: importance of proline spacing and glutamine deamidation. J Immunol. 2005;175(1):254–261. doi: 10.4049/jimmunol.175.1.254. [DOI] [PubMed] [Google Scholar]
- Quarsten H, Molberg O, Fugger L, McAdam SN, Sollid LM. HLA binding and T cell recognition of a tissue transglutaminase-modified gliadin epitope. Eur J Immunol. 1999;29(8):2506–2514. doi: 10.1002/(SICI)1521-4141(199908)29:08<2506::AID-IMMU2506>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Radek JT, Jeong JM, Murthy SNP, Ingham KC, Lorand L. Affinity of human erythrocyte transglutaminase for a 42-kDa gelatin-binding fragment of human plasma fibronectin. Proc Natl Acad Sci. 1993;90:3152–3156. doi: 10.1073/pnas.90.8.3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose DM, Sydlaske AD, Agha-Babakhani A, Johnson K, Terkeltaub R. Transglutaminase 2 limits murine peritoneal acute gout-like inflammation by regulating macrophage clearance of apoptotic neutrophils. Arthritis Rheum. 2006;54(10):3363–3371. doi: 10.1002/art.22137. [DOI] [PubMed] [Google Scholar]
- Ruan Q, Johnson GVW. Transglutaminase 2 in neurodegenerative disorders. Front Biosci. 2007;12:891–904. doi: 10.2741/2111. [DOI] [PubMed] [Google Scholar]
- Sarang Z, Molnar P, Nemeth T, Gomba S, Kardon T, Melino G, et al. Tissue transglutaminase (TG2) acting as G protein protects hepatocytes against Fas-mediated cell death in mice. Hepatology. 2005;42(3):578–587. doi: 10.1002/hep.20812. [DOI] [PubMed] [Google Scholar]
- Sardy M, Odenthal U, Karpati S, Paulsson M, Smyth N. Recombinant human tissue transglutaminase ELISA for the diagnosis of gluten-sensitive enteropathy. Clin Chem. 1999;45(12):2142–2149. [PubMed] [Google Scholar]
- Shan L, Molberg O, Parrot I, Hausch F, Filiz F, Gray GM, et al. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297:2275–2279. doi: 10.1126/science.1074129. [DOI] [PubMed] [Google Scholar]
- Siefring GE, Jr, Apostol AB, Velasco PT, Lorand L. Enzymatic basis for the Ca2+-induced cross-linking of membrane proteins in intact human erythrocytes. Biochemistry. 1978;17(13):2598–2604. doi: 10.1021/bi00606a022. [DOI] [PubMed] [Google Scholar]
- Smethurst PA, Griffin M. Measurement of tissue transglutaminase activity in a permeabilized cell system: its regulation by Ca2+ and nucleotides. Biochem J. 1996;313:803–808. doi: 10.1042/bj3130803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollid LM. Molecular basis of celiac disease. Annu Rev Immunol. 2000;18:53–81. doi: 10.1146/annurev.immunol.18.1.53. [DOI] [PubMed] [Google Scholar]
- Szondy Z, Mastroberardino PG, Varadi J, Farrace MG, Nagy N, Bak I, et al. Tissue transglutaminase (TG2) protects cardiomyocytes against ischemia/reperfusion injury by regulating ATP synthesis. Cell Death Differ. 2006;13(10):1827–1829. doi: 10.1038/sj.cdd.4401889. [DOI] [PubMed] [Google Scholar]
- Szondy Z, Sarang Z, Molnar P, Nemeth T, Piacentini M, Mastroberardino PG, et al. Transglutaminase 2−/− mice reveal a phagocytosis-associated crosstalk between macrophages and apoptotic cells. Proc Natl Acad Sci. 2003;100(13):7812–7817. doi: 10.1073/pnas.0832466100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Thomazy V, Fesus L. Differential expression of tissue transglutaminase in human cells. Cell Tissue Res. 1989;255:215–224. doi: 10.1007/BF00229084. [DOI] [PubMed] [Google Scholar]
- Tso JY, Bower SG, Zalkin H. Mechanism of inactivation of glutamine amidotransferases by the antitumor drug L-(alpha S, 5S)-alpha-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid (AT-125) J Biol Chem. 1980;255(14):6734–6738. [PubMed] [Google Scholar]
- Upchurch HF, Conway E, Patterson MK, Jr, Maxwell MD. Localization of cellular transglutaminase on the extracellular matrix after wounding: characteristics of the matrix bound enzyme. J Cell Physiol. 1991;149(3):375–382. doi: 10.1002/jcp.1041490304. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Pearson J, Bailey CD, Rogers DA, Johnson GV, Hayden MR, et al. Cystamine treatment is neuroprotective in the YAC128 mouse model of Huntington disease. J Neurochem. 2005;95(1):210–220. doi: 10.1111/j.1471-4159.2005.03357.x. [DOI] [PubMed] [Google Scholar]
- Verderio E, Nicholas B, Gross S, Griffin M. Regulated expression of tissue transglutaminase in Swiss 3T3 fibroblasts: effects on the processing of fibronectin, cell attachment, and cell death. Exp Cell Res. 1998;239(1):119–138. doi: 10.1006/excr.1997.3874. [DOI] [PubMed] [Google Scholar]
- Verma A, Wang H, Manavathi B, Fok JY, Mann AP, Kumar R, et al. Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res. 2006;66(21):10525–10533. doi: 10.1158/0008-5472.CAN-06-2387. [DOI] [PubMed] [Google Scholar]
- Watts RE, Siegel M, Khosla C. Structure-activity relationship analysis of the selective inhibition of transglutaminase 2 by dihydroisoxazoles. J Med Chem. 2006;49(25):7493–7501. doi: 10.1021/jm060839a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Siegel M, Bergseng E, Sollid LM, Khosla C. Inhibition of HLA-DQ2-mediated antigen presentation by analogues of a high affinity 33-residue peptide from alpha2-gliadin. J Am Chem Soc. 2006;128(6):1859–1867. doi: 10.1021/ja056423o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Begum S, Hearn JD, Hynes RO. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc Natl Acad Sci. 2006;103(24):9023–9028. doi: 10.1073/pnas.0602681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Choi K, Khosla C, Zheng X, Higashikubo R, Chicoine MR, et al. Tissue transglutaminase 2 inhibition promotes cell death and chemosensitivity in glioblastomas. Mol Cancer Ther. 2005;4(9):1293–1302. doi: 10.1158/1535-7163.MCT-04-0328. [DOI] [PubMed] [Google Scholar]
- Yuan L, Siegel M, Choi K, Khosla C, Miller CR, Jackson EN, et al. Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene. 2006 doi: 10.1038/sj.onc.1210048. [DOI] [PubMed] [Google Scholar]
- Zemskov EA, Janiak A, Hang J, Waghray A, Belkin AM. The role of tissue transglutaminase in cell-matrix interactions. Front Biosci. 2006;11:173–185. doi: 10.2741/1863. [DOI] [PubMed] [Google Scholar]
- Zhang J, Guttmann RP, Johnson GV. Tissue transglutaminase is an in situ substrate of calpain: regulation of activity. J Neurochem. 1998;71(1):240–247. doi: 10.1046/j.1471-4159.1998.71010240.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lesort M, Guttmann RP, Johnson GVW. Modulation of the in Situ Activity of Tissue Transglutaminase by Calcium and GTP. J Biol Chem. 1998;273(4):2288–2295. doi: 10.1074/jbc.273.4.2288. [DOI] [PubMed] [Google Scholar]