

Abstract
Objective
It has been demonstrated that high concentration of the transcription factor PU.1 (encoded by Sfpi1) promotes macrophage development, whereas low concentration induces B cell development in vitro. This has led to the hypothesis that lower levels of PU.1 activity are required for B cell than for macrophage development in vivo. We utilized an allele of Sfpi1 (termed BN) with a mutation in the first coding exon which resulted in a reduction of PU.1 expression in order to test this hypothesis.
Methods
Using gene targeting in ES cells, two ATG-start site codons of PU.1 were mutated, resulting in reduced PU.1 expression originating from a third start codon. Mice were assayed for phenotypic abnormalities using fluorescence activated cell sorting, microscopy, and colony forming ability. In addition, isolated cells were tested for their differentiation potential in vitro and in vivo.
Results
Lymphoid and myeloid cells derived from cultured Sfpi1BN/BN fetal liver cells had reduced levels of PU.1 expression and activity. B cell development was intrinsically blocked in cells isolated from Sfpi1BN/BN mice. In addition, myeloid development was impaired in Sfpi1BN/BN fetal liver. However, neonatal Sfpi1BN/BN mice had a dramatic expansion and infiltration of immature myeloid cells.
Conclusion
Contrary to our original hypothesis, high levels of PU.1 activity are required to induce both myeloid and B cell development. In addition, neonatal mice homozygous for the hypomorphic allele acquire a myeloproliferative disorder and die within 1 month of age.
Introduction
Hematopoiesis is a complex patterning process in which the various distinct cellular lineages of the blood system are generated from a pluripotent stem cell. Many key regulatory steps of hematopoietic development are controlled cell-intrinsically at the level of transcription. The genes involved in this process are regulated by an elaborate network of transcription factors. One transcription factor of particular interest is PU.1 (encoded by the gene Sfpi1), a tissue-specific Ets family member that is expressed in both lymphoid and myeloid cells of the immune system [1,2]. Gene targeting studies have demonstrated that PU.1 is required for the development of both myeloid and lymphoid lineages [3–5]. Previous research has suggested that differences in PU.1 concentration are involved in hematopoietic cell fate decisions. Specifically, PU.1 is expressed at high levels in myeloid lineage cells and at lower levels in B lymphocytes [1,2,6,7]. Based on retroviral gene transfer experiments performed in vitro, it was hypothesized that high concentration of PU.1 promotes differentiation into macrophages, whereas low concentration of PU.1 induces differentiation into B cells [8].
Several recently published studies have utilized a reporter gene encoding green fluorescent protein (GFP) inserted into the Sfpi1 locus to monitor the level of PU.1 transcription in lymphoid and myeloid cells in vivo. These studies suggested that PU.1 is expressed at uniformly high levels in hematopoietic stem cells (HSCs) and common lymphoid progenitor cells (CLPs) [9,10]. During commitment to the B cell lineage, PU.1 expression is reduced to lower levels [10]. Conditional knockout studies demonstrated that deletion of Sfpi1 in committed B cell progenitors minimally affects B cell development [11–13]. An important question that remains unanswered in these studies is whether high PU.1 activity is required for specification to the B lineage.
To test our hypothesis, we generated a novel hypomorphic allele of the Sfpi1 gene generated by mutation of the first coding exon. Mice homozygous for this allele (termed BN) survive 1–3 weeks after birth and are osteopetrotic and runted. Splenic expression of PU.1 is dramatically reduced in Sfpi1BN/BN mice. Furthermore, PU.1 expression and activity are reduced in both lymphoid and myeloid cells cultured from Sfpi1BN/BN fetal liver. Sfpi1BN/BN neonatal mice are B cell deficient. In addition, fetal liver progenitors from Sfpi1BN/BN fetuses cannot reconstitute B cell development upon transplantation into immunodeficient recipient mice. Finally, we demonstrate that myeloid cell differentiation is impaired in Sfpi1BN/BN fetal liver. However, neonatal Sfpi1BN/BN mice have a dramatic proliferation of immature myeloid cells in their spleens and bone marrow. These results suggest that high levels of PU.1 activity are required for both lymphoid and myeloid development. In addition, these results demonstrate that reduced expression of PU.1 results in a neonatal myeloproliferative disorder.
Materials and Methods
Gene targeting and mice
3 kilobases (kb) upstream and 3.5 kb downstream of the two identified ATG-start codons (6.5 kb total) of the Sfpi1 locus were inserted into the osdupdel2 vector along with E. coli TEM-1 β-lactamase coding sequence, obtained by digestion of BlaM(−)/pUC plasmid (Invitrogen, Carlsbad, CA, USA) with SalI and KpnI, to generate the 53Bladd targeting vector. Primers used to amplify the Sfpi1 sequence from genomic DNA from 129SV mice were as follows: 5′-arm (5′-TCAGCACCACAGCAATAGTCTCGGG-3′ and 5′-TCCAGGTTGGTCAGATCCCCTGCTTCGGTG-3′), 3′-arm (5′-GAAGGGTTTTCCCTCACCGCCCCTGTGAG-3′ and 5′-GGCCAAGCAGAACGCAACACTGATGAACTC-3′). 129SV ES cells were electroporated with linearized targeting vector DNA and positive/negative selection of clones was performed using standard methods. Recombination events were screened by PCR and confirmed by Southern blot analysis on HindIII digested DNA and a probe directed against the neomycin gene. Additionally, DNA sequencing was performed on PCR-amplified genomic DNA. The mice used in this study were a primarily outbred mixture of C57Bl/6 and Black/Swiss (Taconic, Germantown, NY). For genotyping, PCR was performed with the following genotyping primers: 1 (5′-CGGCCAGAGACTTCCTGTAG-3′), 2 (5′-AAGTTGGCCGCAGTGTTATC-3′), 3 (5′-GCTCTTCGTCCAGATCATCC-3′), 4 (5′-ATGGTCACACATCCCAAAGC-3′).
Construction of Retroviral Vectors
MSCV-IRES-EGFP (MigR1) and MSCV-PU.1-IRES-EGFP (MigPU.1, HA-PU.1wt) retroviruses have been described previously [14]. Retroviral vectors encoding wild-type or Sfpi1BN transcripts were constructed by inserting either transcript between the XhoI and EcoRI sites of MigR1. Transcripts were amplified from cDNA prepared from pro-B cells using LA-TAQ (TaKaRa Bio Inc., Otsu Shiga, Japan) and cloned using the topoisomerase cloning kit (Stratagene, La Jolla CA). Primers used were as follows: wild-type forward (5′-ACCTGGAGCTCAGCTGGATGTTA-3′), BN forward (5′-CGTATCGATTGTACACTTAAGGGCCG-3′), common reverse (5′GGGCGACGGGTTAATGCTAT-3′). HA-PU.1ΔN31 retrovirus was constructed by insertion of ΔN31 transcript into the Not I and Apa I sites to replace the Spi-C sequence of MIG-Spi-C vector [15]. ΔN31 transcripts were amplified from pBluescript plasmid containing PU.1 cDNA lacking the N-terminal 31 amino acids and cloned as above. Primers used were as follows: DN31 forward (5′-CGGCGGCCGCATGCATGACTACTACTCCTTC-3′) and DN31 reverse (5′-CCGGGCCCTCAGTGGGGCGGGAGGCGCCG-3′)
Cell Culture
Cell culture conditions and generation of pro-B cell lines have been described previously [16]. Cultured IL-3 myeloid cells were generated by culturing fetal liver progenitors (FLPs) in complete IMDM with IL-3 (5 ng/ml), IL-6 (10 ng/ml), and SCF (100 ng/ml) (Peprotech, Rocky Hill, NJ) for two days, followed by passage in complete IMDM containing IL-3 (5 ng/ml).
Day 14.5 Sfpi1+/+, Sfpi1−/−, Sfpi1+/BN, or Sfpi1BN/BN FLPs were generated by timed matings of Sfpi1+/BN or Sfpi1+/− mice as previously described [17]. For rescue experiments, Sfpi1−/−FLPs were infected by spin-infection and analyzed by flow cytometry after two days of culture, as described [15]. Colony forming assays were performed as previously described [16] using recombinant murine GM-CSF (1 ng/ml), G-CSF (10 ng/ml), M-CSF (10 ng/ml) or IL-3 (5 ng/ml) + IL-6 (10 ng/ml) + SCF (100 ng/ml) (Peprotech).
Morphological, Flow Cytometric, and Immunoblot Analysis
For morphological analysis, 105 washed cells were cytospun onto glass slides and stained with Wright stain. Cells were viewed using an Olympus BH2 light microscope and photographed using an Olympus AX-70 microscope with a Photometrics digital camera. Flow cytometric analysis was performed on cells stained with the biotin-conjugated monoclonal antibodies (mAb) M1/70 (CD11b), RB6-8C5 (Gr-1), 2.4G2 (FcγRII/III); antibodies conjugated -Kit); orto antibodies conjugated to allophycocyanin (APC): A3-1 (F4/80), RA3-6B2 (B220) (BD Pharmingen). Biotin-conjugated antibodies were visualized by secondary staining with streptavidin-APC conjugate or streptavidin-PE conjugate (BD Pharmingen, San Diego, CA). Cells were analyzed using a BD FACSCalibur system. Immunoblotting was performed as previously described [13] using rabbit anti-PU.1 peptide Ab (Santa Cruz Biotechnology, Santa Cruz, CA) and HRP-conjugated anti-rabbit secondary Ab (Pierce, Rockford, IL).
Transfection and Reporter Assays
An 1817 bp FcγRIIb promoter fragment was PCR amplified from C57Bl/6 genomic DNA and cloned into the pGL3-basic luciferase reporter vector (Promega, Madison, WI) as previously described [15]. A single nucleotide in the PU.1 binding site was mutated using the Stratagene QuickChange XL kit (Stratagene), using the oligonucleotides 5′-GACCGTTTCTTTTCACGTCCCCATTTGGACTTCA-3′ and 5′-TGAAGTCCAAATGGGGACGTGAAAAGAAACGGTC-3′ (PU.1 binding site italicized, mutated nucleotide underlined). 38B9 and WEHI-231 cells were transfected with reporter DNA as well as a pRL-TK control vector using electroporation as previously described [15]. RAW264.7 cells were transfected using lipofectamine (Invitrogen). Lysates were prepared 24 hours after transfection and normalized luciferase activity was obtained using a Dual luciferase kit (Promega).
Electrophoretic Mobility Shift Assay (EMSA)
EMSAs were performed as previously described [15]. Synthetic complementary oligonucleotides were annealed and end-labeled with [α-32P]dCTP using the Klenow fragment. Double-stranded oligonucleotide probes contained the PU.1-binding site in the FcγRIIb promoter, 5′-TCGATTCTTTTCACTTCCCCAT-3′ or a mutated PU.1-binding site (mutant FcγRIIb), 5′-TCGATTCTTTTCACGTCCCCAT-3′. An E2A consensus site oligonucleotide was used as a control, 5′-GATCTACATCTGTGCAGTTCGCTG-3′.
Transplantation into Immunodeficient Recipient Mice
Fetal liver cells (FLCs) were erythrocyte-depleted and two million FLCs were injected into the tail vein of sublethally irradiated (400 Rad, single dose) Rag-2−/− Il2rg−/−immunodeficient recipient mice (Taconic emerging models program) [18]. Injections were performed within 4 hours of irradiation.
RT-PCR Analysis
RT-PCR was performed as previously described [13]. Primer sequences used are listed as follows: exon 1 forward (5′-TTTCCCTCACCGCCC-3′), exon 3 reverse (5′-TATCGAGGACGTGCATCTGTTCCA-3′), exon 3 forward (5′-ATCACTACTGGGATTTCTCCGCAC-3′), exon 4 reverse (5′-ATCAGCTTCTCCACACAGACACCTC-3′), exon 4 forward (5′-CGGATGTGCTTCCCTTATCAAAC-3′), exon 5 reverse (5′-TTTGTCCTTGTCCACCCACCAGAT-3′), fcγRIIb (5′-AAGCCTGTCACCATCACTGTCCAA-3′ and 5′-AGGGTTTCTCCCATTTCCCTGTGA-3′), β-actin (5′-TCCTTCGTTGCCGGTCCACA-3′ and 5′-CGTCTCCGGAGTCCATCACA-3′), g6pdh (5′-GAACATCATCCCTGCATCCA-3′ and 5′-CCAGTGAGCTTCCCGTTCA-3′). Real-time PCR was performed as previously described using a Cepheid SmartCycler system [13,19].
RESULTS
Generation of mice with a mutant Sfpi1 allele
The first exon of Sfpi1 has been reported to contain two translation start codons [1]. To generate a reporter allele for Sfpi1 transcription, we designed a gene targeting strategy to replace both translation start codons in exon 1 with the E. coli TEM-1β-lactamase gene (Fig. 1A). Correctly targeted ES cells were injected into C57Bl/6 blastocysts to generate chimeric mice. One founder successfully transmitted the targeted allele (termed BN) to F1 offspring. Mice with this genotype were termed Sfpi1+/BN. Genotypes were confirmed by PCR, Southern blotting, and DNA sequencing (Fig. 1B, 1C, and data not shown). Sfpi1+/BN mice were born at expected Mendelian frequencies (Table 1), had no phenotypic abnormalities, and were fertile.
Figure 1. Generation of Sfpi1+/BN mice.
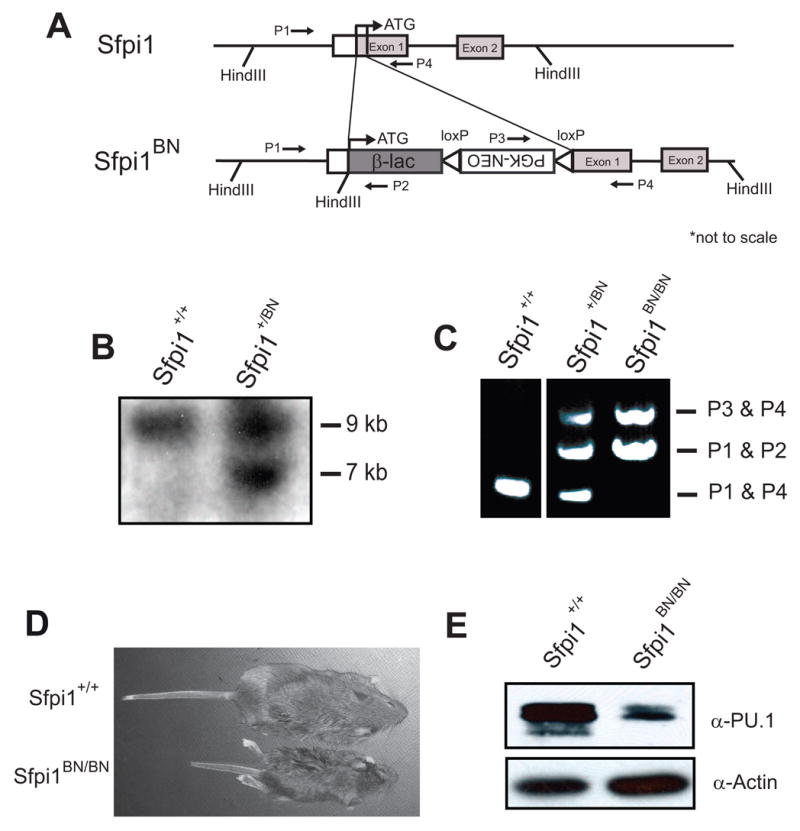
(A) Targeting strategy used to replace the translation start sites of Sfpi1 with β-lactamase (β-lac). LoxP sites are represented by open arrowheads. The locations of primers used in genotyping are indicated. (B) Southern blot analysis on HindIII-digested ES cell genomic DNA. The DNA was hybridized with a probe complementary to the PGK-NEO gene, which recognizes a 10.5 kb wild-type product and a 6.4 kb targeted fragment. The 9 kb product is due to the presence of the Neomycin gene in the feeder cell layer. (C) PCR results of a representative genotyping analysis using primers indicated above. (D) Photograph of 21 day-old littermates from Sfpi1+/BN heterozygous mating. (E) Western blot using α-PU.1 antibody demonstrating reduced levels of PU.1 in the spleens of 21-day Sfpi1BN/BN mice compared to wild-type mice.
Table 1.
Frequency of Live Births from Matings of Sfpi1+/BN Mice BN Matings
| AGE | Total | PU.1 +/+ | PU.1 +/BN | PU.1 BN/BN |
|---|---|---|---|---|
| E 14.5 | 32 | 9 | 14 | 9 |
| Day 1 | 119 | 29 | 61 | 29 |
| Day 8 | 165 | 54 | 88 | 23 |
|
| ||||
| Day 21 | 259 | 104 | 146 | 9 |
Genomic DNA was prepared from d14.5 fetuses, from newborns one day after birth, from mice one week after birth, or at the time of weaning (21 days). Genomic DNA was used to identify genotypes by PCR as described in Materials and Methods.
We attempted to detect β-lactamase activity in Sfpi1+/BN mice using flow cytometry or immunoblotting [20]. However, to date we have not detected β-lactamase activity in cells or tissues prepared from these mice. RT-PCR analysis of transcription in the targeted allele suggests that RNA splicing occurs at high frequency from several consensus splice donor sites within the β-lactamase cDNA. The data suggest that there are likely various splice acceptor sites within the PGK-Neo cassette and within the Sfpi1 gene (data not shown).
We mated Sfpi1+/BN heterozygous mice to generate Sfpi1+/+, Sfpi1+/BN and Sfpi1BN/BN offspring. Sfpi1BN/BN mice were born at near Mendelian frequency and survived between 1 and 3 weeks, with an occasional mouse reaching weaning age (~3% of total mice at 21 days of age, Table 1). At 2–3 weeks of age, Sfpi1BN/BN mice were runted and had multiple symptoms of osteopetrosis, including failure of tooth eruption and partially occluded femurs (Fig. 1D and data not shown). These characteristics have been reported for Sfpi1−/− mice [21] and suggest that these mice are macrophage-deficient. To determine if any PU.1 protein was generated by the BN allele, we prepared lysates from the spleens of two week-old wild-type or Sfpi1BN/BN mice. As shown in Fig. 1E, Sfpi1BN/BN mice express reduced levels of PU.1 protein compared to wild-type. In summary, these data suggest that the Sfpi1BN allele represents a hypomorphic, and not a null, allele.
Sfpi1BN allele results in reduced expression of PU.1
In order to analyze transcription of the Sfpi1BN allele in distinct lineages, we established IL-7 dependent pro-B cell or IL-3 dependent myeloid cell lines from mouse fetal liver as described in Materials and Methods. Using RNA prepared from these cells, we performed RT-PCR across all Sfpi1 exons to examine the integrity of the Sfpi1 transcript. Sfpi1 transcripts were intact but at reduced levels in Sfpi1BN/BN pro-B cells compared to wild-type cells (Fig. 2A). To determine the levels of Sfpi1 transcription, we performed real-time RT-PCR using primers specific to exons 3 and 4 of Sfpi1. Mutation of the Sfpi1 allele resulted in a 6.7-fold reduction in transcripts from myeloid cells and a 2-fold reduction in transcripts from pro-B cells (Fig. 2B and 2C). PU.1 protein was expressed at 20% of wild-type levels in Sfpi1BN/BN pro-B cells, as revealed by immunoblotting of serial dilutions of nuclear lysates (Fig. 2D and data not shown). These results suggest that transcription of the Sfpi1BN allele is reduced in both lymphoid and myeloid cell lineages.
Figure 2. PU.1 expression is reduced by the BN mutation in both myeloid and lymphoid lineages.
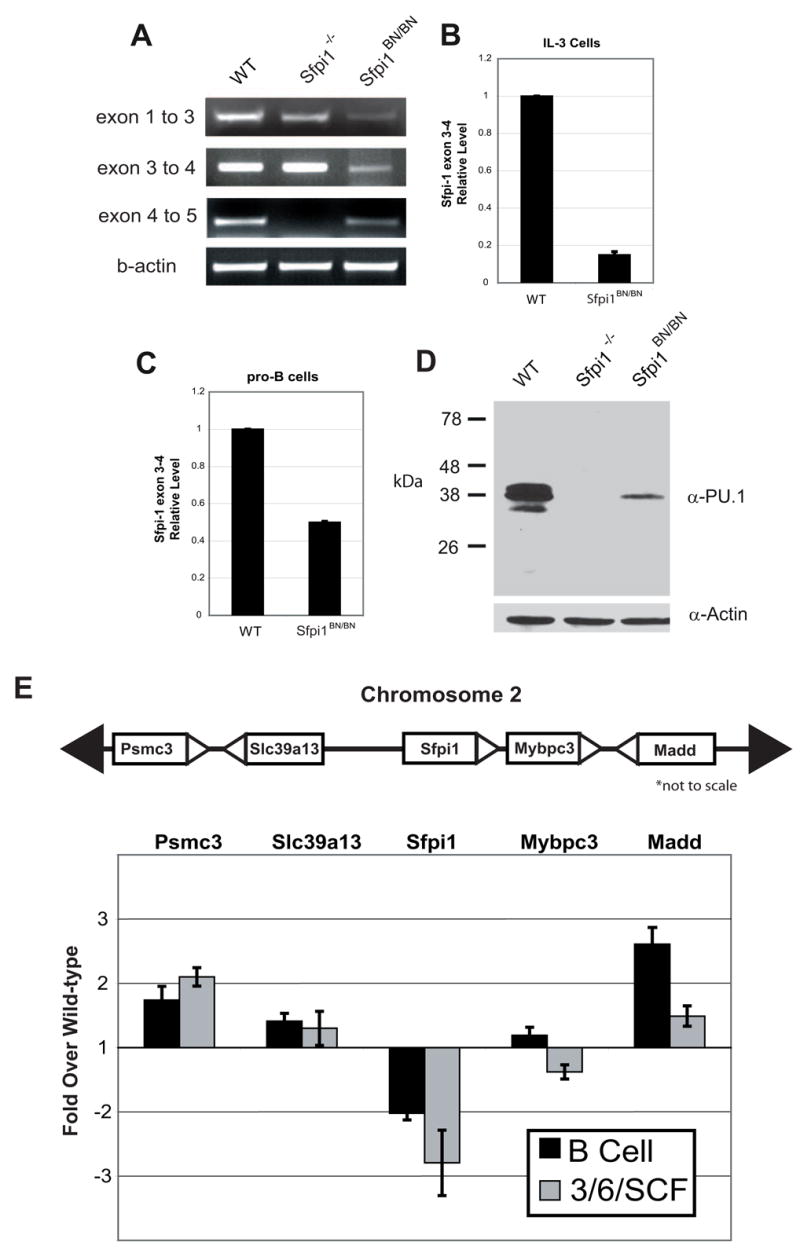
(A) RT-PCR analysis of transcription across the Sfpi1 locus. PCR was performed using primers that reside within the exons indicated. The lack of product between exons 4 and 5 in the Sfpi1−/− cells is due to the partial deletion of exon 5 [4]. (B, C) Real-time RT-PCR was used to measure Sfpi1 exon 3 to 4 transcripts in cultured IL-3 cells (B) or cultured IL-7 and stromal cell pro-B cells (C) from wild-type or Sfpi1BN/BN fetal liver. (D) Western blot for PU.1 protein expression in nuclear lysates of pro-B cells of the indicated genotype. Quantitative analysis demonstrated a 5-fold reduction in PU.1 expression. (E) Real-time RT-PCR analysis of RNA transcripts from pro-B cells or fetal liver progenitors cultured for 2 days in IL-3, IL-6, and SCF. Bars indicate the fold change in transcriptional expression in Sfpi1BN/BN cells compared to wild-type cells using primers that recognize the genes indicated. Gene location in relation to Sfpi1 is shown above. Error bars indicate the standard error.
To determine whether the Sfpi1BN allele affects transcription of neighboring genes, we examined the expression of Psmc3, Slc39a13, Mybpc3, and Madd using real-time RT-PCR (Fig. 2E). Transcripts of Madd and Psmc3 were slightly increased (~2 fold) in either cultured Sfpi1BN/BN pro-B cells or progenitors expanded in IL-3, IL-6, and SCF, relative to wild-type cells. Transcripts of Slc39a13 or Mybpc3 were not significantly altered. These changes are small and are likely to be a consequence of the phenotype imposed by hypomorphic PU.1.
The Sfpi1BN allele generates functional PU.1 protein
Upon further examination of the DNA sequence of the targeted allele, we identified a third potential ATG-start codon that might account for PU.1 expression (Fig. 3A). To determine if the mutated transcript has biological activity, we generated retroviruses containing either the wild-type or Sfpi1BN full-length transcripts and tested their ability to rescue Sfpi1−/− fetal liver progenitor cells (FLPs) in M-CSF-dependent colony-forming assays (Fig. 3B) [8]. Expression of PU.1 from the cloned BN transcript was substantially reduced compared to wild-type (Fig. 3C and 3D). Sfpi1−/− FLPs infected with the cloned BN retrovirus generated colonies in response to M-CSF; however, the number of colonies was reduced compared to the number generated by infection with the cloned wild-type retrovirus (18 colonies per 105 cells plated compared to 345 colonies per 105 cells) (Fig. 3E). To determine whether this reduction in the ability of the Sfpi1BN transcript-encoding retrovirus to rescue Sfpi1−/− FLPs was due to reduced expression or altered function, we constructed an additional retrovirus encoding a truncated PU.1 protein (HA-PU.1ΔN31, Fig 3B). To optimize expression, this retroviral vector contains an HA-tag and Kozak sequence identical to a vector encoding full-length PU.1 protein (HA-PU.1wt, [14]). As expected, expression of PU.1 was readily detectable in western blots of HA-PU.1ΔN31-transfected cells (Fig. 3C and 3D). Furthermore, infection of Sfpi1−/− FLPs with either HA-PU.1wt or HA-PU.1ΔN31 rescued M-CSF-dependent colony formation at near equivalent efficiency (643 and 711 colonies per 105 cells plated, respectively, Fig. 3E). Macrophages from HA-PU.1ΔN31-infected Sfpi1−/−FLPs resembled wild-type-rescued macrophages phenotypically when cytospun to glass slides and Wright stained (Fig. 2H). These results suggest that the reduction in PU.1 activity caused by the Sfpi1BN allele is due to a reduction in expression and not altered function.
Figure 3. PU.1 activity is reduced by the BN mutation in both myeloid and lymphoid lineages.
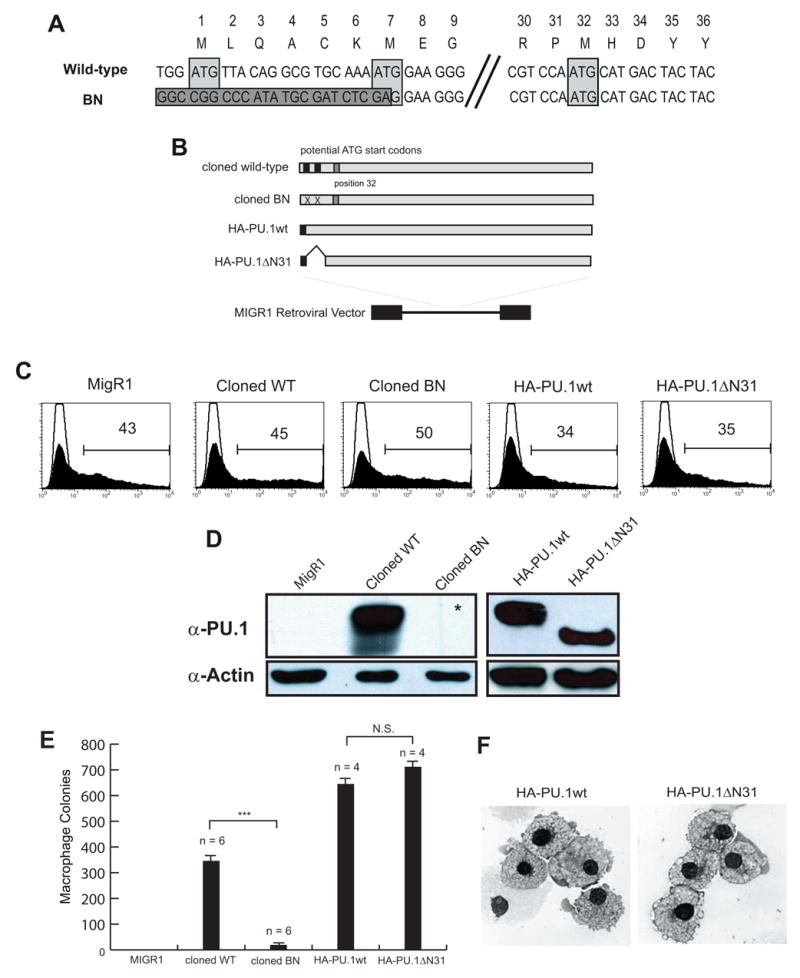
(A) Alignment of genomic DNA sequences of wild-type and targeted Sfpi1BN/BN including previously identified codon locations and their equivalent amino acids. The dark gray box indicates sequence following genomic DNA targeting resulting in the removal of both previously identified ATG-start codons (light gray boxes). Position 32 contains an in-frame potential ATG-start codon (3rd light gray box). (B) Diagram of cloning strategy to generate retroviruses. (C) Flow cytometric analysis of transfected Plat-E cells measuring GFP expression and demonstrating equivalent transfection efficiency of the indicated retroviruses. (D) Western blot analysis of transfected cells analyzed in (B) showing reduced expression of PU.1 protein from the cloned BN retrovirus. The asterisk indicates that a band was seen upon longer exposure. (E) Colony forming ability of retrovirally-infected Sfpi1−/− fetal liver progenitors (FLPs). Sfpi1−/−FLPs were isolated according to Materials and Methods and spin-infected with the indicated retrovirus. Cells were plated in methylcellulose containing M-CSF and colonies were counted after 5 days. Numbers correspond to number of colonies per 105 infected cells plated (*** - p <= .001). (F) Cytospins of cells from methylcellulose plates from (E). Cytospun and Wright stained cells were visualized using an Olympus BH2 light microscope and photographed as described in Materials and Methods.
Reduction in PU.1 activity results in reduction of target gene expression
In order to determine whether hypomorphic expression of PU.1 affects transcription of target genes, we analyzed expression of a gene recently identified as PU.1-dependent, FcγRIIb, which encodes the low-affinity IgG Fc receptor [13,22]. We performed real-time RT-PCR analysis to measure steady-state levels of FcγRIIb transcripts in cells cultured from wild-type, Sfpi1−/−, and Sfpi1BN/BN mice. FcγRIIb transcripts were reduced 7.8-fold in IL-3-dependent myeloid cells and 4-fold in IL-7-dependent pro-B cells. As expected, FcγRIIb transcripts were further reduced in Sfpi1−/−IL-3 and pro-B cells (Fig. 4A and 4B).
Figure 4. Reduction in PU.1 Activity as Measured by Transcription of the FcγRIIb gene.
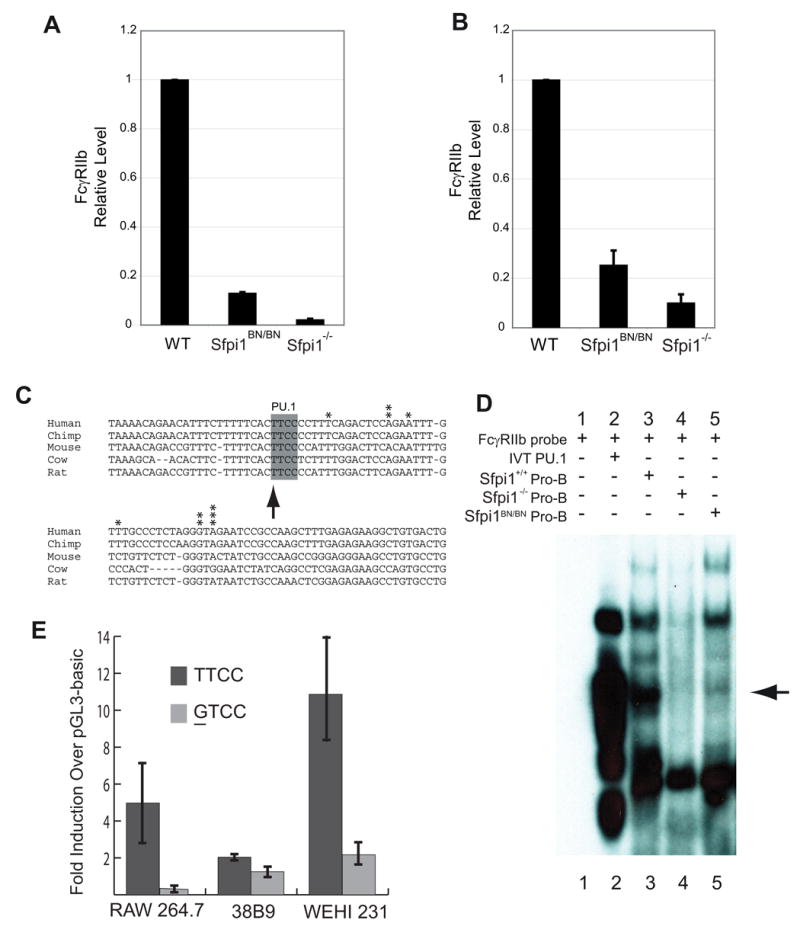
(A) Real-time RT-PCR analysis of FcγRIIb expression from RNA prepared from cultured IL-3-dependent myeloid cells of the indicated genotype. (B) Real-time RT-PCR analysis of FcγRIIb expression from RNA samples obtained from cultured IL-7-dependent pro-B cells of the indicated genotype. (C) Sequence comparison of the FcγRIIb promoter among various species. Arrow indicates PU.1-binding site. Asterisks indicate identified transcription start sites. (D) Electrophoretic mobility shift assay of nuclear lysates using an FcγRIIb probe. Arrow indicates the band corresponding to PU.1 interacting with the DNA probe. (E) Luciferase reporter assays using wild-type (TTCC) or mutated (GTCC) PU.1 binding site indicated in (C). Cell lines used include RAW264.7 (macrophage), 38B9 (pro-B), WEHI-231 (B cells).
To determine if PU.1 regulates the FcγRIIb gene directly or indirectly, we characterized the promoter of the gene. Using 5′ RACE analysis, we identified a number of transcription start sites in the region 50–70 bp upstream of the first coding exon (Fig. 4C). A predicted PU.1 binding site was located immediately upstream of the transcription start sites within a block of conserved DNA sequence (Fig. 4C). Electrophoretic mobility shift analysis (EMSA) showed that PU.1 in nuclear extracts from wild-type pro-B cells could interact with this site (Fig. 4D). Importantly, Sfpi1BN/BN pro-B cells contained reduced levels of PU.1 capable of interacting with this probe. No similar complex was formed using nuclear extracts from Sfpi1−/− pro-B cells. These complexes were demonstrated to contain PU.1 by supershift analysis with an anti-PU.1 antibody (data not shown). Furthermore, mutation of the PU.1 binding site reduced activity of a cloned segment of the FcγRIIb promoter as tested by transient transfection analysis of RAW264.7 macrophages, 38B9 pro-B cells, and WEHI-231 B cells (Fig. 4E). In summary, these data suggest that the FcγRIIb gene is directly regulated by PU.1. In addition, these data demonstrate that the Sfpi1BN allele results in a reduction in PU.1 activity.
B cell development is blocked in Sfpi1BN/BN mice
To determine whether B cell development is affected by the BN mutation, we examined fetal liver or spleen cells of wild-type or Sfpi1BN/BN mice at various ages by flow cytometry. The spleens and fetal livers of Sfpi1BN/BN mice had a dramatic reduction in the number of B220+CD19+ cells relative to wild-type mice that was apparent from embryonic day 16.5 (E.D. 16.5) (Fig. 5A and 5B). To determine whether this defect is cell-intrinsic, we transplanted wild-type or Sfpi1BN/BN E.D.14.5 FLPs into sublethally irradiated immunodeficient mice lacking both Rag-2 and common gamma chain (Rag2−/−Il2rg−/− mice) [18]. Whereas wild-type FLPs generated B cells efficiently in recipient mice six weeks after transplantation, Sfpi1BN/BN FLPs failed to generate any detectable B lymphocytes in either spleen or bone marrow after six or nine weeks (Fig. 5C). These results suggest that Sfpi1BN/BN FLPs possess a cell-intrinsic block to B cell development.
Figure 5. Cell Intrinsic Block to B Cell Development in Sfpi1BN/BN mice.
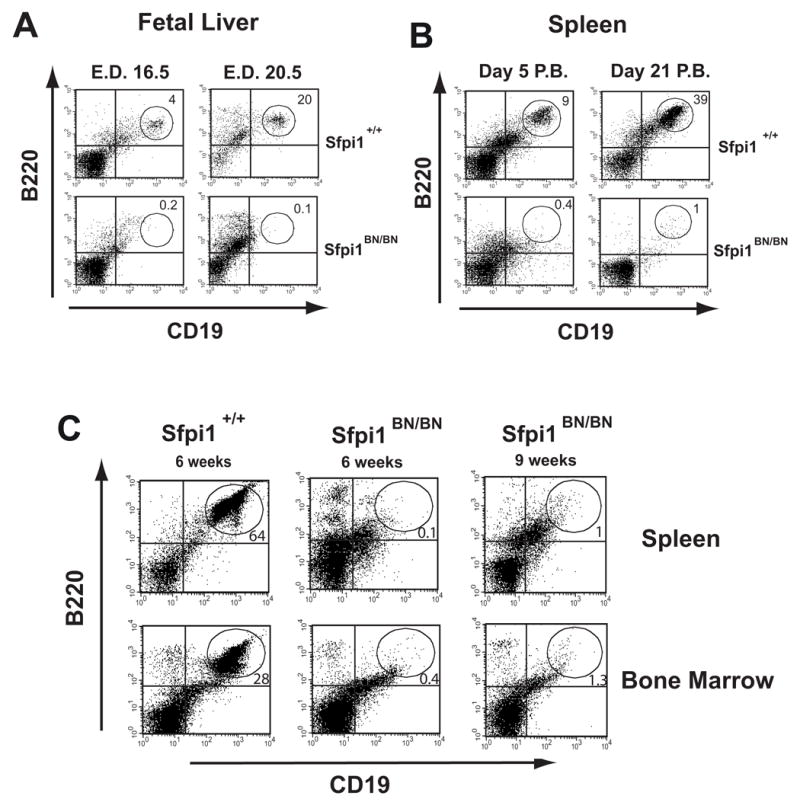
(A, B) Flow cytometric analysis of single-cell suspensions from either fetal livers or spleens of wild-type and Sfpi1BN/BN mice at the indicated age. Isolated spleen cells were analyzed by flow cytometry using gating for size and granularity and antibodies to the indicated cell surface markers. Numbers indicate percentage of gated cells in the indicated region. E.D. = emybryonic day, P.B. = post birth. (C) Flow cytometric analysis of single-cell suspensions from spleens or bone marrow of recipient mice transplanted with wild-type or Sfpi1BN/BN fetal liver cells. Fetal liver cells were isolated from wild-type or Sfpi1BN/BN mice and transplanted by tail vein injection into sublethally irradiated Rag2−/− Il2rg−/−recipient mice. Analysis was performed after the indicated time following transplantation.
Sfpi1BN/BN fetuses have reduced fetal myelopoiesis
Sfpi1−/− mice have a multilineage defect in the generation of monocytes, granulocytes, and B and T lymphocytes that is apparent by day 14.5 of fetal development [4]. Flow cytometric analysis of fetal livers from E.D. 14.5 Sfpi1BN/BN fetuses revealed a reduction in cells expressing the myeloid cell surface markers FcγRIIb/III, Gr-1, or CD11b (Fig. 6A), whereas the total cellularity of the fetal livers was increased (Fig. 6B). To determine the frequency of cytokine-responsive myeloid progenitor cells, we performed colony-forming assays on fetal liver cells (FLCs) from wild-type and Sfpi1BN/BN E.D. 14.5 fetuses. As shown in Fig. 6C, Sfpi1BN/BN fetal livers had a severe reduction in the frequency of progenitor cells able to respond to G-CSF (0.86 colonies/105 cells plated), M-CSF (0.11 colonies/105 cells plated), GM-CSF (0.58 colonies/105 cells plated), and IL-3/IL-6/SCF (11.7 colonies/105 cells plated) compared to wild-type (36, 15, 41.5, 58.3 colonies/105 cells plated, respectively).
Figure 6. – Fetal Liver Myelopoiesis is Severely Reduced in Sfpi1BN/BN Mice.
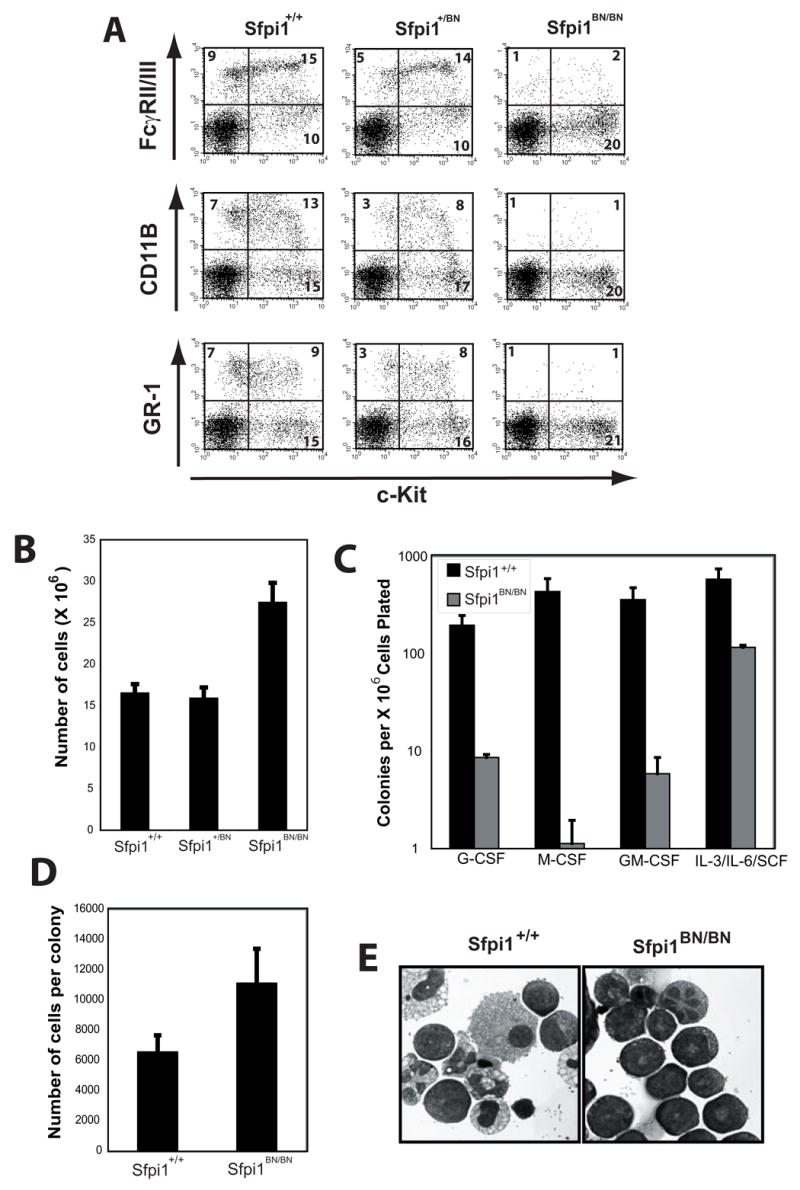
(A) Flow cytometric analysis of single-cell suspensions from day 14.5 fetal liver cells. The cell surface markers analyzed are shown. Numbers indicate the percentage of total cells. (B) Total number of cells per fetal liver of wild-type, heterozygous, and Sfpi1BN/BN mice. (C) Colony assays from fetal liver progenitors isolated from wild-type and Sfpi1BN/BN mice. Fetal liver cells were isolated from the indicated mouse and plated at various concentrations in methylcellulose with the indicated cytokines. Colonies were scored following 7 days of culture. Error bars represent the standard error of a minimum of three plates. (D) Calculated cells per colony of either wild-type or Sfpi1BN/BN fetal liver progenitors. Cells in methylcellulose cultures from (C) were washed and counted. (E) Cytospins of cells from methylcellulose plates described in (C and D).
Due to the difference in colony generating ability of Sfpi1BN/BN FLCs placed in IL-3/IL-6/SCF compared to other tested cytokines, we examined the nature of the colonies that grew under these conditions. Colonies that formed from Sfpi1BN/BN FLCs in IL-3/IL-6/SCF had a nearly 2-fold increase in the number of cells per colony compared to wild-type (Fig. 6D). Additionally, wild-type colonies in IL-3/IL-6/SCF contained mature macrophages and granulocytes, whereas Sfpi1BN/BN colonies contained promyelocytes and blast-like cells but no mature myeloid cells (Fig. 6E). These data therefore demonstrate that Sfpi1BN/BN fetuses have a substantial reduction in myelopoiesis relative to wild-type fetuses. In addition, Sfpi1BN/BN FLCs are capable of proliferating in response to IL-3/IL-6/SCF but are incapable of differentiation.
Neonatal Sfpi1BN/BN mice acquire a myeloproliferative disorder
As mentioned above, Sfpi1BN/BN mice are born at the expected Mendelian frequency and occasionally survive until weaning age. The spleens of 2–3 week old Sfpi1BN/BN mice were moderately enlarged compared to wild-type littermates. Flow cytometric analysis of Sfpi1BN/BN fetal liver, spleen, and bone marrow cells at various time points revealed a dramatic expansion of cells co-expressing the stem and progenitor cell marker c-Kit with the myeloid cell-surface markers FcγRII/III, Gr-1, and CD11b (Fig. 7A, 7B, and data not shown). This expansion was not apparent until after birth and was detectable as early as day 5 post birth (P.B.) (Fig. 7A). The percentage of cells containing the mature myeloid marker F4/80 was not increased (Fig. 7B). Cytospin analysis revealed that Sfpi1BN/BN spleens contained almost exclusively immature myeloid cells (Fig. 7C). These results demonstrate that reduced PU.1 activity results in proliferation of immature myeloid cells in neonatal mice, but not in fetal liver.
Figure 7. Myeloproliferative Disorder in Sfpi1BN/BN neonates.
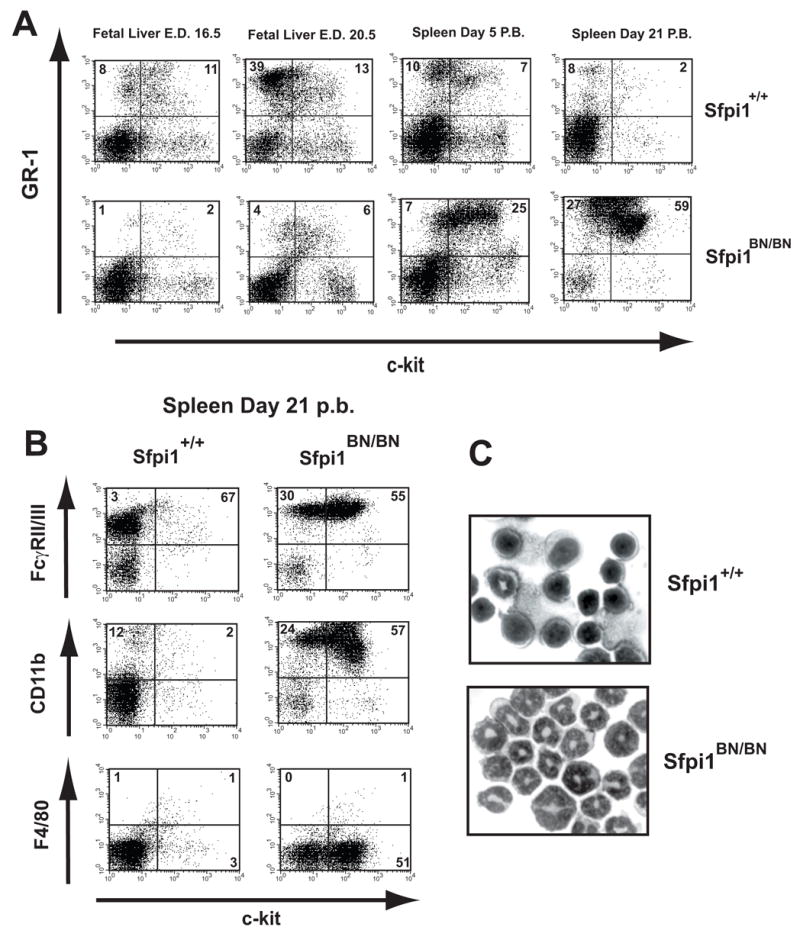
(A) Flow cytometric analysis of single-cell suspensions from either fetal liver or spleens of mice at the age indicated. Isolated cells were gated for proper size and granularity and analyzed by flow cytometry using antibodies to the indicated cell surface markers. Numbers indicate the percentage of gated cells. (B) Flow cytometric analysis of single-cell suspensions from spleens of 21 day-old wild-type and Sfpi1BN/BN mice, performed as described in (A). (C) Cytospins of spleen cells from 18 day-old littermates demonstrating infiltration of immature myeloid cells in the spleens of Sfpi1BN/BN mice. Isolated spleen cells were cytospun onto glass slides, Wright stained, and photographed as described in Materials and Methods.
Discussion
In this study, we addressed the hypothesis that lower levels of PU.1 activity might be required for B cell than for macrophage development in vivo. If this hypothesis is true, then system-wide reduction of PU.1 activity in vivo would be expected to induce B cell development while failing to promote macrophage development. We have tested this hypothesis by examining mice homozygous for a hypomorphic allele (termed BN) of the Sfpi1 gene. Analysis of Sfpi1BN/BN mice demonstrates that both B cell and macrophage development are impaired at an early stage. The block to B cell development appears more severe than the block to myeloid development, as an abnormal expansion of immature granulocytic precursors is apparent in the spleen and bone marrow of neonatal Sfpi1BN/BN mice.
Studies utilizing a green florescent protein as a reporter gene “knocked-in” to the Sfpi1 locus have revealed that PU.1 is expressed at uniformly high levels in HSCs as well as CLPs [9,10]. However, upon commitment to the B cell lineage, PU.1 expression is reduced to eight-fold lower levels in B cells than in Mac-1+ myeloid cells [10]. Conditional knockout studies of the Sfpi1 gene have also provided important insights about the role of PU.1 in B cell development. When Sfpi1 is inactivated in HSCs, B cell development is profoundly blocked [5]. In contrast, when Sfpi1 is inactivated in CLPs using a retrovirally expressed Cre recombinase or in committed B cells using Cre recombinase under control of the CD19 promoter, B cell development is minimally affected [5,11,12]. Taken together with our studies, these data suggest that high levels of PU.1 are essential for B cell development. However, these high levels are essential only at the earliest stages of B cell specification and may be dispensable after commitment to the B cell lineage. We expect that high levels of PU.1 are required to activate genes involved in B lineage specification such as IL-7Rα[14] and early B cell factor (EBF) [23].
Reporter gene studies show that PU.1 is expressed at high levels in a subset of common myeloid progenitors (CMPs) that is fated to develop into macrophages and granulocytes [10] and maintains high levels of expression throughout differentiation [9,10]. Studies of conditional Sfpi1 knockout mice show that high levels of PU.1 expression are essential for proper differentiation of macrophages and granulocytes at all stages of development [5]. However, the results we have presented in this paper, as well as those published by others, suggest that reduced levels of PU.1 still permit specification to the myeloid lineages. Interestingly, reduced levels of PU.1 promote hyperproliferation of immature myeloid precursor cells during neonatal but not fetal development. Another study also concluded that inactivation of PU.1 in adult hematopoiesis led to increased proliferation of immature myeloid cells, and eventually acute myeloid leukemia [24,25]. An important future area of investigation will be to determine why low levels of PU.1 activity result in a hyperproliferation of immature myeloid cells in vivo.
Recently, studies have been published in which an upstream regulatory element (URE) in the Sfpi1 locus was deleted using homologous recombination in ES cells [26–28]. The phenotype reported in these publications is quite distinct from the one that we report. Whereas Sfpi1BN/BN mice die within 1 month of age,ΔURE mice are healthy and phenotypically normal until 2–3 months of age [28]. However, between 3–8 months of age, ΔURE mice develop an aggressive acute myeloid leukemia. During the preleukemic phase, ΔURE mice have severely impaired B-2 B cell and myeloid development. The differences between these results and the ones we report might be due to differences in regulation of the mutated Sfpi1 alleles.
In conclusion, the original hypothesis as presented above must be reconsidered. Our results suggest that both B cell and myeloid development require high levels of PU.1 activity in vivo. However, we suggest that the requirement for high levels of PU.1 during B cell development is during specification to the B cell lineage, and is not essential to maintain B cell identity after commitment. In contrast, in the myeloid lineage, high levels of PU.1 do not appear to be essential for specification, but are required for terminal differentiation. Finally, we demonstrate that reduction in PU.1 activity results in a myeloproliferative disorder in neonates. We expect that the hypomorphic allele of Sfpi1 that we have characterized in this study will be of value to identify PU.1 target genes that mediate hematopoietic specification and commitment.
Acknowledgments
We would like to acknowledge Phil Sanford of the Gene Targeted Mouse Service (University of Cincinnati) for useful advice regarding gene targeting, and Lara Strittmatter (Miami University) for assistance in microscopy. We would like to thank H. Leighton Grimes for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Klemsz MJ, McKercher SR, Celada A, Van Beveren C, Maki RA. The macrophage and B cell-specific transcription factor PU.1 is related to the ets oncogene. Cell. 1990;61:113–124. doi: 10.1016/0092-8674(90)90219-5. [DOI] [PubMed] [Google Scholar]
- 2.Hromas R, Orazi A, Neiman RS, Maki R, Van Beveran C, Moore J, Klemsz M. Hematopoietic lineage- and stage-restricted expression of the ETS oncogene family member PU.1. Blood. 1993;82:2998–3004. [PubMed] [Google Scholar]
- 3.McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, Maki RA. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. Embo J. 1996;15:5647–5658. [PMC free article] [PubMed] [Google Scholar]
- 4.Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- 5.Iwasaki H, Somoza C, Shigematsu H, Duprez EA, Iwasaki-Arai J, Mizuno S, Arinobu Y, Geary K, Zhang P, Dayaram T, Fenyus ML, Elf S, Chan S, Kastner P, Huettner CS, Murray R, Tenen DG, Akashi K. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood. 2005;106:1590–1600. doi: 10.1182/blood-2005-03-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelsen B, Tian G, Erman B, Gregoire J, Maki R, Graves B, Sen R. Regulation of lymphoid-specific immunoglobulin mu heavy chain gene enhancer by ETS-domain proteins. Science. 1993;261:82–86. doi: 10.1126/science.8316859. [DOI] [PubMed] [Google Scholar]
- 7.Ross IL, Dunn TL, Yue X, Roy S, Barnett CJ, Hume DA. Comparison of the expression and function of the transcription factor PU.1 (Spi-1 proto-oncogene) between murine macrophages and B lymphocytes. Oncogene. 1994;9:121–132. [PubMed] [Google Scholar]
- 8.DeKoter RP, Singh H. Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science. 2000;288:1439–1441. doi: 10.1126/science.288.5470.1439. [DOI] [PubMed] [Google Scholar]
- 9.Back J, Allman D, Chan S, Kastner P. Visualizing PU.1 activity during hematopoiesis. Exp Hematol. 2005;33:395–402. doi: 10.1016/j.exphem.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 10.Nutt SL, Metcalf D, D’Amico A, Polli M, Wu L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J Exp Med. 2005;201:221–231. doi: 10.1084/jem.20041535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polli M, Dakic A, Light A, Wu L, Tarlinton DM, Nutt SL. The development of functional B lymphocytes in conditional PU.1 knock-out mice. Blood. 2005;106:2083–2090. doi: 10.1182/blood-2005-01-0283. [DOI] [PubMed] [Google Scholar]
- 12.Ye M, Ermakova O, Graf T. PU.1 is not strictly required for B cell development and its absence induces a B-2 to B-1 cell switch. J Exp Med. 2005;202:1411–1422. doi: 10.1084/jem.20051089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schweitzer BL, DeKoter RP. Analysis of gene expression and Ig transcription in PU.1/Spi-B-deficient progenitor B cell lines. J Immunol. 2004;172:144–154. doi: 10.4049/jimmunol.172.1.144. [DOI] [PubMed] [Google Scholar]
- 14.DeKoter RP, Lee HJ, Singh H. PU.1 regulates expression of the interleukin-7 receptor in lymphoid progenitors. Immunity. 2002;16:297–309. doi: 10.1016/s1074-7613(02)00269-8. [DOI] [PubMed] [Google Scholar]
- 15.Schweitzer BL, Huang KJ, Kamath MB, Emelyanov AV, Birshtein BK, DeKoter RP. Spi-C has opposing effects to PU.1 on gene expression in progenitor B cells. J Immunol. 2006;177:2195–2207. doi: 10.4049/jimmunol.177.4.2195. [DOI] [PubMed] [Google Scholar]
- 16.Houston IB, Huang KJ, Jennings SR, Dekoter RP. PU.1 immortalizes hematopoietic progenitors in a GM-CSF-dependent manner. Exp Hematol. 2007;35:374–384. e371. doi: 10.1016/j.exphem.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 17.DeKoter RP, Walsh JC, Singh H. PU.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progenitors. Embo J. 1998;17:4456–4468. doi: 10.1093/emboj/17.15.4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Croy BA, Di Santo JP, Greenwood JD, Chantakru S, Ashkar AA. Transplantation into genetically alymphoid mice as an approach to dissect the roles of uterine natural killer cells during pregnancy--a review. Placenta. 2000;21 (Suppl A):S77–80. doi: 10.1053/plac.1999.0518. [DOI] [PubMed] [Google Scholar]
- 19.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zlokarnik G, Negulescu PA, Knapp TE, Mere L, Burres N, Feng L, Whitney M, Roemer K, Tsien RY. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science. 1998;279:84–88. doi: 10.1126/science.279.5347.84. [DOI] [PubMed] [Google Scholar]
- 21.Tondravi MM, McKercher SR, Anderson K, Erdmann JM, Quiroz M, Maki R, Teitelbaum SL. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature. 1997;386:81–84. doi: 10.1038/386081a0. [DOI] [PubMed] [Google Scholar]
- 22.Berclaz PY, Shibata Y, Whitsett JA, Trapnell BC. GM-CSF, via PU.1, regulates alveolar macrophage Fcgamma R-mediated phagocytosis and the IL-18/IFN-gamma -mediated molecular connection between innate and adaptive immunity in the lung. Blood. 2002;100:4193–4200. doi: 10.1182/blood-2002-04-1102. [DOI] [PubMed] [Google Scholar]
- 23.Medina KL, Pongubala JM, Reddy KL, Lancki DW, Dekoter R, Kieslinger M, Grosschedl R, Singh H. Assembling a gene regulatory network for specification of the B cell fate. Dev Cell. 2004;7:607–617. doi: 10.1016/j.devcel.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Dakic A, Metcalf D, Di Rago L, Mifsud S, Wu L, Nutt SL. PU.1 regulates the commitment of adult hematopoietic progenitors and restricts granulopoiesis. J Exp Med. 2005;201:1487–1502. doi: 10.1084/jem.20050075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metcalf D, Dakic A, Mifsud S, Di Rago L, Wu L, Nutt S. Inactivation of PU.1 in adult mice leads to the development of myeloid leukemia. Proc Natl Acad Sci U S A. 2006;103:1486–1491. doi: 10.1073/pnas.0510616103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okuno Y, Huang G, Rosenbauer F, Evans EK, Radomska HS, Iwasaki H, Akashi K, Moreau-Gachelin F, Li Y, Zhang P, Gottgens B, Tenen DG. Potential autoregulation of transcription factor PU.1 by an upstream regulatory element. Mol Cell Biol. 2005;25:2832–2845. doi: 10.1128/MCB.25.7.2832-2845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenbauer F, Owens BM, Yu L, Tumang JR, Steidl U, Kutok JL, Clayton LK, Wagner K, Scheller M, Iwasaki H, Liu C, Hackanson B, Akashi K, Leutz A, Rothstein TL, Plass C, Tenen DG. Lymphoid cell growth and transformation are suppressed by a key regulatory element of the gene encoding PU.1. Nat Genet. 2006;38:27–37. doi: 10.1038/ng1679. [DOI] [PubMed] [Google Scholar]
- 28.Rosenbauer F, Wagner K, Kutok JL, Iwasaki H, Le Beau MM, Okuno Y, Akashi K, Fiering S, Tenen DG. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat Genet. 2004;36:624–630. doi: 10.1038/ng1361. [DOI] [PubMed] [Google Scholar]