

Abstract
[(pF)Phe4Aib7Arg14Lys15]N/OFQ-NH2 (UFP-112) has been designed as a novel ligand for the nociceptin/orphanin FQ (N/OFQ) peptide receptor (NOP) by combining into the same peptide different chemical modifications reported to increase N/OFQ potency. In vitro data obtained in the electrically stimulated mouse vas deferens demonstrated that UFP-112 behaved as a high potency (pEC50 9.43) full agonist at the NOP receptor. UFP-112 effects were sensitive to the NOP antagonist UFP-101 but not to naloxone and no longer evident in tissues taken from NOP−/− mice. In vitro half life of UFP-112 in mouse plasma and brain homogenate was 2.6 and 3.5 fold higher than that of N/OFQ. In vivo, in the mouse tail withdrawal assay, UFP-112 (1–100 pmol, i.c.v.) mimicked the actions of N/OFQ producing pronociceptive effects after i.c.v. administration and antinociceptive effects when given i.t; in both cases, UFP-112 was approx. 100 fold more potent than the natural peptide and produced longer lasting effects. UFP-112 also mimicked the hyperphagic effect of N/OFQ producing a bell shaped dose response curve with the maximum reached at 10 pmol. The hyperphagic effects of N/OFQ and UFP-112 were absent in NOP−/− mice. Equi-effective high doses of UFP-112 (0.1 nmol) and N/OFQ (10 nmol) were injected i.c.v. in mice and spontaneous locomotor activity recorded for 16 h. N/OFQ produced a clear inhibitory effect which lasted for 60 min while UFP-112 elicited longer lasting effects (> 6h). In conscious rats, UFP-112 (0.1 and 10 nmol/kg, i.v.) produced a marked and sustained decrease in heart rate, blood pressure, and urinary sodium excretion and a profound increase in urine flow. Collectively, these findings demonstrate that UFP-112 behaves in vitro and in vivo as a highly potent and selective ligand able to produce full and long lasting activation of NOP receptors.
Keywords: nociceptin/orphanin, FQ, NOP receptor, UFP-112, isolated tissues, tail withdrawal assay, locomotor activity, food intake, cardiovascular and renal functions
1. Introduction
About ten years ago, the heptadecapeptide nociceptin/orphanin FQ (N/OFQ) was identified as the endogenous ligand of a previously orphan G-protein coupled receptor [41,49] now named N/OFQ peptide (NOP) receptor [17]. Both the peptide and its receptor are widely distributed in the brain, spinal cord and peripheral nervous system, and modulate a large variety of biological functions including pain transmission, anxiety, memory, food intake, locomotor activity and the functions of some peripheral systems such as the airways, the cardiovascular, gastrointestinal and genitourinary systems [7,42].
Potent and selective ligands are required for investigating in detail the functions regulated by the N/OFQ–NOP receptor system and ultimately for identifying the therapeutic indications for NOP receptor agonists and antagonists. The following NOP ligands are currently available: i) small non peptide molecules, e.g. the agonist Ro 64-6198 [29] and the antagonists J-113397 [46] and SB-612111 [52]; ii) small peptides such as the hexapeptides Ac-RYYRWK-NH2 and Ac-RYYRIK-NH2 [19] and the pseudopentadapeptide III-BTD [2]; iii) a large series of N/OFQ related peptides chemically modified for increasing agonist potency or for reducing or eliminating efficacy. Details about these three classes of NOP ligands can be found in recent review articles such as [53] and [3]. Our research group has substantially contributed to the identification of N/OFQ related NOP ligands by performing a series of structure activity studies which allow the identification of the following useful chemical modifications: C-terminus amidation that increases agonist potency and reduces susceptibility to peptidases [23], reduction of the first peptide bond of N/OFQ which reduces ligand efficacy [8], shift of the Phe1 side chain to the nitrogen atom that eliminates efficacy [25], and substitutions on the Phe4 side chain (e.g. (pF)Phe4) that increase peptide potency [24]. Other groups identified different chemical modifications able to increase peptide potency including the Arg14Lys15 substitution [45] or that with the unnatural residue Aib in position 7 and/or 11 [55]. Recently we combined into the N/OFQ sequence all the above mentioned chemical modifications thus generating a new series of NOP receptor ligands [1]. The most interesting peptides generated in this study were the antagonist [Nphe1Aib7Arg14Lys15]N/OFQ-NH2, the partial agonist [Phe1Ψ(CH2-NH)Gly2(pF)Phe4 Aib7Arg14Lys15]N/OFQ-NH2, and the full agonist [(pF)Phe4Aib7Arg14Lys15]N/OFQ-NH2 (coded as UFP-112). These peptides were characterized pharmacologically in a series of in vitro assays including receptor binding and functional [35S]GTPγS binding in membranes from CHO cells stably expressing the human NOP receptor and in bioassays experiments performed in the electrically stimulated mouse and rat vas deferens and guinea pig ileum tissues, where they displayed very high affinity/potency and NOP selectivity [1].
Here we present a detailed evaluation of the pharmacological profile of UFP-112. In vitro the peptide was evaluated in the electrically stimulated mouse vas deferens, challenged with antagonists and reassessed in tissues taken from NOP receptor gene knockout mice (NOP−/−), moreover the metabolic stability of UFP-112 was compared to that of N/OFQ in mouse brain homogenate and plasma.
In vivo UFP-112 was studied in rodents by assessing its effects on various biological functions known to be modulated by N/OFQ, including supraspinal and spinal pain transmission, food intake, locomotor activity, as well as cardiovascular and renal functions.
2. Material and methods
2.1 Animals
Male Swiss albino (Morini, Reggio Emilia, Italy, 25–30 g) were housed in 425 x 266 x 155 mm cages (Tecniplast, MN, Italy), 8 animals / cage, under standard conditions (22°C, 55% humidity, 12-h light-dark cycle, light on at 7.00 AM) with food (MIL, standard diet, Morini, RE, Italy) and water ad libitum for at least 5 days before experiments commenced. CD1/C57BL6/J-129 wild type (NOP+/+) and knockout for the NOP receptor gene (NOP−/−) mice were genotyped by PCR. Details of the generation and breeding of mutant mice have been previously published [22,44]. Male Sprague-Dawley rats (Harlan, Indianapolis, IN) weighing 300–350g were used for the cardiovascular and renal function studies. Rats were housed in groups of two and were maintained on a 12 h light/dark cycle (lights on at 7 am) with free access to a normal sodium diet (sodium content, 163 mEq/Kg) and tap water ad libitum. All procedures were conducted in accordance to the guidelines published in the European Communities Council directives (86/609/EEC) and those of the National Institutes of Health for the Care and Use of Animals approved by the Louisiana State University Health Sciences Center Institutional Animal Care and Use Committee.
2.2 Electrically stimulated mouse vas deferens
Vas deferens taken from Swiss or CD1/C57BL6/J-129 NOP+/+ and NOP−/− mice were prepared as previously described [4]. Tissues were continuously stimulated through two platinum ring electrodes with supramaximal voltage rectangular pulses of 1 msec duration and 0.05 Hz frequency. The electrically evoked contractions (twitches) were measured isotonically with a strain gauge transducer (Basile 7006) and recorded with the PC based acquisition system PowerLab 4/25 (model ML845, ADInstruments, Australia). After an equilibration period of about 60 min, the contractions induced by electrical field stimulation were stable; at this time, cumulative concentration-response curves to N/OFQ or UFP-112 were performed (0.5 log-unit step) in the absence or in presence of antagonists (the selective NOP receptor antagonist UFP-101[11], or the non selective classical opioid receptor antagonist naloxone, (15 min pre-incubation time).
2.3 Peptide half-life in mouse plasma and brain homogenate
Plasma and brain tissue samples were obtained from male Swiss mice. The animals, after being sacrificed under ether anesthesia, were perfused with physiological heparin solution injected through a needle placed in the left ventricle. Blood was then withdrawn and centrifuged at 14000 xg for 2 minutes at room temperature. After separation from the pellet, the plasma was aliquoted and stocked at −80° C. After blood withdrawal, the animal was further perfused with a physiological solution for 2 minutes before brain removal. The brain tissue was homogenized in 5 vol. (w/v) of Tris/HCl (50 mM, pH 7.4, 0°C) with a ultra-Turrax (Janke Kunkel, Staufen, FRG) 3 times for 15 seconds each. The supernatant obtained by centrifugation (3000 xg for 15 min at 4°C) was decanted and then stocked at −80°C. The protein content of the preparations, determined by the Bradford method [5], was approximately 8 μg/μl for the brain homogenate and 17 μg/μl for the plasma. An aliquot of 100 μl solution of each peptide (3 mg/500 μl Tris) was incubated (at a final concentration of 6 μg/μl) with brain homogenate or plasma (450 μl) in a total volume of 1 ml, containing Tris/HCl 50 mM pH 7.4 buffer. Incubation of the aliquots was carried out at 37 °C for various periods up to 240 min. At different incubation times, an aliquot of the solution (100 μl) was removed and peptide degradation was blocked by addition of 4.5% TFA solution (200 μl). After centrifugation (3000 rpm for 15 minutes) an aliquot (100 μl) of supernatant was injected into RP-HPLC. HPLC analysis was performed in a Kromasil 100-5C18 column (4.6 x 250 mm) using a Beckman System Gold chromatography system equipped with a variable wave length UV detector. The experimental conditions for elution included a gradient analysis with water (solvent A) and acetonitrile (solvent B), both containing 0.1% TFA, at a flow rate of 0.7 ml/min. The following protocol was used for gradient analysis: linear gradient from 5% to 40% B in 20 minutes; linear gradient from 40% to 60% B in 5 minutes; linear gradient from 60% to 5% B in 5 minutes. The elute was monitored at 220 nm. The half life (T1/2) was obtained by linear regression with the least square method, diagramming the peak areas of each derivative as a function of the incubation times. In particular, the time points for determining the T1/2 of N/OFQ and UFP-112 were the following: 0, 5, 10, 20, 30, 60, 120 and 240 min in the plasma; 0, 1, 2, 5, 7, 10, 15 and 20 min in the brain. 2.4 Intracerebroventricularly (i.c.v.) or intrathecally (i.t.) injections Mice were i.c.v. (2 μl/mouse) or i.t. (5 μl/mouse) injected, under light ether anesthesia (i.e. sufficient for losing the righting reflex), according to the procedure described by [36] and [27], respectively.
2.5 Tail-withdrawal assay
All experiments were started at 10.00 a.m. and performed according to the procedure previously described in detail [9]. Briefly, the mice were placed in a holder and the distal half of the tail was immersed in water at 48 °C. Withdrawal latency time was measured by an experienced observer blind to drug treatment. A cut off time of 20 s was chosen to avoid tissue damage. For each series of experiments at least 12 mice were randomly assigned to each treatment. Tail-withdrawal latency was determined immediately before and 5, 15, 30, 60, 90 and 120 min after i.c.v. or i.t injection of saline (control) or UFP-112 (1-100 pmol).
2.6 Food Intake
The day before the experiment mice were individually housed in cages with food and water ad libitum. Mice were i.c.v. injected with saline, N/OFQ (0.1 – 10 nmol) or UFP-112 (1 – 100 pmol). The effects of 1 nmol of N/OFQ and 0.01 nmol of UFP-112 were assessed in NOP+/+ and NOP−/− mice. Food intake was measured after 30 and 60 min from drug injection and expressed as g/kg of body weight. All experiments were started at 10:00 a.m.
2.7 Locomotor activity assay
Equieffective doses of UPF-112 (0.1 nmol) and N/OFQ (10 nmol) were injected i.c.v. (at 17.00 PM) and locomotor activity was recorded for 16h. Experiments were carried following the procedure described in [50]. Locomotor activity was assessed using Basile activity cages, which consist of a four-channel resistance detector circuit which converts the bridges “broken” by the animal paws into pulses that are summed by an electronic counter every 5 min. Total number of impulses were recorded every 30 min for 16 hours. Mice were not accustomed to the cages before drug treatment.
2.8 Cardiovascular and renal functions in conscious male Sprague-Dawley rats
On the day of the study, rats were anesthetized with sodium methohexital (75 mg/kg i.p. and supplemented with 10 mg/kg i.v. as needed; King Pharmaceuticals, Bristol, TN) and instrumented with left femoral artery (blood pressure measurement), vein (isotonic saline infusion), and urinary bladder (urine collection) catheters using standard techniques described previously [31,32]. Following surgical preparation rats were placed in a rat holder (a chamber with plexiglass ends connected by stainless steel rods; the metal rods formed an inverted U shape and a flat base in which the rat would sit), which permits forward and backward movement of the rat while minimizing movement during surgical recovery and allows for collection of urine. An i.v. infusion of isotonic saline (55 μl/min) was started and continued for the duration of the experiment. The experimental protocol commenced after rats regained full consciousness and cardiovascular, renal and excretory functions stabilized. After collection of baseline control measurements for each parameter, UFP-112 (0.1 nmol; n=6, 10 nmol; n=6) or N/OFQ (10 nmol; n=6) were administered by an i.v. bolus injection. Immediately after i.v. administration, experimental urine samples (10-min consecutive periods) were collected for 80 min. Heart rate was derived from the pulse pressure with a tachograph (model 7 P4H; Grass Instruments, Quincy, MA). Heart rate and arterial pressure were continuously monitored and recorded on a Grass polygraph (model 7). Urine volume was determined gravimetrically. Urine sodium concentration was measured by flame photometry (model 943; Instrumental Laboratories, Lexington, MA).
2.9 Chemicals
The peptides used in this study were prepared and purified as recently described [1]. For in vitro experiments, the compounds were solubilized in H2O and stock solutions (2 mM) were stored at–70 °C until use; for in vivo studies, the substances were solubilized in physiological medium just before performing the experiment.
2.10 Statistical analysis
All data are expressed as mean ± s.e.m. and the number of separate experiments is reported for each series of data. Data have been analyzed statistically with the Student’s t-test or one way ANOVA followed by the Dunnett’s test, as specified in table and figure legends; p values less than 0.05 were considered statistically significant. Curve fitting was performed using PRISM 4.0 (GraphPad Software In., San Diego, U.S.A.). Agonist potencies were expressed as pEC50, which is the negative logarithm to base 10 of the agonist molar concentration that produces 50% of the maximal possible effect of that agonist. The Emax is the maximal effect that an agonist can elicit in a given tissue/preparation. pA2 values were calculated using the Gaddum Schild equation pKB = −log((CR-1)/[Antagonist]), assuming a slope equal to unity.
3. Results
3.1 Electrically stimulated mouse vas deferens
In the electrically stimulated mouse vas deferens, N/OFQ and UFP-112 concentration dependently inhibited electrically induced twitches with similar maximal effects. However, UFP-112 was approximately 100 fold more potent than N/OFQ (pEC50 values 9.43 and 7.47, respectively). Single concentrations of the NOP selective antagonist UFP-101 (1 μM) and of the non-selective opioid receptor antagonist naloxone (1 μM) were challenged against the inhibitory effects induced by N/OFQ and UFP-112 (figure 1) in this preparation. Both the antagonists did not modify per se the control twitches and naloxone did not affect the concentration response curve to either N/OFQ or UFP-112. In contrast UFP-101 displaced to the right the concentration response curve to N/OFQ and UFP-112 to a similar extent, therefore showing similar pKB values (6.81 and 6.91 respectively). The effects of the NOP agonists UFP-112 and N/OFQ, and those elicited by the selective DOP agonist [D-Pen2,D-Pen5]-Enkephalin (DPDPE) were investigated in the electrically stimulated mouse vas deferens taken from wild type (NOP+/+) and NOP receptor knockout (NOP−/−) mice (Figure 2). In NOP+/+ tissues UFP-112 mimicked the inhibitory effects of N/OFQ (Emax 91 ± 1%; pEC50 7.62), showing similar maximal effects (86 ± 2%) but higher potency (pEC50 9.40) (Figure 2, left panel). In tissues taken from NOP−/− mice, N/OFQ and UFP-112 were virtually inactive up to micromolar concentrations (figure 2, right panel). In the same series of experiments, the DOP receptor selective agonist DPDPE displayed similar potency (pEC50 8.49 and 8.17) and efficacy (97 ± 2% and 95 ± 1%) in tissues taken from NOP+/+ and NOP−/− mice (Figure 2).
Figure 1.
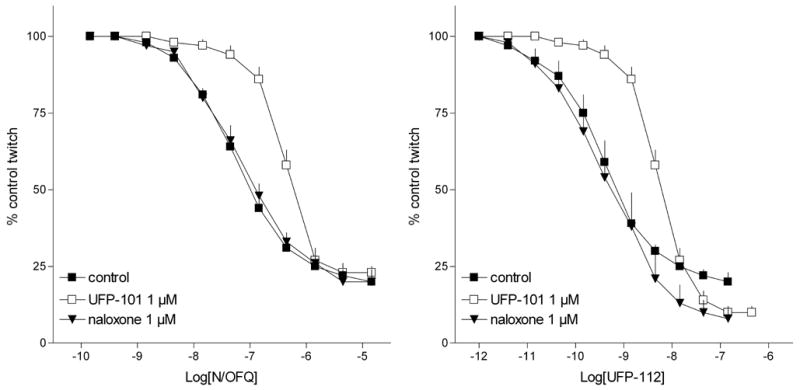
Electrically stimulated mouse vas deferens. Concentration response curve to N/OFQ (left panel) or UFP-112 (right panel) obtained in the absence (control) and in presence of UFP-101 and naloxone. Points indicate the means and vertical lines the s.e.m. of four separate experiments.
Figure 2.
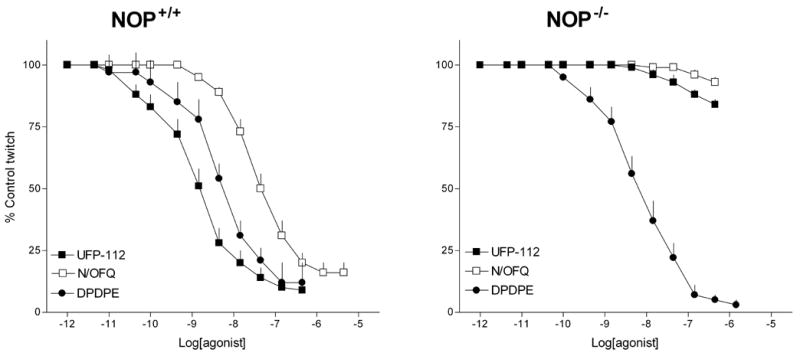
Electrically stimulated mouse vas deferens. Concentration response curve to DPDPE, N/OFQ and UFP-112 in tissues taken from wild type (NOP+/+) (left panel) and NOP receptor knockout (NOP−/−) mice (right panel). Points indicate the means and vertical lines the s.e.m of four separate experiments.
3.2 Peptide half-life in mouse plasma and brain homogenate
The degradation half-life (T1/2) of N/OFQ and UFP-112 in mouse plasma and brain homogenate was obtained by least-square linear regression analysis of peptide pick area versus time as shown in table 1. Results of this analysis indicate that N/OFQ showed a relatively long half live in the plasma (about 1 h) compared to that obtained in the brain homogenate (approx 3 min). UFP-112 exhibited significantly longer half lives compared to the natural peptide. In particular, the plasma T½ of UFP-112 is about 3-fold longer than that of N/OFQ and this difference was even more pronounced in the mouse brain homogenate.
Table 1.
T½ (min) of N/OFQ and UFP-112 in the mouse plasma and brain homogenate.
The data are s.e.m of three separate experiments.
p<0.05 vs N/OFQ according to the Student’s t-test.
3.3 Tail-withdrawal assay in mice
The i.t. injection of UFP-112 (1–100 pmol) and the i.c.v. injection of 1 pmol of peptide in mice did not induce any effect on animal gross behavior. In contrast, mice treated with UFP-112 at 10 pmol and particularly at 100 pmol i.c.v. showed a decrease in locomotor activity, ataxia and loss of the righting reflex, in a similar manner to what occurs after i.c.v. injection of high doses of N/OFQ (i.e. 10 nmol) [9, 49, 50]. However, while the N/OFQ effects appeared immediately after i.c.v. injection, those produced by UFP-112 were only evident after at least 30 min. Results summarized in figure 3 show that the tail withdrawal latencies of saline injected mice were stable at 4–5 s over the time course of the experiment. UFP-112 (1–100 pmol) produced dose dependent pronociceptive effects after i.c.v. administration (figure 3, left panel) and antinociceptive effects when given i.t. (figure 3, right panel). Thus, UFP-112 mimicked the actions of N/OFQ, approximately 100-fold more potent than the natural peptide, and produced longer lasting effects (see for comparison the dose response curves to N/OFQ previously published in [9] and [43]). In fact, the effects induced by UFP-112 in the tail-withdrawal assay were still evident at least 120 min after the i.c.v. or i.t. injection of the peptide, while those elicited by N/OFQ lasted for about 30 min.
Figure 3.
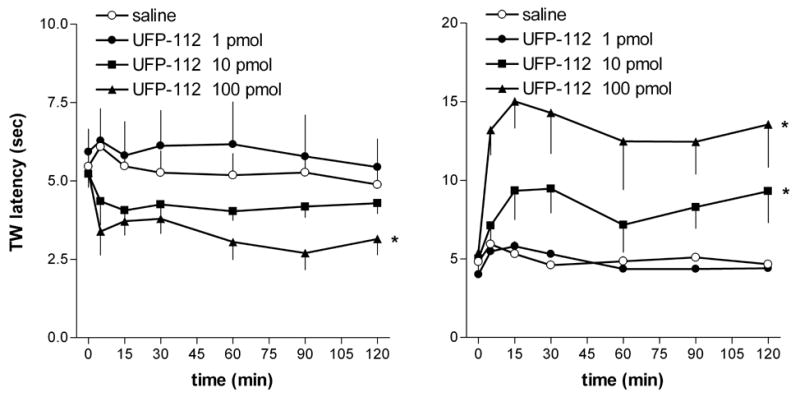
Mouse tail withdrawal assay. Dose response curve to UFP-112 (i.c.v., left panel) (i.t., right panel). Points indicate the means and vertical lines the s.e.m of five separate experiments. * p < 0.05 vs saline according to ANOVA followed by the Dunnett’s test.
3.4 Food intake studies in mice
Under the present experimental conditions, mice treated with saline eat 1.63 ± 0.61 and 4.50 ± 0.96 g/kg at 30 and 60 min, respectively. The i.c.v. injection of N/OFQ induced a dose dependent hyperphagic effect: the peptide was inactive at 0.1 nmol, while at 1 nmol produced a statistically significant orexygenic effect both at 30 and 60 min (figure 4, left panel). Increasing the dose of peptide to 10 nmol resulted into a loss of effect, thus making the dose response curve to N/OFQ bell shaped. It is worthy of mention that the administration of such a high dose of peptide was associated with evident sedative effects (i.e. decrease in locomotor activity, reduction of muscle tone, ataxia and impairment of the righting reflex) as previously reported by our group and others [7]. UFP-112 mimicked the hyperphagic effect of N/OFQ producing a bell shaped dose response curve with the maximum reached at 10 pmol (figure 4, right panel). Thus UFP-112 was found to be approx 100 fold more potent than N/OFQ also in this assay. In addition, it is worthy of mention that the amount of the maximal orexygenic effect elicited by UFP-112 was approximately double that evoked by N/OFQ.
Figure 4.
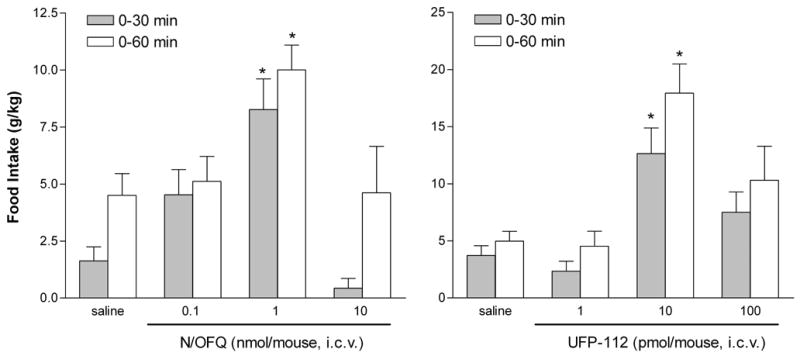
Cumulative 30 and 60 min food intake following i.c.v. injection of N/OFQ (0.1 - 10 nmol) (left panel) or UFP-112 (1–100 pmol) (right panel). Values are means ± s.e.m. of 12–15 mice. Ordinate: g / kg body weight. *p < 0.05 vs saline according to ANOVA followed by the Dunnett’s t test
NOP receptor knockout mice were used for characterizing the hyperphagic effect induced by N/OFQ and UFP-112. There were no differences in food consumption between NOP+/+ and NOP−/− mice. 1 nmol N/OFQ and 10 pmol UFP-112 (figure 5) elicited a robust hyperphagic effect in NOP+/+ mice while the two peptides were found inactive in NOP−/− animals. It is worthy of mention that the orexygenic effects of both N/OFQ and UFP-112 were much more pronounced in CD1/C57BL6/J-129 than in Swiss mice, and that the effect of UFP-112 in CD1/C57-BL6J-129 mice was higher that that elicited by N/OFQ.
Figure 5.
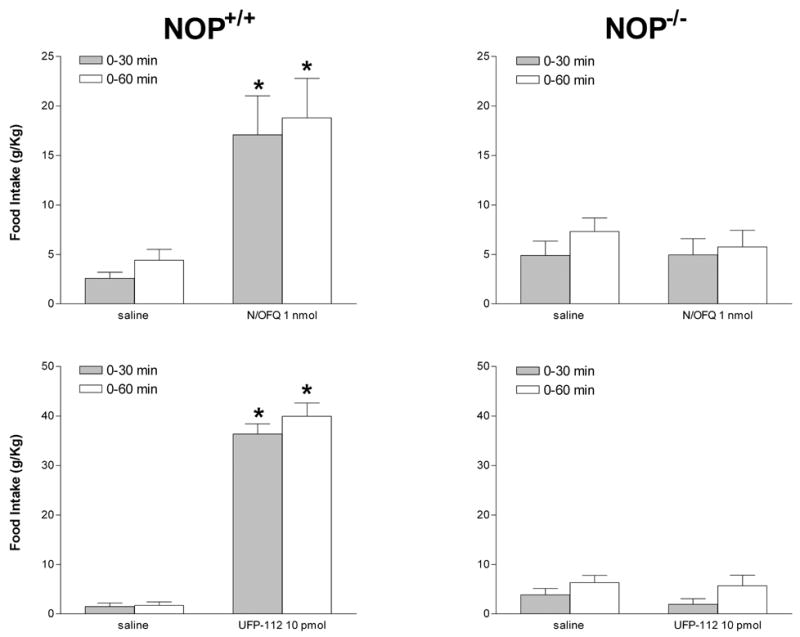
Cumulative 30 and 60 min food intake following i.c.v. injection of saline, 1 nmol N/OFQ (top panels) and 10 pmol UFP-112 (bottom panels) in NOP+/+ (left panels) and NOP−/− (right panels) mice. Values are means ± s.e.m. of 9–12 mice. Ordinate: g / kg body weight. *p < 0.05 vs saline according to the Student t test.
3.5 Locomotor activity studies in mice
As shown in figure 6, mice injected with saline showed a progressive reduction of spontaneous locomotor activity from 950 ± 160 at 5:30 pm to 105 ± 58 at 7:00 pm. However, when the light was turned off (at 7:00 pm), there was a clear increase in the spontaneous locomotor activity which progressively returned to the baseline over the time course of the experiment, as expected based on the rodent nocturnal activity preference. Under these experimental conditions, 10 nmol N/OFQ given i.c.v. produced a clear inhibitory effect on spontaneous locomotor activity which lasted for only 60 min while UFP-112 at 100 fold lower doses (i.e. 0.1 nmol) elicited longer lasting effects by inhibiting locomotor activity for more than 6 hours (figure 6).
Figure 6.
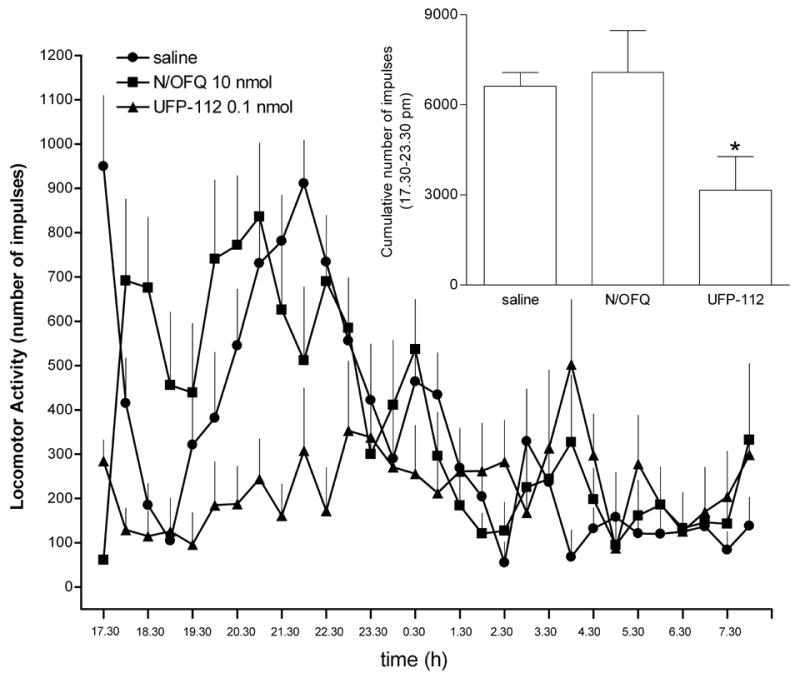
Locomotor activity. Time course of the inhibitory effect on spontaneous locomotion induced by i.c.v. injection of equieffective doses of N/OFQ and UFP-112. Values are means ± s.e.m. of at 8–10 mice. *p < 0.05 vs saline according to ANOVA followed by the Dunnett’s t test
3.6 Cardiovascular and renal responses in rats
The cardiovascular responses produced by i.v. bolus injection of N/OFQ and UFP-112 in conscious rats are shown in figures 7 (peak changes) and 8 (time course). As shown in Figure 7, in contrast to isotonic saline vehicle, i.v. injection of 10 nmol/kg N/OFQ produced a slight but significant reduction in mean arterial pressure (basal, 123 ± 2 mmHg; peak, 116 ± 5 mmHg), and a slight non-significant reduction in heart rate (basal, 388 ± 16 bpm; peak, 376 ± 6 bpm). Similarly, immediately following drug injection a 100-fold lower dose of UFP-112 (0.1 nmol/kg, i.v.) produced a comparably small but significant hypotension and bradycardia (figure 7). However, when administered at 10 nmol/kg, UFP-112 profoundly reduced mean arterial pressure (basal, 127 ± 2 mmHg; peak, 92 ± 2 mmHg) and heart rate (basal, 364 ± 9 bpm; peak, 255 ± 12 bpm) (figure 7). In addition to increased potency, UFP-112 also produced markedly longer cardiovascular responses than N/OFQ. In this regard, the changes in mean arterial pressure and heart rate produced by N/OFQ at 10 nmol/kg (present study; figures 7 and 8) or higher (30 and 100 nmol/kg; [33]) recovered within 10-min post-drug injection. At the 10 nmol/kg dose UFP-112 significantly decreased heart rate and mean arterial pressure throughout the 80-min protocol (figure 8). In two of these animals it was observed that these cardiovascular depressor responses persisted for at least 120-min (further time points not studied; Wainford and Kapusta personal observations). In addition to changes in cardiovascular function, in these same animals i.v. bolus injection of UFP-112 (0.1 and 10 nmol/kg) also produced a concurrent diuretic response (figure 8). At the higher dose tested (10 nmol/kg, i.v.) the UFP-112-induced increase in urine flow rate was delayed in onset (approximately 30-min), of relatively long duration (50-60 min), and associated with a reduction in urinary sodium excretion (not statistically significant). Of note, when administered at 10 nmol/kg N/OFQ did not alter urine flow rate (figure 8), and instead at higher doses (30, 100, 300, 1000 nmol/kg, i.v) produced antidiuresis ([33] and Kapusta personal observations). Finally, it should be noted that following i.v. administration of UFP-112 (10 nmol/kg) the rats appeared sedated and calm (e.g., no exploratory behaviors).
Figure 7.
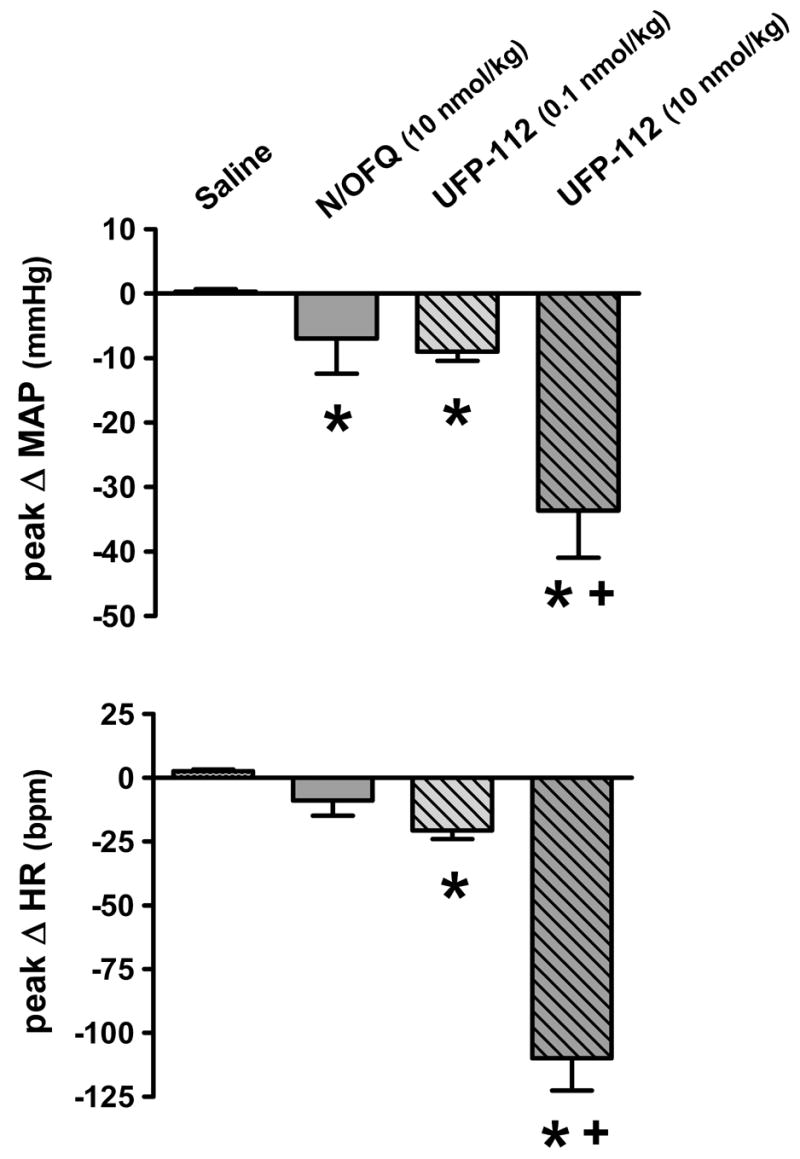
Peak changes in mean arterial pressure and heart rate produced immediately following i.v. bolus injection of N/OFQ (10 nmol/kg) and UFP-112 (0.1 and 10 nmol/kg). Values are mean ± s.e.m. of six separate experiments. MAP, mean arterial pressure; HR, heart rate. *p < 0.05, statistically different from saline control group (ANOVA followed by Dunnett’s test). tp < 0.05 statistically different from corresponding N/OFQ treatment at the same dose (unpaired Student’s t test).
Figure 8.
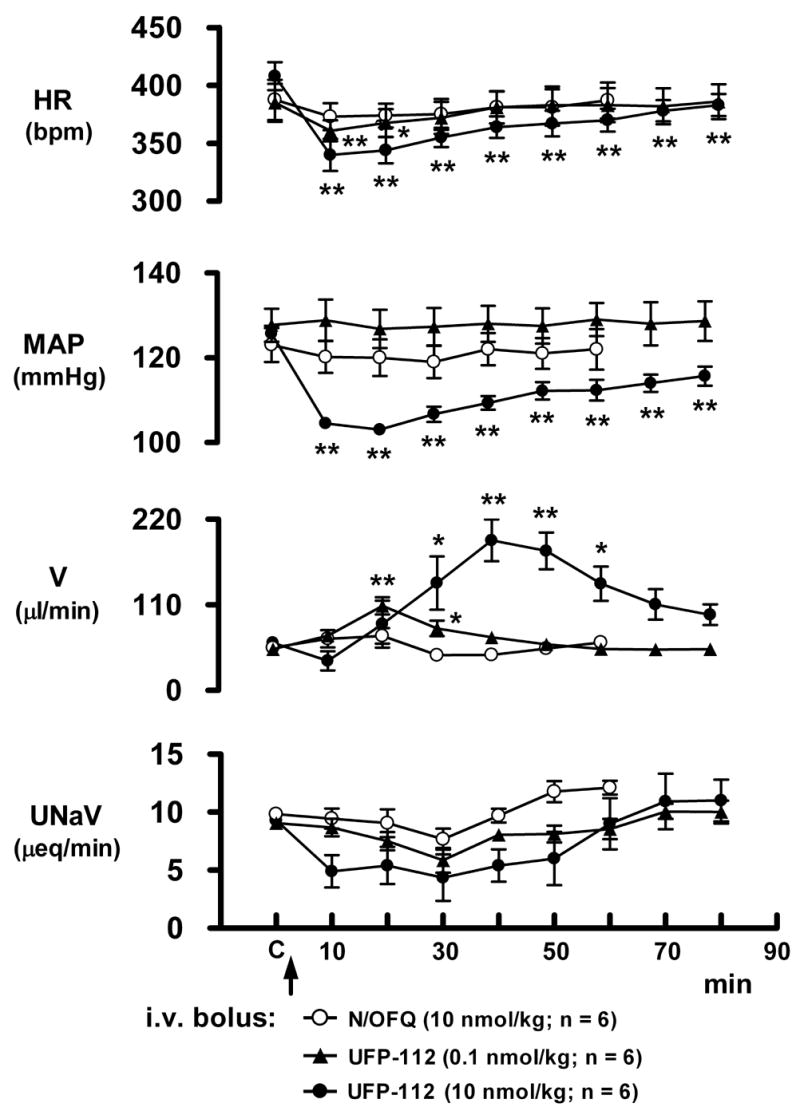
Cardiovascular and renal excretory responses produced by i.v. bolus N/OFQ (10 nmol/kg) and UFP-112 (0.1 nmol/kg or 10 nmol/kg) in conscious male Sprague-Dawley rats. Values are mean ± s.e.m. of six separate experiments. Urine samples (consecutive 10-min periods) were collected during control (C, 20 min) and immediately after drug administration for 60 min (N/OFQ), 80 min (UFP-112; 0.1 nmol/kg or 10 nmol/kg). HR, heart rate; MAP, mean arterial pressure; UNaV, urinary sodium excretion; V, urine flow rate; ** p < 0.01, * p < 0.05, statistically different from corresponding group control (C) value (repeated measures ANOVA followed by Dunnett’s test).
4. Discussion
The in vitro and in vivo findings summarized in the present study demonstrate that UFP-112 is a novel NOP receptor ligand that behaves as a highly potent and selective full agonist and produces long lasting effects in vivo. These pharmacological features make UFP-112 an extremely useful tool for future studies investigating the physiological role of the N/OFQ–NOP receptor system and the therapeutic possibilities of innovative drugs interacting with the NOP receptor.
As far as ligand efficacy is concerned, previous studies performed in a panel of in vitro assays [1] demonstrated that UFP-112 behaves as a full agonist at NOP receptors. In fact, in GTPγS] binding experiments performed on membranes prepared from CHO cells expressing the human NOP receptor, N/OFQ and UFP-112 induced similar maximal effects corresponding to approximately 8 times the basal values [1]. Similarly, in tissues from various species (mouse and rat vas deferens, guinea pig ileum) both peptides inhibited the electrically stimulated twitch response showing superimposable maximal effects [1]. This has been confirmed in the present experiments performed on vas deferent tissues taken from Swiss and CD1/C57BL6/J-129 mice. The full agonist pharmacological activity of UFP-112 was confirmed in vivo in the present series of experiments where UFP-112 mimicked the actions elicited by N/OFQ after i.c.v. (pronociceptive and orexigenic effects, inhibition of locomotor activity), i.t. (antinociceptive action), and i.v. (hypotensive and bradycardic effects) administration producing maximal effects similar or even greater than those of natural ligand. However, it should be mentioned that the evaluation of ligand efficacy in in vivo experiments cannot be as detailed as in in vitro studies, the former being possibly biased by a series of factors including differences in the onset and duration of action, in the ability to reach the target receptors, in the resistance to enzymatic degradation, etc. Despite these considerations, in vitro and in vivo data converge indicating that UFP-112 is able to evoke, via NOP receptor activation, maximal effect similar to those of N/OFQ, thus behaving as a full agonist.
Very high potency was consistently recorded when investigating the actions of UFP-112. In fact, UFP-112 bound the NOP receptor with a pKi of 10.55, 10 times higher than that of N/OFQ (pKi 9.50), and displayed pEC50 values in isolated tissues (range 8.34 – 9.24) approximately 30 times higher than N/OFQ (range 6.83 – 8.05). This difference in potency was even more pronounced in vivo, where UFP-112 mimicked N/OFQ actions at doses 100 fold lower. Thus, the difference in potency between N/OFQ and UFP-112 seems to increase from 10 to 30 and 100 in membranes, tissues and whole animals, respectively. This suggests that other factors are involved apart from higher affinity to the NOP receptor, for instance the susceptibility to enzymatic degradation which has been demonstrated to be significantly lower (by approximately 3 fold) for UFP-112 compared to N/OFQ both in mouse plasma and brain homogenate.
Another pharmacological feature critical in determining the usefulness of a given ligand is the selectivity of action. The following lines of evidence demonstrated that UFP-112 is highly selective for the NOP receptor: i) in binding experiments UFP-112 displayed affinity for NOP sites at concentrations at least 100 fold lower than those required to bind classical opioid sites [1]; ii) in bioassay experiments performed in the mouse vas deferens, the action of UFP-112 was resistant to naloxone and prevented by the selective NOP-receptor antagonist UFP-101 [10,11] which displayed superimposable pKB values against UFP-112 and N/OFQ; iii) in the same preparation, the inhibitory effects of both UFP-112 and N/OFQ were no longer present in tissues taken from NOP−/− mice, while those of the DOP selective agonist DPDPE were similar in tissues from NOP+/+and NOP−/− animals (thus ruling out the possibility that the lack of NOP receptor produces a non specific loss of sensitivity to inhibitory signals), iv) the in vivo orexygenic action of UFP-112 (and N/OFQ) was evident in NOP+/+ but not in NOP−/− mice. Altogether these series of data demonstrated that the in vitro and in vivo actions of UFP-112 are solely due to the selective activation of the NOP receptor.
The analysis of the in vivo actions of UFP-112 also demonstrated that this ligand produces long lasting effects compared to N/OFQ. This was consistently observed among a large series of assays/actions. In fact, the supraspinal pronociceptive effect of N/OFQ lasted for about 15–30 min [9], while that of UFP-112 was still evident after 2 hours from i.c.v. injection; the spinal antinociceptive action of N/OFQ disappeared after 60 min from i.t. injection [43] while those of UFP-112 were still present and statistically significant after 2 hours. Similar results were obtained measuring the hypotensive and bradycardic effects evoked by N/OFQ and UFP-112 after i.v. administration in rats, where the action of N/OFQ was short lasting (about 10 min) while that of UFP-112 persisted for the time of the experiment (i.e. 80 min). In addition, a specific series of experiments was performed to investigate the duration of action of UFP-112 in comparison with N/OFQ. Mice were i.c.v. injected with equieffective doses of N/OFQ (10 nmol) and UFP-112 (0.1 nmol) and their locomotor activity followed overnight. N/OFQ evoked a clear inhibitory effect for 60 min while the action of UFP-112 persisted for more than 5 h. Finally, in both Swiss and CD1/C57BL6/J-129 mice the amount of food eaten in response to UFP-112 was approximately double that stimulated by N/OFQ; this can be interpreted by assuming a more prolonged stimulation of the NOP receptor by the former peptide.
In all assays performed, apart from the higher potency and longer duration of action, UFP-112 exclusively mimicked N/OFQ actions without producing any other additional effects. However, there was an exception to this rule which deserves further comment. Concurrent with the changes in cardiovascular function, i.v. injection of UFP-112 in rats also produced a pronounced diuresis and tendency for antinatriuresis. This is of considerable interest since this is the first time that a purported NOP receptor full agonist has produced a diuretic response following i.v. bolus administration. These responses to UFP-112 are different to those to the natural ligand N/OFQ, which at all previously tested doses up to 1000 nmol/kg i.v. bolus has not produced diuresis ([33] and Kapusta et al., unpublished results). There are two possible explanations for the ability of UFP-112 to produce a diuretic response after bolus injection i.v. Although not tested, UFP-112 may produce diuresis by activating a potential peripheral (e.g. kidney; [26]) diuretic pathway similar to that previously proposed for NOP receptor partial agonists [34]. The ability of UFP-112 (but not N/OFQ) to reach these sites after i.v. bolus injection may derive from its relatively low susceptibility to enzymatic degradation. Alternatively, following i.v. injection and systemic circulation UFP-112 may have crossed the blood brain barrier and entered the brain to elicit renal responses through a CNS controlled pathway. In fact, a number of NOP receptor ligands (full and partial agonists) including N/OFQ have been shown to elicit diuresis (and antinatriuresis) following i.c.v. administration [30–32]. Although the high molecular weight associated with the presence of several positively charged residues makes this latter possibility unlikely, the observed sedation which occurred in rats following i.v. administration of this drug may support the proposal that UFP-112 may indeed be able to cross the blood-brain barrier to access the CNS. Experiments are underway in our laboratories to specifically address this issue.
The UFP-112 pharmacological profile described in the present report, high potency associated with high NOP selectivity and long duration of action, is corroborated by a number of findings obtained in other laboratories. In fact, results superimposable to those presented here have been obtained by comparing UFP-112 and N/OFQ for their ability to i) inhibit capsaicin induced bronchoconstriction in vitro in the isolated and perfused mouse lung (D’Agostino et al., personal communication), ii) stimulating food intake in rats (Cifani et al., personal communication), iii) reducing gastric acid secretion and preventing ethanol induced gastric lesions after i.c.v. administration in rats [6], and i.v.) reducing alcohol consumption in genetically selected alcohol preferring rats[20]. In addition to these findings, the Chinese group of research of Prof Wang independently identified and pharmacologically characterized a peptide ligand for the NOP receptor, [(pF)Phe4Aib7Aib11Arg14Lys15]N/OFQ-NH2, very similar to UFP-112. Data obtained with this peptide in vitro in rat brain binding and bioassay experiments [13] and in vivo investigating its ability to evoke pronociceptive effects in mice after i.c.v. administration and hypotensive effects in rats after i.v. injection [47] are again very similar to what we describe here using UFP-112. Finally the features of UFP-112 should be compared to those of the best NOP receptor full agonists previously described in literature the peptide UFP-102 [12] and the non peptide Ro 64-6198 [29]. This latter molecule has the obvious pharmacokinetic advantages deriving from its non peptide nature (including the ability of crossing the blood brain barrier) which facilitate the design and realization of chronic studies (see for instance [18] and [51]). However, findings obtained with Ro 64-6198 can be biased by its ability to bind classical opioid receptors (at least in rats [20]) and to recognize only a subset of NOP receptors (at least in some brain areas, e.g. the periaqueductal gray [14]). As far as UFP-102 is concerned, this peptide displays an in vitro pharmacological profile very similar to that of UFP-112 ([1,12] and present data) but its in vivo potency seems to be 3-10 fold lower than that of UFP-112 ([12,20] and present data).
5. Summary
In conclusion the present experiments suggest that UFP-112 is a highly potent and selective full agonist for the NOP receptor, partially resistant to enzymatic degradation, and able to produce long lasting effects in vivo. These pharmacological features are highly desirable especially for investigating those conditions and states in which a selective and prolonged stimulation of NOP receptor is beneficial, including anxiety states [21,28], drug addiction [15,48], anorectic conditions [16], spinal analgesia (as indicated by laboratory animals [54] as well as primate [35] studies), cough and possibly other respiratory diseases [40], acute heart failure [26,34], and, as indicated by clinical studies, urinary incontinence due to overactive bladder [37–39].
Acknowledgments
This work was financially supported by the University of Ferrara (FAR grant to GC and SS), by the Italian Ministry of University (PRIN 2006 grant to DR), and by the National Institute of Health (Heart Blood and Lung Institute, grant HL71212 to DRK and DR; National Institute of Diabetes and Digestive and Kidney Diseases grants DK-43337 to DRK; and by the NHI National Center for Research Resources (NCRR) COBRE Grant P20 RR018766 to DRK).
Abbreviations
- N/OFQ
nociceptin/orphanin FQ
- UFP-112
[(pF)Phe4,Aib7,Arg14,Lys15]N/OFQ-NH2
- UFP-101
[Nphe1,Arg14,Lys15]N/OFQ-NH2
- DPDPE
[D-Pen2,D-Pen5]-Enkephalin
- i.c.v
intracerebroventricular
- i.t
intrathecal
- ANOVA
analysis of variance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arduin M, Spagnolo B, Calo' G, Guerrini R, Carra' G, Fischetti C, Trapella C, Marzola E, McDonald J, Lambert DG, Regoli D, Salvadori S. Synthesis and biological activity of nociceptin/orphanin FQ analogues substituted in position 7 or 11 with Calpha-alpha-dialkylated amino acids. J Bioorg Med Chem. 2007 doi: 10.1016/j.bmc.2007.04.026. in press. [DOI] [PubMed] [Google Scholar]
- 2.Becker JA, Wallace A, Garzon A, Ingallinella P, Bianchi E, Cortese R, Simonin F, Kieffer BL, Pessi A. Ligands for kappa-opioid and ORL1 receptors identified from a conformationally constrained peptide combinatorial library. J Biol Chem. 1999;274:27513–22. doi: 10.1074/jbc.274.39.27513. [DOI] [PubMed] [Google Scholar]
- 3.Bignan GC, Connolly PJ, Middleton SA. Recent advances towards the discovery of ORL-1 receptor agonists and antagonists. Expert Opin Ther Patents. 2005;15:357–88. [Google Scholar]
- 4.Bigoni R, Giuliani S, Calo G, Rizzi A, Guerrini R, Salvadori S, Regoli D, Maggi CA. Characterization of nociceptin receptors in the periphery: in vitro and in vivo studies. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:160–67. doi: 10.1007/pl00005338. [DOI] [PubMed] [Google Scholar]
- 5.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye-binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 6.Broccardo M, Guerrini R, Morini G, Agostini S, Petrella C, Improta G. The gastric effects of UFP 112, a new agonist of the NOP receptor, in physiological and pathological conditions. Peptides. 2007 doi: 10.1016/j.peptides.2007.07.021. submitted for publication. [DOI] [PubMed] [Google Scholar]
- 7.Calo G, Guerrini R, Rizzi A, Salvadori S, Regoli D. Pharmacology of nociceptin and its receptor: a novel therapeutic target. Br J Pharmacol. 2000;129:1261–83. doi: 10.1038/sj.bjp.0703219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calo G, Guerrini R, Bigoni R, Rizzi A, Bianchi C, Regoli D, Salvadori S. Structure-activity study of the nociceptin(1–13)-NH2 N-terminal tetrapeptide and discovery of a nociceptin receptor antagonist. J Med Chem. 1998;41:3360–6. doi: 10.1021/jm970805q. [DOI] [PubMed] [Google Scholar]
- 9.Calo G, Rizzi A, Marzola G, Guerrini R, Salvadori S, Beani L, Regoli D, Bianchi C. Pharmacological characterization of the nociceptin receptor mediating hyperalgesia in the mouse tail withdrawal assay. Br J Pharmacol. 1998;125:373–8. doi: 10.1038/sj.bjp.0702087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calo G, Guerrini R, Rizzi A, Salvadori S, Burmeister M, Kapusta DR, Lambert DG, Regoli D. UFP-101, a Peptide Antagonist Selective for the Nociceptin/Orphanin FQ Receptor. CNS Drug Rev. 2005;11:97–112. doi: 10.1111/j.1527-3458.2005.tb00264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calo G, Rizzi A, Rizzi D, Bigoni R, Guerrini R, Marzola G, Marti M, McDonald J, Morari M, Lambert DG, Salvadori S, Regoli D. [Nphe(1),Arg(14),Lys(15)]nociceptin-NH(2), a novel potent and selective antagonist of the nociceptin/orphanin FQ receptor. Br J Pharmacol. 2002;136:303–11. doi: 10.1038/sj.bjp.0704706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carra G, Rizzi A, Guerrini R, Barnes TA, McDonald J, Hebbes CP, Mela F, Kenigs VA, Marzola G, Rizzi D, Gavioli E, Zucchini S, Regoli D, Morari M, Salvadori S, Rowbotham DJ, Lambert DG, Kapusta DR, Calo G. [(pF)Phe4,Arg14,Lys15]N/OFQ-NH2 (UFP-102), a highly potent and selective agonist of the nociceptin/orphanin FQ receptor. J Pharmacol Exp Ther. 2005;312:1114–23. doi: 10.1124/jpet.104.077339. [DOI] [PubMed] [Google Scholar]
- 13.Chang M, Peng YL, Dong SL, Han RW, Li W, Yang DJ, Chen Q, Wang R. Structure-activity studies on different modifications of nociceptin/orphanin FQ: identification of highly potent agonists and antagonists of its receptor. Regul Pept. 2005;130:116–22. doi: 10.1016/j.regpep.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 14.Chiou LC, Chuang KC, Wichmann J, Adam G. Ro 64-6198 [(1S,3aS)-8-(2,3,3a,4,5,6-Hexahydro-1H-phenalen-1-yl)-1-phenyl-1,3,8-triaz a-spiro[4.5]decan-4-one] acts differently from nociceptin/orphanin FQ in rat periaqueductal gray slices. J Pharmacol Exp Ther. 2004;311:645–51. doi: 10.1124/jpet.104.070219. [DOI] [PubMed] [Google Scholar]
- 15.Ciccocioppo R, Angeletti S, Panocka I, Massi M. Nociceptin/orphanin FQ and drugs of abuse. Peptides. 2000;21:1071–80. doi: 10.1016/s0196-9781(00)00245-x. [DOI] [PubMed] [Google Scholar]
- 16.Ciccocioppo R, Cippitelli A, Economidou D, Fedeli A, Massi M. Nociceptin/orphanin FQ acts as a functional antagonist of corticotropin-releasing factor to inhibit its anorectic effect. Physiol Behav. 2004;82:63–8. doi: 10.1016/j.physbeh.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 17.Cox BM, Chavkin C, Christie MJ, Civelli O, Evans C, Hamon MD, Hoellt V, Kieffer B, Kitchen I, McKnight AT, Meunier JC, Portoghese PS. Opioid receptors. In: Girdlestone D, editor. The IUPHAR Compendium of Receptor Characterization and Classification. IUPHAR Media Ltd; London: 2000. pp. 321–33. [Google Scholar]
- 18.Dautzenberg FM, Wichmann J, Higelin J, Py-Lang G, Kratzeisen C, Malherbe P, Kilpatrick GJ, Jenck F. Pharmacological characterization of the novel nonpeptide orphanin FQ/nociceptin receptor agonist Ro 64-6198: rapid and reversible desensitization of the ORL1 receptor in vitro and lack of tolerance in vivo. J Pharmacol Exp Ther. 2001;298:812–9. [PubMed] [Google Scholar]
- 19.Dooley CT, Spaeth CG, Berzetei-Gurske IP, Craymer K, Adapa ID, Brandt SR, Houghten RA, Toll L. Binding and in vitro activities of peptides with high affinity for the nociceptin/orphanin FQ receptor, ORL1. J Pharmacol Exp Ther. 1997;283:735–41. [PubMed] [Google Scholar]
- 20.Economidou D, Fedeli A, Fardon RM, Weiss F, Massi M, Ciccocioppo R. Effect of novel nociceptin/orphanin FQ-NOP receptor ligands on ethanol drinking in alcohol-preferring msP rats. Peptides. 2006;27:3299–306. doi: 10.1016/j.peptides.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gavioli EC, Calo G. Antidepressant- and anxiolytic-like effects of nociceptin/orphanin FQ receptor ligands. Naunyn Schmiedebergs Arch Pharmacol. 2006;372:319–30. doi: 10.1007/s00210-006-0035-8. [DOI] [PubMed] [Google Scholar]
- 22.Gavioli EC, Marzola G, Guerrini R, Bertorelli R, Zucchini S, De Lima TC, Rae GA, Salvadori S, Regoli D, Calo G. Blockade of nociceptin/orphanin FQ-NOP receptor signalling produces antidepressant-like effects: pharmacological and genetic evidences from the mouse forced swimming test. Eur J Neurosci. 2003;17:1987–90. doi: 10.1046/j.1460-9568.2003.02603.x. [DOI] [PubMed] [Google Scholar]
- 23.Guerrini R, Calo G, Rizzi A, Bianchi C, Lazarus LH, Salvadori S, Temussi PA, Regoli D. Address and message sequences for the nociceptin receptor: a structure-activity study of nociceptin-(1–13)-peptide amide. J Med Chem. 1997;40:1789–93. doi: 10.1021/jm970011b. [DOI] [PubMed] [Google Scholar]
- 24.Guerrini R, Calo G, Bigoni R, Rizzi D, Rizzi A, Zucchini M, Varani K, Hashiba E, Lambert DG, Toth G, Borea PA, Salvadori S, Regoli D. Structure-activity studies of the Phe(4) residue of nociceptin(1–13)-NH(2): identification of highly potent agonists of the nociceptin/orphanin FQ receptor. J Med Chem. 2001;44:3956–64. doi: 10.1021/jm010221v. [DOI] [PubMed] [Google Scholar]
- 25.Guerrini R, Calo G, Bigoni R, Rizzi A, Varani K, Toth G, Gessi S, Hashiba E, Hashimoto Y, Lambert DG, Borea PA, Tomatis R, Salvadori S, Regoli D. Further studies on nociceptin-related peptides: discovery of a new chemical template with antagonist activity on the nociceptin receptor. J Med Chem. 2000;43:2805–13. doi: 10.1021/jm990075h. [DOI] [PubMed] [Google Scholar]
- 26.Hadrup M, Peterson JS, Praetorius J, Meier E, Graebe M, Brond L, Staahltoft D, Nielsen S, Christensen S, Kapusta DR, Jonassen TEN. Opioid receptor-like 1 stimulation in the collecting duct induces aquaresis through vasopressin-independent aquaporin-2 downregulation. Am J Physiol Renal Physiol. 2004;287:F160–F168. doi: 10.1152/ajprenal.00329.2003. [DOI] [PubMed] [Google Scholar]
- 27.Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;17:313–6. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- 28.Jenck F, Moreau JL, Martin JR, Kilpatrick GJ, Reinscheid RK, Monsma FJ, Jr, Nothacker HP, Civelli O. Orphanin FQ acts as an anxiolytic to attenuate behavioral responses to stress. Proc Natl Acad Sci U S A. 1997;94:14854–8. doi: 10.1073/pnas.94.26.14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenck F, Wichmann J, Dautzenberg FM, Moreau JL, Ouagazzal AM, Martin JR, Lundstrom K, Cesura AM, Poli SM, Roever S, Kolczewski S, Adam G, Kilpatrick G. A synthetic agonist at the orphanin FQ/nociceptin receptor ORL1: anxiolytic profile in the rat. Proc Natl Acad Sci U S A. 2000;97:4938–43. doi: 10.1073/pnas.090514397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kapusta DR. Neurohumoral effects of orphanin FQ/nociceptin: relevance to cardiovascular and renal function. Peptides. 2000;21:1081–99. doi: 10.1016/s0196-9781(00)00246-1. [DOI] [PubMed] [Google Scholar]
- 31.Kapusta DR, Kenigs VA. Cardiovascular and renal responses produced by central orphanin FQ/nociceptin occur independent of renal nerves. Am J Physiol. 1999;277:R987–95. doi: 10.1152/ajpregu.1999.277.4.R987. [DOI] [PubMed] [Google Scholar]
- 32.Kapusta DR, Sezen SF, Chang JK, Lippton H, Kenigs VA. Diuretic and antinatriuretic responses produced by the endogenous opioid-like peptide, nociceptin (orphanin FQ) Life Sci. 1997;60:PL15–21. doi: 10.1016/s0024-3205(96)00593-0. [DOI] [PubMed] [Google Scholar]
- 33.Kapusta DR, Burmeister MA, Calo G, Guerrini R, Gottlieb HB, Kenigs VA. Functional selectivity of Nociceptin/orphanin FQ peptide receptor partial agonists on cardiovascular and renal function. J Pharm Exp Ther. 2005;314:643–51. doi: 10.1124/jpet.104.082768. [DOI] [PubMed] [Google Scholar]
- 34.Kapusta DR, Thorkildsen C, Kenigs VA, Meier E, Vinge MM, Quist C, Peterson JS. Pharmacodynamic characterization of ZP120 (Ac-RYYRWKKKKKKK-NH2), a novel, functionally selective Nociceptin/orphanin FQ peptide receptor partial agonist with sodium-potassium-sparing aquaretic activity. J Pharm Exp Ther. 2005;314:652–60. doi: 10.1124/jpet.105.083436. [DOI] [PubMed] [Google Scholar]
- 35.Ko MC, Wei H, Woods JH, Kennedy RT. Effects of Intrathecally Administered Nociceptin/Orphanin FQ in Monkeys: Behavioral and Mass Spectrometric Studies. J Pharmacol Exp Ther. 2006;318:1257–64. doi: 10.1124/jpet.106.106120. [DOI] [PubMed] [Google Scholar]
- 36.Laursen SE, Belknap JK. Intracerebroventricular injections in mice. Some methodological refinements. J Pharmacol Methods. 1986;16:355–7. doi: 10.1016/0160-5402(86)90038-0. [DOI] [PubMed] [Google Scholar]
- 37.Lazzeri M, Calo G, Spinelli M, Guerrini R, Beneforti P, Sandri S, Zanollo A, Regoli D, Turini D. Urodynamic and clinical evidence of acute inhibitory effects of intravesical nociceptin/orphanin FQ on detrusor overactivity in humans: a pilot study. J Urol. 2001;166:2237–40. [PubMed] [Google Scholar]
- 38.Lazzeri M, Calo G, Spinelli M, Guerrini R, Salvadori S, Beneforti P, Sandri S, Regoli D, Turini D. Urodynamic effects of intravesical nociceptin/orphanin FQ in neurogenic detrusor overactivity: a randomized, placebo-controlled, double-blind study. Urology. 2003;61:946–50. doi: 10.1016/s0090-4295(02)02587-6. [DOI] [PubMed] [Google Scholar]
- 39.Lazzeri M, Calo' G, Spinelli M, Malaguti S, Guerrini R, Salvadori S, Beneforti P, Regoli D, Turini D. Daily intravescical instillation of 1 mg nociceptin/orphanin FQ for the control of neurogenic detrusor overactivity - a multicenter, placebo controlled, randomized exploratory study. J Urol. 2006;176:2098–2102. doi: 10.1016/j.juro.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 40.McLeod RL, Bolser DC, Jia Y, Parra LE, Mutter JC, Wang X, Tulshian DB, Egan RW, Hey JA. Antitussive effect of nociceptin/orphanin FQ in experimental cough models. Pulm Pharmacol Ther. 2002;15:213–6. doi: 10.1006/pupt.2002.0357. [DOI] [PubMed] [Google Scholar]
- 41.Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monserrat B, Mazarguil H, Vassart G, Parmentier M, Costentin J. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–5. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- 42.Mogil JS, Pasternak GW. The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacol Rev. 2001;53:381–415. [PubMed] [Google Scholar]
- 43.Nazzaro C, Rizzi A, Salvadori S, Guerrini R, Regoli D, Zeilhofer HU, Calo' G. UFP-101 antagonizes the spinal antinociceptive effects of nociceptin/orphanin FQ: behavioral and electrophysiological studies in mice. Peptides. 2006;28:663–9. doi: 10.1016/j.peptides.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 44.Nishi M, Houtani T, Noda Y, Mamiya T, Sato K, Doi T, Kuno J, Takeshima H, Nukada T, Nabeshima T, Yamashita T, Noda T, Sugimoto T. Unrestrained nociceptive response and disregulation of hearing ability in mice lacking the nociceptin/orphaninFQ receptor. Embo J. 1997;16:1858–64. doi: 10.1093/emboj/16.8.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okada K, Sujaku T, Chuman Y, Nakashima R, Nose T, Costa T, Yamada Y, Yokoyama M, Nagahisa A, Shimohigashi Y. Highly potent nociceptin analog containing the Arg-Lys triple repeat. Biochem Biophys Res Commun. 2000;278:493–8. doi: 10.1006/bbrc.2000.3822. [DOI] [PubMed] [Google Scholar]
- 46.Ozaki S, Kawamoto H, Itoh Y, Miyaji M, Azuma T, Ichikawa D, Nambu H, Iguchi T, Iwasawa Y, Ohta H. In vitro and in vivo pharmacological characterization of J-113397, a potent and selective non-peptidyl ORL1 receptor antagonist. Eur J Pharmacol. 2000;402:45–53. doi: 10.1016/s0014-2999(00)00520-3. [DOI] [PubMed] [Google Scholar]
- 47.Peng YL, Chang M, Dong SL, Li W, Han RW, Fu GX, Chen Q, Wang R. Novel potent agonist [(pF)Phe4,Aib7,Aib11,Arg14,Lys15]N/OFQ-NH2 and antagonist [Nphe1,(pF)Phe4,Aib7,Aib11,Arg14,Lys15]N/OFQ-NH2 of nociceptin/orphanin FQ receptor. Regul Pep. 2006;134:75–81. doi: 10.1016/j.regpep.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 48.Reinscheid RK. The Orphanin FQ / Nociceptin receptor as a novel drug target in psychiatric disorders. CNS Neurol Disord Drug Targets. 2006;5:219–24. doi: 10.2174/187152706776359628. [DOI] [PubMed] [Google Scholar]
- 49.Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Jr, Civelli O. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–4. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- 50.Rizzi A, Bigoni R, Marzola G, Guerrini R, Salvadori S, Regoli D, Calo G. Characterization of the locomotor activity-inhibiting effect of nociceptin/orphanin FQ in mice. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:161–5. doi: 10.1007/s002100000358. [DOI] [PubMed] [Google Scholar]
- 51.Shoblock JR, Wichmann J, Maidment NT. The effect of a systemically active ORL-1 agonist, Ro 64-6198, on the acquisition, expression, extinction, and reinstatement of morphine conditioned place preference. Neuropharmacology. 2005;49:439–46. doi: 10.1016/j.neuropharm.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 52.Zaratin PF, Petrone G, Sbacchi M, Garnier M, Fossati C, Petrillo P, Ronzoni S, Giardina GA, Scheideler MA. Modification of nociception and morphine tolerance by the selective opiate receptor-like orphan receptor antagonist (-)-cis-1-methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9- tetrahydro-5H-benzocyclohepten-5-ol (SB-612111) J Pharmacol Exp Ther. 2004;308:454–61. doi: 10.1124/jpet.103.055848. [DOI] [PubMed] [Google Scholar]
- 53.Zaveri N. Peptide and nonpeptide ligands for the nociceptin/orphanin FQ receptor ORL1: research tools and potential therapeutic agents. Life Sci. 2003;73:663–78. doi: 10.1016/s0024-3205(03)00387-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zeilhofer HU, Calo G. Nociceptin/Orphanin FQ and its receptor - Potential targets for pain therapy? J Pharmacol Exp Ther. 2003;306:423–9. doi: 10.1124/jpet.102.046979. [DOI] [PubMed] [Google Scholar]
- 55.Zhang C, Miller W, Valenzano KJ, Kyle DJ. Novel, potent ORL-1 receptor agonist peptides containing alpha-Helix-promoting conformational constraints. J Med Chem. 2002;45:5280–6. doi: 10.1021/jm0202021. [DOI] [PubMed] [Google Scholar]