

Abstract
PURPOSE.
Docosahexaenoic acid (DHA22:6,n3) is the principal n3 polyunsaturated fatty acid (PUFA) in the retina. The authors previously demonstrated that DHA22:6,n3 inhibited cytokine-induced adhesion molecule expression in primary human retinal vascular endothelial (hRVE) cells, the target tissue affected by diabetic retinopathy. Despite the importance of vascular inflammation in diabetic retinopathy, the mechanisms underlying anti-inflammatory effects of DHA22:6,n3 in vascular endothelial cells are not understood. In this study the authors address the hypothesis that DHA22:6,n3 acts through modifying lipid composition of caveolae/lipid rafts, thereby changing the outcome of important signaling events in these specialized plasma membrane microdomains.
METHODS.
hRVE cells were cultured in the presence or absence of DHA22:6,n3. Isolated caveolae/lipid raft–enriched detergent-resistant membrane domains were prepared using sucrose gradient ultracentrifugation. Fatty acid composition and cholesterol content of caveolae/lipid rafts before and after treatment were measured by HPLC. The expression of Src family kinases was assayed by Western blotting and immunohistochemistry.
RESULTS.
Disruption of the caveolae/lipid raft structure with a cholesterol-depleting agent, methyl-cyclodextrin (MCD), diminished cytokine-induced signaling in hRVE cells. Growth of hRVE cells in media enriched in DHA22:6,n3 resulted in significant incorporation of DHA22:6,n3 into the major phospholipids of caveolae/lipid rafts, causing an increase in the unsaturation index in the membrane microdomain. DHA22:6,n3 enrichment in the caveolae/raft was accompanied by a 70% depletion of cholesterol from caveolae/lipid rafts and displacement of the SFK, Fyn, and c-Yes from caveolae/lipid rafts. Adding water-soluble cholesterol to DHA22:6,n3-treated cells replenished cholesterol in caveolae/lipid rafts and reversed the effect of DHA22:6,n3 on cytokine-induced signaling.
CONCLUSIONS.
Incorporation of DHA22:6,n3 into fatty acyl chains of phospholipids in caveolae/lipid rafts, followed by cholesterol depletion and displacement of important signaling molecules, provides a potential mechanism for anti-inflammatory effect of DHA22:6,n3 in hRVE cells.
Polyunsaturated fatty acids (PUFAs), especially n3-PUFAs such as eicosapentaenoic acid (EPA20:5n3) and DHA22:6,n3, are well known to have anti-inflammatory properties.1,2 Vascular inflammation is a process that involves the activation of vascular endothelium and circulating leukocytes. The anti-inflammatory action of n-3 PUFAs has been studied in association with their ability to suppress leukocyte activation and function.3-11 However, the effect of PUFAs on the vascular component of inflammation has received less attention. hRVE cells are the target vascular tissue affected by diabetic retinopathy, a condition that has been recently associated with low-grade inflammation in the retina. We have previously demonstrated that the growth of human retinal vascular endothelial cells in media enriched in DHA22:6,n3 resulted in the inhibition of IL-1β, TNF-α, and VEGF-induced intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 expression.12 The mechanism of anti-inflammatory action of DHA22:6,n3 in hRVE cells is unknown.
Recent studies strongly suggest that the inhibitory effects of n3-PUFA on T-lymphocyte signal transduction are, at least in part, caused by the modification of functional membrane lipid microdomains known as lipid rafts.3,5,7,9,10,13 Vascular endothelial cells, such as hRVE cells, contain lipid rafts and a second type of specialized micromembrane domain known as caveolae. The lipid composition of caveolae is similar to that of lipid rafts, but caveolae are stabilized by the caveolin family of structural and regulatory proteins, which give them a characteristic flasklike shape.14-17 Caveolae are important in regulating vascular permeability, vesicle trafficking, cholesterol homeostasis, and, in particular, signal transduction. Despite debate regarding the spatial and temporal aspects of lipid rafts and the initiating factors in lipid raft assembly,16,18-22 it is well accepted that caveolae and lipid rafts are enriched in cholesterol and sphingolipids and depleted in glycerophospholipids, resulting in a glycerophospholipid/sphingolipid/cholesterol molar ratio of 1:1:1.23 Furthermore, the glycerophospholipids in lipid rafts are highly enriched in saturated fatty acyl chains. Highly saturated structures in the lipid raft membranes stabilized by cholesterol form lipid-ordered domains that govern lipid–protein interactions.23-26
A number of signaling molecules are attracted by the highly ordered raft lipid domain, including glycosylphosphatidylinositol (GPI)-anchored proteins in the exoplasmic domain and dually acylated nonreceptor tyrosine kinases in the cytoplasmic domain.27-31 Most of the Srk family kinases, such as Fyn, Lck, and c-Yes, are myristoylated on the amino-terminal glycine residue and palmitoylated on amino-terminal cysteine3 residue.32,33 Dual acylation with saturated fatty acids is proposed to increase the affinity of Src family kinases to caveolae and lipid rafts.
Cytokine signaling in endothelial cells is initiated at the specific receptors localized in the exoplasmic leaflet of the plasma membrane. Many cytokine receptors are GPI-linked proteins that localize in caveolae/lipid rafts, such as TNF-R130,31 and VEGF-R2 in endothelial cells.27,28 Although the direct connection between Src family kinases and cytokine receptors signaling remains to be elucidated, the proximity of Src family kinases localized to the endoplasmic domain of lipid rafts to TNF-R,34,35 IL-1βR,36,37 and VEGF-R238-41 was documented in several cell types. PUFA treatment was shown to inhibit cytokine signal transduction in T lymphocytes through selective displacement of dually acylated proteins from lipid rafts.3,5,7,9,42
In this study we demonstrate that treating human vascular endothelial cells with DHA22:6,n3 greatly increased the caveolae/lipid raft unsaturation index by enriching fatty acyl chains of phospholipids in caveolae/lipid rafts with this highly unsaturated PUFA. Moreover, DHA22:6,n3 incorporation into caveolae/lipid rafts caused cholesterol displacement from these specialized membrane microdomains. Changes in lipid composition were associated with decreased affinity of the SFK constitutively expressed in the caveolae/lipid rafts in hRVE cells, Fyn, and c-Yes for the raft fraction. The modification of caveolae/lipid raft lipid composition with PUFA, followed by the displacement of important signaling proteins resident in caveolae/lipid rafts, provides a potential mechanism for the inhibition of cytokine-induced inflammatory signaling by DHA22:6,n3 in hRVE cells.
MATERIALS AND METHODS
Reagents and Antibodies
HPLC-grade acetonitrile, acetic acid, methanol, chloroform, methyl-cyclodextrin (MCD), and water-soluble cholesterol were purchased (Sigma, St. Louis, MO), and the antibodies mouse anti–caveolin-1 and flotillin-1 (Upstate Biotechnology, Inc., Lake Placid, NY) and mouse anti–c-Src, Fyn, rabbit antibodies against c-Yes, and CD36 (Santa Cruz Biotechnology, Santa Cruz, CA) were used.
Cell Culture
Primary cultures of hRVE cells were prepared from tissue (National Disease Research Interchange, Philadelphia, PA) cultured as previously described. Passages 1 to 6 were used in the experiments. For experimental treatment, cells were transferred to serum-free medium for 14 to 24 hours before stimulatory agents were added.
Fatty Acid Treatment
Fatty acid stocks were prepared by dissolving fatty acids (NuCheck Prep, Inc., Elysian, MN) in ethanol to a final concentration of 100 mM fatty acid, as described previously.43 The fatty acid stock solutions were diluted to 50 to 100 μM in serum-free medium containing 10 to 20 μM charcoal-treated, solvent-extracted, fatty acid–free bovine serum albumin (BSA; Serologica Inc., Norcross, GA) as a fatty acid carrier. The fatty acid/albumin molar ratio was maintained at 5:1. The final concentration of ethanol in the media was less than 0.1%. Cells were incubated for the times indicated in Results. Equivalent amounts of BSA and ethanol were added to control plates.
Fatty acids at concentrations higher than 200 μM were shown to induce apoptosis in human umbilical vein endothelial cells.44 Concentrations of fatty acids used in this study were well below the proapoptotic range; however, we routinely verified, according to manufacturer’s protocol (Vybrant Apoptosis Assay Kit 4; Invitrogen, Carlsbad, CA), that the rate of apoptosis in fatty acid–treated hRVE cells was not higher than in control cells.
MCD and Cholesterol Treatment
To deplete cholesterol from the membranes, cells were treated with 8 mM MCD for 30 minutes. To replenish cholesterol, cells were treated with 25 μM water-soluble cholesterol (25 μM cholesterol complexed with 250 μM MCD; Sigma) for 30 minutes. After MCD or water-soluble cholesterol treatment, the cells were washed twice with PBS containing 40 μM BSA before stimulation with cytokines for the time indicated in Results.
SDS-PAGE and Western Blot Analysis
Cells were lysed in the lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X-100, and 10% glycerol) with freshly added protease inhibitor cocktail (Sigma) and phosphatase inhibitors (1 mM Na3VO4, 100 μM glycerophosphate, 10 mM NaF, 1 mM Na4PPi). Proteins were resolved by SDS-PAGE, transferred to nitrocellulose, and immunoblotted using appropriate antibodies followed by secondary horseradish peroxidase–conjugated antibody (Bio-Rad, Hercules, CA). Immunoreactive bands were visualized by enhanced chemiluminescence (ECL kit; Amersham Pharmacia Biotech, Piscataway, NJ). Blots were quantitated by scanning densitometry (ImageJ software, version 1.29; available by ftp at zippy.nimh.nih.gov or at http://rsb.info.nih.gov/nih-image; developed by Wayne Rasband, National Institutes of Health, Bethesda, MD).
Subcellular Fractionation
Postnuclear supernatants were prepared as described previously.45 Cells were lysed in hypotonic buffer (10 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM MgCl2 freshly added with protease inhibitors) for 30 minutes on ice, then homogenized with 20 strokes in a nonstick (Teflon; DuPont, Wilmington, DE)/glass homogenizer and spun down at 500g for 5 minutes. Supernatants were subjected to centrifugation (16,900g, 60 minutes, 4°C), and pellets were collected as plasma membrane–enriched fractions.
Isolation of caveolae/lipid raft–enriched detergent-resistant membrane domains were prepared using a slightly modified sucrose gradient ultracentrifugation protocol.46 Briefly, 5 × 106 hRVE cells were washed twice with cold PBS, lysed in 0.8 mL MNE buffer (25 mM MES, pH 6.5, 0.15 M NaCl, and 5 mM EDTA) containing 1% Triton X-100, fresh protease, and phosphatase inhibitors, and kept on ice for 20 minutes. Lysates were then homogenized with 10 strokes of a tight-fitting Dounce homogenizer and spun down at 4000g at 4°C for 10 minutes. Supernatant (0.8 mL) was mixed with the same volume of 80% sucrose prepared in MNE buffer and was placed at the bottom of an ultracentrifuge tube. Then 1.6 mL of 30% sucrose and 0.8 mL of 5% sucrose were overlaid on top of the sample to form a 5% to 30% discontinuous sucrose gradient. After 16 hours of centrifugation at 200,000g and 4°C in a swinging bucket rotor, 0.4-mL samples were collected carefully from the top of each fraction. A band confined to fractions 2 through 4 was designated as caveolae/lipid raft–enriched membrane domains. The combined 2 to 4 fractions were diluted 3 times in MNE buffer and spun at 200,000g and 4°C for another 2 hours to obtain the caveolae/lipid rafts. Fractions 6 to 10 were also combined and designated as nonraft membranes.
Fatty Acid Metabolism
hRVE cells were plated at 0.12 × 106 cells/6-cm plate and cultured as indicated to 90% confluence. Cells were then serum starved overnight and treated with 100 μM DHA22:6,n3/20μM BSA containing tracer amounts of 14C-DHA22:6,n3 (0.5 μCi in 3 mL media) for 1.5 and 24 hours. 14C-DHA22:6,n3 (1.7 Ci/mol) was purchased (PerkinElmer Life Sciences, Wellesley, MA). At harvest, cells were washed once with PBS + 20 μM BSA and once by PBS alone. Cells were then resuspended in 500 μL of 40% methanol, and lipids were extracted with chloroform/methanol (2:1); dried under nitrogen, and dissolved in chloroform for storage at −80°C. Total lipids were separated by thin-layer chromatography (LK6D Silica G 60A; Whatman, Clifton, NJ) in hexane/diethyl ether/acetic acid (90:30:1). Polar lipids were separated in chloroform/methanol/acetic acid (30:20:4). TLC plates were dried, and radioactivity was detected and quantified by phosphor imaging. Location of total lipids was compared with authentic standards for triacylglycerol (TAG), diacylglycerol (DAG), cholesterol esters (CEs), and fatty acids (Sigma). Location of phospholipids was also compared with authentic standards for phosphatidylcholine (PC), phosphatidylinositol (PI), phosphatidylserine (PS), and phosphatidylethanolamine (PE; Avanti Polar Lipids, Alabaster, AL).
Fatty Acid and Cholesterol Analysis
Total lipids from caveolae/lipid rafts and plasma membranes corresponding to equal amounts of protein (measured by Bradford assay [BioRad]) were extracted with chloroform–methanol (2:1), dried, and stored in chloroform at −80°C. Internal standards consisting of 19:1 (1 μmol/mg protein) and 14:1/14:1 PC (70 nmol/mg protein) were used. A fraction of the total lipids was further fractionated on an aminopropyl (Alltech, Lexington, KY) column to obtain neutral lipids, neutral phospholipids, acidic phospholipids, and nonesterified fatty acids (NEFAs), as described.47 Neutral lipids were separated by a 5-μM, 250 × 4.6-mm column (YMC-Diol-120NP; Waters, Milford, MA) using evaporative light scatter to detect cholesterol. Pure lipid standards for cholesterol, triacylglycerol, and diacylglycerol (Avanti Polar Lipids) were used after each experiment to confirm retention times and purity. To quantify the data, calibration curves were prepared for each class of lipids at the end of each experiment.
A fraction of neutral phospholipids and acidic phospholipids were saponified (0.4 n KOH in 80% methanol, 50°C for 1 hour) for fatty acid composition analysis. After saponification, lipids were acidified and extracted with diethyl ether with 0.1% acetic acid, dried under nitrogen, and stored in methanol. Saponified lipids were fractionated and quantitated by reverse-phase HPLC (RP-HPLC) with a separation technology column (J’sphere ODS-H80; YMC Co., Ltd., Kyoto, Japan) and a sigmoidal gradient starting at 86.5% acetonitrile + acetic acid (0.1%) and ending at 100% acetonitrile + acetic acid (0.1%) over 50 minutes with a flow rate of 1.0 mL/min and a controller (model 600; Waters). Fatty acids were detected using ultraviolet absorbance at 192 nm (model 2487; Waters) and evaporative light scatter (model 2420; Waters). Samples were normalized using recovery standards. Calibration curves were prepared for each fatty acid at the end of the experiment in accordance with fatty acid standards for RP-HPLC (Nu-Chek Prep, Elysian, MN).
Statistical Analysis
Data are expressed as the mean ± SD. Factorial ANOVA with post hoc Tukey test (calculated using http://faculty.vassar.edu/lowry/VassarStats.html) was used for comparing data obtained from independent samples. Significance was established at P < 0.05.
RESULTS
Incorporation of DHA22:6,n3 into Phospholipids in hRVE Cells
Exogenously supplied [14C]-DHA22:6,n3 was taken up by the cells and rapidly incorporated into different cellular lipids, including DAG, NEFA, triglycerides, and, to a large extent, polar lipids (Fig. 1A). Further separation of polar lipids demonstrated that [14C]-DHA22:6,n3 was incorporated mainly into the phosphatidylcholine (PC) fraction (Fig. 1B).
FIGURE 1.
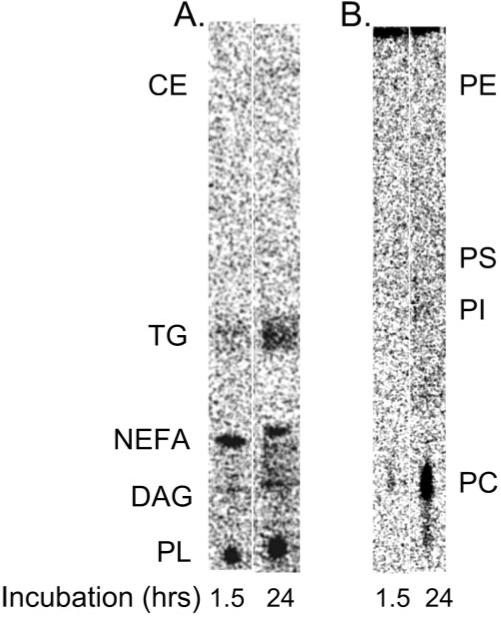
Incorporation of 14C- DHA22:6,n3 into hRVE cellular lipids. hRVE cells were treated with 14C-DHA22:6,n3 for the time periods indicated. Total lipids were extracted and subjected to thin-layer chromatography (TLC) analysis. Radioactivity was detected and quantified by phosphorimaging. A phosphorimage of total lipid TLC analysis is presented (A), as is phospholipid TLC analysis (B). Location of the authentic standards for polar lipid (PL), diacylglycerol (DAG), nonesterified fatty acid (NEFA), triacylglycerol (TG), and cholesterol ester (CE) is shown on the total lipid TLC (A). Location of the authentic standards for phosphatidylcholine (PC), phosphatidylinositol (PI), phosphatidylserine (PS), and phosphatidylethanolamine (PE) are shown on the phospholipid TLC (B).
Characterization of Caveolae/Lipid Rafts from hRVE Cells
Because phospholipids are important structural components of caveolae/lipid rafts, we examined whether the incorporation of DHA22:6,n3 affected the signaling environment of caveolae/lipid rafts. Caveolae/lipid raft fractions were isolated by sucrose discontinuous gradient ultracentrifugation based on their insolubility in Triton X-100 at 4°C (Fig. 2). Confirmation of isolated lipid rafts was achieved by demonstrating enrichment of caveolin-1 (a caveolae marker) and flotillin-1 (a lipid raft marker) in density gradient fractions 2 to 4. In contrast, the plasma membrane marker Na+/K+-ATPase was excluded from the lipid raft fractions. PKC-α and ERK1/2 were found in the nonraft membrane fraction (fractions 6–9) under basal conditions (Fig. 2). These results demonstrate that hRVE cells contain caveolae and lipid raft structures and that a caveolae/lipid raft–enriched fraction can be isolated using methods involving detergent insolubility and density gradient fractionation.
FIGURE 2.
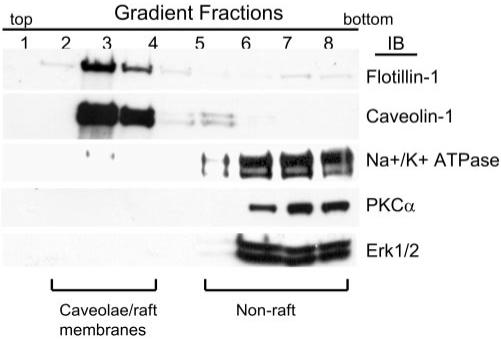
Characterization of caveolae/lipid raft protein components in hRVE cells. Membrane lipids were fractionated to enrich for caveolae/lipid rafts and nonraft components. Each membrane fraction was separated on SDS-PAGE. Western blot analysis for the caveolae marker caveolin-1, the lipid raft marker flotillin-1, the plasma membrane marker Na+/K+ATPase, PKC-α, and Erk1/2 was performed to analyze protein distribution in each fraction. Fractions designated caveolae/raft and nonraft membrane are indicated.
Incorporation of DHA22:6,n3 into Phospholipids of Caveolae/Lipid Rafts
To determine the effect of exogenously added fatty acids on total plasma membrane and caveolae/lipid raft lipid composition, the fractions were isolated from control cells or cells treated with 100 μM palmitic acid16:0 (lipid control) or DHA22:6,n3 for 24 hours. After treatment, total lipids were extracted and separated by an amino-propyl column to enrich for polar lipids. The method yields two phospholipid fractions, neutral phospholipids (containing PC, PE, and SM) and acidic phospholipid (containing PI, PS, and PA).47 Because most (more than 70% in total plasma membrane and more than 85% in caveolae/lipid raft fraction) phospholipids were recovered in the neutral phospholipid fraction, only this fraction was saponified and analyzed for the fatty acyl composition of phospholipids by RP-HPLC (Fig. 3). As expected, the phospholipids from caveolae/lipid raft fractions from control and palmitate-treated cells were particularly enriched in saturated palmitic16:0 and stearic18:0 acids. Monounsaturated oleic18:1n9, polyunsaturated linoleic18:2n6, and arachidonic20:4n6 fatty acids were less abundant in caveolae/lipid raft phospholipids than in total plasma membrane phospholipids. Although treating cells with palmitic acid16:0 had modest effects on membrane lipid composition, treating cells with DHA22:6,n3 significantly enriched membrane lipids, both in the total plasma membranes and the caveolae/lipid raft fractions (Figs. 3A, 3B). The unsaturation index—i.e., the number of double bonds per fatty acyl residue—was significantly lower in caveolae/lipid rafts than in total plasma membranes, confirming a high lipid–ordered state in caveolae/lipid rafts. DHA22:6,n3 treatment caused a significant (35%) increase in the unsaturation index in neutral phospholipids (0.44 vs. 0.29 double bonds per fatty acyl residue) in caveolae/lipid rafts compared with that of control hRVE cells.
FIGURE 3.
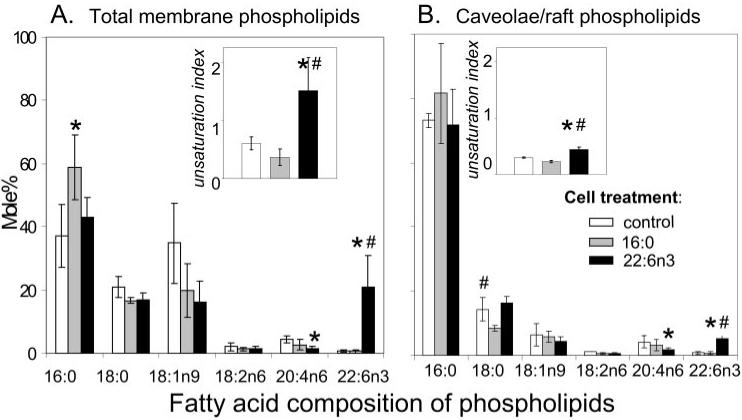
Modification of phospholipid fatty acyl composition of caveolae/lipid raft and total plasma membrane of hRVE cells by incubation with palmitic acid16:0 or DHA22:6,n3. hRVE cells were treated with 100 μM palmitic acid16:0 (gray bars) or DHA22: 6,n3 (black bars) for 24 hours. Vehicle (20 μM BSA)-treated cells (white bars) were used as a control. Caveolae/lipid raft–enriched and nonenriched fractions were purified as described in Experimental Procedures. Lipids were extracted, and phospholipids and sphingolipids were enriched by aminopropyl column fractionation. Neutral phospholipids (PC, PE, SM) and acidic phospholipids (PI, PS, PA) were saponified and analyzed by RP-HPLC. The concentration for each fatty acid was obtained by normalizing to standards and was presented as the mole percent of total fatty acids in each fraction. The unsaturation index was calculated as the average number of double bonds per fatty acyl residue. Given that most phospholipids were in the neutral phospholipid fraction, only the data for neutral phospholipid fraction are presented. Data are the mean ± SD of results in three experiments. *P < 0.05 compared with control. #P < 0.05 compared with palmitic acid16:0-treated cells.
In plasma membranes, the effect of DHA22:6,n3 on the unsaturation index was even more apparent (1.52 vs. 0.61 in DHA22:6,n3-treated vs. control cells; Figs. 3A, 3B). Taken together, these data show that DHA22:6,n3 is effectively incorporated into phospholipids of caveolae/lipid rafts and significantly alters the lipid environment of these specialized membrane microdomains.
Cholesterol Depletion in Caveolae/Lipid Rafts by DHA22:6,n3
Cholesterol content in caveolae/lipid rafts was approximately 10 times higher than in total plasma membranes, confirming that caveolae/lipid rafts are highly enriched in cholesterol (Fig. 4). Introduction of bulky acyl chains such as DHA22:6,n3 into phospholipids, which normally contain saturated fatty acids, can affect fatty acyl chain-cholesterol interaction in caveolae/lipid rafts.20 Indeed, DHA22:6,n3 enrichment was associated with a 70% decrease in the cholesterol level in caveolae/lipid rafts compared with control, whereas palmitic acid16:0 treatment had no significant impact on cholesterol levels. Moreover, the depletion of cholesterol associated with DHA22:6,n3 enrichment only occurred in caveolae/lipid rafts, with no significant effect on cholesterol levels in total plasma membranes (Fig. 4). Thus, enrichment of caveolae/lipid raft phospholipid with DHA22:6,n3 (Fig. 3) results in cholesterol depletion from these structures (Fig. 4). Adding 25 μM water-soluble cholesterol for 30 minutes replenished the cholesterol content of caveolae/lipid rafts in DHA22:6,n3-treated cells (Fig. 4)
FIGURE 4.
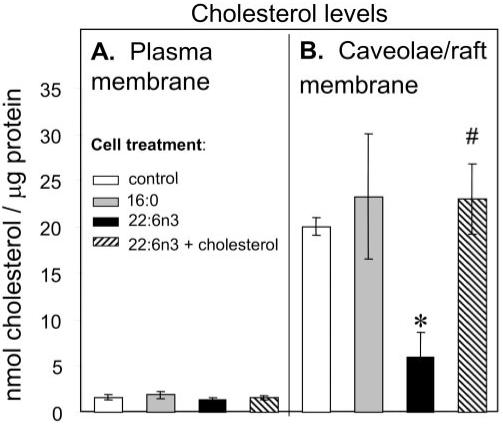
DHA22:6,n3 treatment depletes cholesterol in caveolae/lipid rafts. hRVE cells were treated with 100 μM palmitic acid16:0 (gray bars) or DHA22:6,n3 (black bars) for 24 hours. Vehicle (20 μM BSA)–treated cells were used as a control (white bars). Water-soluble cholesterol (25 μM cholesterol complexed with 250 μM MCD) was added to DHA22:6,n3-treated cells for 30 minutes (striped bars). Caveolae/lipid raft–enriched domains were purified as described in Experimental Procedures and submitted to total lipid extraction and amino-propyl column fractionation. Neutral lipids were fractionated by normal-phase HPLC analyses. The amount of cholesterol was presented as nmol/μg protein. Data are the mean ± SD of results in three experiments. *P < 0.05 compared with control. #P < 0.05 compared with DHA22:6,n3-treated cells without cholesterol.
Inhibition of TNF-α– and IL-1β–Induced CAM Expression by Cholesterol Depletion
The cholesterol-depleting agent MCD, known to disrupt the structure of caveolae/lipid rafts, was used to assess the role of caveolae/lipid rafts in cytokine-induced CAM expression in retinal endothelial cells. MCD pretreatment of hRVE cells downregulated TNF-α–induced phosphorylation of inhibitor of NF-kappa B alpha (IκBα) (Fig. 5A) and ICAM-1 expression (Fig. 5B). In contrast, TNF-α–triggered ERK phosphorylation was not affected. A similar effect was observed with IL-1β–mediated IκBβ phosphorylation and ICAM-1 expression in response to MCD treatment (data not shown). These data suggest that the integrity of caveolae/lipid rafts is required for cytokine–induced NF-κB signaling and downstream cell adhesion molecules (CAM) expression.
FIGURE 5.
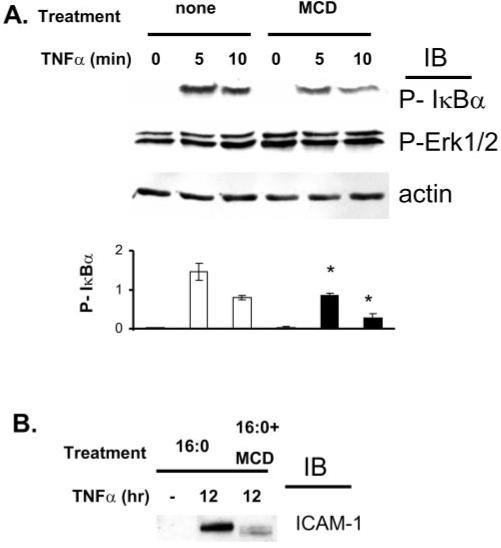
Inhibition of TNF-α–induced NF-κB signaling by (MCD) pretreatment of hRVE cells. hRVE cells were pretreated with 8 mM MCD for 30 minutes before stimulation with TNF-α (20 ng/mL) for the indicated time. IκBα and ERK phosphorylation levels (A) and ICAM-1 expression levels (B) were assessed by immunoblot analyses. Equal amounts of protein were added to each lane, as confirmed by actin levels. Data are the mean ± SD of results in three experiments. *P < 0.05.
Reversal of DHA22:6,n3 Inhibition of TNF-α–Induced ICAM-1 Expression by Cholesterol Replenishment
As we have previously reported,48 the pretreatment of hRVE cells with DHA22:6,n3 significantly inhibits TNF-α–induced ICAM-1 expression compared with TNF-α–induced ICAM-1 expression in control (palmitic16:0 acid–treated) cells (Fig. 6). Replenishing cholesterol reverses the inhibitory effect of DHA22:6,n3 on TNF-α–induced ICAM-1 expression (Fig. 6).
FIGURE 6.
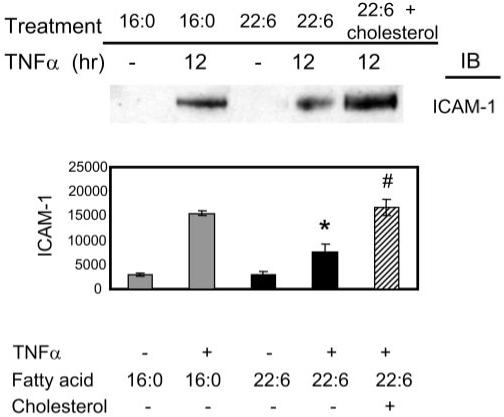
Cholesterol replenishment in caveolae/lipid rafts reverses DHA22:6,n3 inhibition of TNF-α induced ICAM-1 expression. hRVE cells treated with 100 μM DHA22:6,n3 (black bars) for 12 hours followed by TNF-α stimulation for 12 hours. Cells treated with palmitic acid16:0 were used as a control (gray bars). Water-soluble cholesterol (25 μM cholesterol complexed with 250 μM MCD) was added to DHA22:6,n3-treated cells for 30 minutes before TNF-α stimulation (striped bar). ICAM-1 expression levels were assessed by immunoblot analyses. Equal amounts of protein were added to each lane, as confirmed by actin levels. Data are the mean ± SD of results in three experiments, *P < 0.05 compared with palmitic acid16:0 treated cells. #P < 0.05 compared with DHA22:6,n3-treated cells without cholesterol.
Src Family Kinase Displacement from Caveolae/Lipid Rafts in Endothelial Cells in Response to DHA22:6,n3-Induced Change in Lipid Composition
To determine whether DHA-induced changes in lipid composition would lead to the alteration of caveolae/lipid raft signaling components, we first characterized Src family kinase localization in hRVE cells. The dual-acylated Src family kinases Fyn and c-Yes were exclusively localized in caveolae/lipid rafts, whereas only myristoylated c-Src was excluded (Fig. 7). To examine whether the inhibition of SFKs localized in caveolae/lipid rafts would lead to a decrease of cytokine-induced inflammatory response, PP2, a specific inhibitor of SFKs (c-Src, Fyn, c-Yes) was used (Fig. 8). PP2 diminished TNF-α–induced VCAM-1 expression in hRVE cells (Fig. 8), suggesting that SFKs localized in caveolae/lipid rafts could be important players in the cytokine-induced inflammatory response.
FIGURE 7.
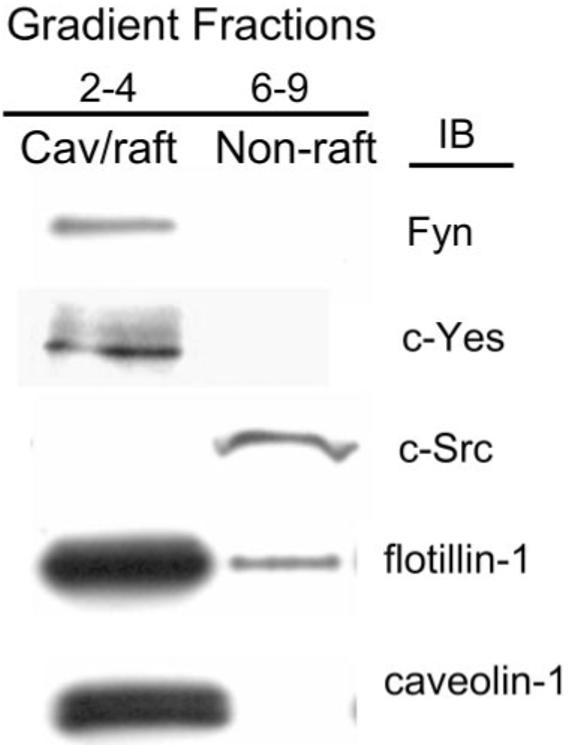
Src family kinase enrichment in hRVE caveolae/lipid rafts. Caveolae/lipid raft fractions were purified, as described in Materials and Methods. Fractions 2 to 4 were combined and designated the caveolae/lipid raft (cav/raft) fractions, and fractions 5 to 9 were combined and designated the nonraft membrane fraction. Fractions were separated on SDS-PAGE and analyzed by Western blot. Results are representative of seven experiments.
FIGURE 8.
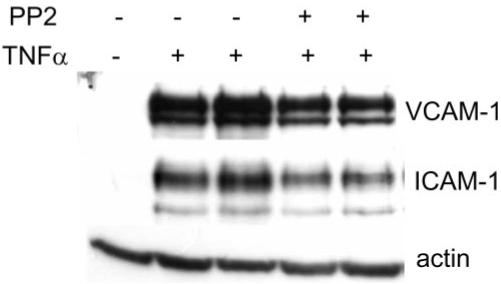
Effect of SFK inhibition on TNF-α–induced VCAM-1 expression. hRVE cells were pretreated with PP2 (10 μM) for 30 minutes and stimulated with TNF-α (5 ng/mL) for 6 hours. VCAM-1 and ICAM-1 expression levels were assessed by immunoblot analyses. Equal amounts of protein were added to each lane, as confirmed by actin levels. Results are representative of three experiments.
We next determined whether DHA22:6,n3 had effects on SFK interaction with caveolae/lipid rafts. Treatment of hRVE cells with 100 μM DHA22:6,n3 significantly enriched lipid raft DHA22:6,n3 content (Fig. 3). DHA22:6,n3 enrichment in the raft correlated with a 60% displacement of Fyn and a 35% displacement of c-Yes from the caveolae/lipid raft fraction (Fig. 9). DHA22:6,n3 treatment did not disrupt caveolae/lipid raft structure because the major structural proteins of caveolae and lipid rafts—caveolin-1 and flotillin-1—were not affected by the increased enrichment with DHA22:6,n3. Thus, DHA22:6,n3 enrichment of caveolae/rafts depletes cholesterol and at least two SFKs, Fyn and c-Yes, from these structures.
FIGURE 9.
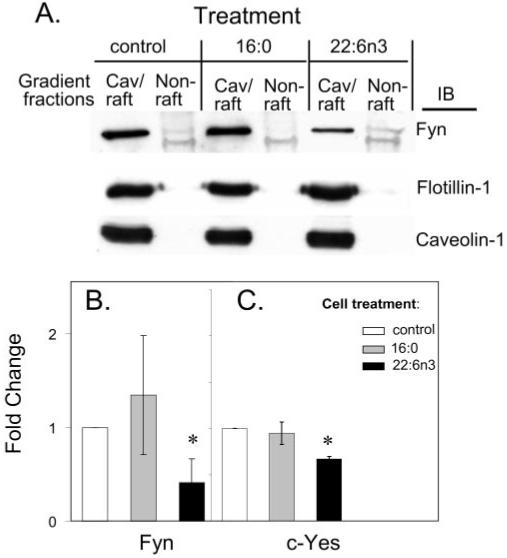
DHA22:6,n3 displaces Fyn and c-Yes from hRVE caveolae/lipid rafts. (A) hRVE cells were treated with 100 μM palmitic acid16:0 or DHA22:6,n3 for 24 hours. Vehicle (20 μM BSA)-treated cells were used as a control. Caveolae/lipid raft fractions were isolated and analyzed by Western blot. Amounts of Fyn (B) and c-Yes (C), localized in caveolae/lipid rafts isolated from fatty acid–treated cells, were quantified and presented as a fold change. Data are the mean ± SD of results in three experiments. *P < 0.05.
DISCUSSION
Ample experimental evidence suggests that early diabetic retinopathy is a low-grade chronic inflammatory disease.49-56 Under diabetic conditions, retinal endothelial cells were shown to be activated to produce adhesion molecules such as ICAM-1 and VCAM-1. Adhesion molecules interact with their counterpart receptors on activated leukocytes to govern the adherence and transmigration of leukocytes to the endothelium and to mediate inflammation. Although n-3 PUFAs suppress cytokine induced adhesion molecule expression in endothelial cells,48,57-61 the functional mechanism for this suppression is not well understood. Our results demonstrate that the anti-inflammatory effect of the most abundant n-3 PUFA in the retina, DHA22:6,n3, on retinal endothelial cells may be mediated through modification of the important membrane signaling microdomains, caveolae/lipid rafts.
Caveolae/lipids rafts are important in mediating cytokine-induced inflammatory signaling.31,62-66 Several signaling molecules implicated in the development of diabetic retinopathy, such as VEGFR2,27,28,67 TNF-R1,30,31 and IL-2R,68 have been shown to localize to the caveolae/lipid raft fractions in various cell types, including endothelial cells. Diabetes and hypertension lead to increased caveolin-1 expression in retinal pericytes and endothelial cells.69 Moreover, hyperglycemia has been shown to induce caveolin-1 expression in a bovine aortic endothelial cell model.70 We have previously demonstrated that DHA22:6,n3 inhibits IL-1β–, TNF-α–, and VEGF-induced inflammatory signaling through inhibition of the NF-κB pathway upstream of IκBα phosphorylation and degradation in hRVE cells.48 The data provided in this study show that the lipid integrity of caveolae/lipid rafts is related to the intactness of cytokine-induced NF-κB activation (Fig. 5). Thus, DHA22:6,n3 could oppose hyperglycemia-induced proinflammatory changes by modifying caveolae/lipid raft structure and function in the diabetic retina.
The Src family kinase inhibitor PP2 inhibited the cytokine-induced expression of adhesion molecules, implicating the involvement of Src kinases in cytokine-induced inflammatory signaling. In fact, a growing body of literature has demonstrated the requirement of SFKs in TNF receptor–mediated34,35 and IL-1β receptor–mediated36,37 inflammatory signaling. The recruitment of specific SFKs to VEGFR2 on VEGF binding was also documented in several cell types.38-41 Although the detailed evaluation of the role of Src family kinases in cytokine signaling is beyond the scope of this study, we used Src family kinases as a model to study the effect of DHA22:6,n3 on signaling components of the caveolae/lipid rafts in hRVE cells.
In agreement with previous studies, dual-acylated SFKs such as Fyn and c-Yes were localized to the caveolae/lipid raft fraction. c-Srk itself, which is only myristoylated because it is missing the cysteine3 palmitoylation site, was excluded from caveolae/lipid rafts.32,33 Thus, the highly ordered lipid structure of the caveolae/raft and the posttranslational modification of proteins that direct protein into or out of rafts are likely important regulatory features controlling signal transduction. In this study, we demonstrate that DHA22:6,n3 enrichment in hRVE cells is associated with displacement of SFKs Fyn and c-Yes from caveolae/lipid rafts.
Altered lipid environment and altered protein acylation are two possible routes by which PUFA can affect protein targeting to lipid rafts. In hRVE cells, DHA22:6,n3 treatment resulted in its incorporation into phospholipids in the caveolae/lipid rafts. These data are in agreement with reports in T lymphocytes, suggesting that the substitution of fatty acyl chains by PUFA in phospholipids of lipid rafts could change the lipid environment and thus affect the association of proteins dually acylated by saturated fatty acids.9,10,13 Moreover, in vivo feeding studies demonstrated increased n-3 PUFAs in the lipid rafts from T lymphocytes and colons of mice fed a diet enriched in n-3 PUFAs collectively with displacement of important signaling molecules, such as Ras, caveolin-1, and eNOS.5,71,72
DHA22:6,n3 incorporated primarily into the phosphatidylcholine fraction of phospholipids in hRVE cells. This is different from the reports in T cells showing EPA20:5n3 and DHA22:6,n3 incorporating into phosphatidylcholine and phosphatidylethanolamine,9,73 underscoring the difference in lipid metabolism between retinal vascular endothelial cells and other cell types. Substitution of phospholipid fatty acyl chains by DHA22:6,n3 in total plasma membranes was much higher than in caveolae/lipid rafts, suggesting that cells tend to maintain the higher lipid-ordered structure of caveolae/lipid rafts by maintaining a saturated fatty acyl environment. Nevertheless, incorporation of DHA22:6,n3 resulted in a considerable (35%) increase in unsaturation in acyl chains of neutral phospholipids in caveolae/lipid rafts of DHA22:6,n3-treated hRVE cells (Fig. 3B).
The enrichment of DHA22:6,n3 in raft phospholipids is associated with a decline in arachidonic acid20:4,n6 (Fig. 3). When released from membranes, arachidonic acid20:4,n6 serves as a precursor of inflammatory mediators, including leukotrienes, thromboxanes, and prostaglandins, and other bioactive lipid mediators, such as hydroxy and epoxy fatty acids.74-76 Our previous studies indicated that treatment of hRVE cells with arachidonic acid20:4,n6 leads to a lipoxygenase-dependent increase in ICAM/VCAM expression.43 This effect of arachidonic acid20:4,n6 was observed after 12-hour treatment of hRVE cells. The fact that the effects of cholesterol depletion/replenishment on NF-κB signaling and adhesion molecule expression are obvious in 30 minutes argues against a decline in arachidonic acid20:4,n6 as a primary mechanism for the inhibitory effect of arachidonic acid20:4,n6 on caveolae/lipid raft signaling. However, a reduction of arachidonic acid20:4,n6 phospholipid content by DHA22:6,n3 could represent an additional mechanism of anti-inflammatory effect of DHA22:6,n3 in hRVE cells.
Enrichment of phospholipids with DHA22:6,n3 dramatically altered the lipid environment in caveolae/lipid rafts. This is particularly evident in the 70% decrease in caveolae/lipid raft–associated cholesterol. The effect of DHA22:6,n3 on raft cholesterol is specific because significant changes in the overall plasma membrane cholesterol content were not detected (Fig. 4). Cholesterol depletion using MCD mimicked the inhibitory effects on DHA22:6,n3 on cytokine-induced IκBα phosphorylation and ICAM-1 expression. Moreover, cholesterol replenishment reversed the inhibitory effect of DHA22:6,n3 on cytokine signaling in hRVE cells.
Cholesterol is the major structural lipid in caveolae/lipid rafts that is required to maintain the ordered state of the raft membrane. Cholesterol sterol rings tightly bind the ceramide moiety of sphingomyelin in the exoplasmic leaflet of caveolae/lipid rafts, promoting caveolae/lipid raft lateral separation.77-79 In the cytoplasmic leaflet that lacks sphingolipids, the interaction of phospholipid-saturated acyl chains with cholesterol is required to maintain organization of caveolae/lipid rafts in a liquid-ordered phase.77,80 Cholesterol interacts differentially with different membrane lipids and has a particularly strong association with saturated phospholipids and sphingolipids and a weak association with highly unsaturated lipid species (for a review, see Silvius21). Thus, substitutions of acyl chains of the phospholipids by DHA22:6,n3 could dramatically affect cholesterol interaction and cause a decreased lipid order in the caveolae/lipid rafts. The mechanism of specific cholesterol depletion in caveolae/lipid rafts induced by DHA22:6,n3 enrichment is unclear. It may involve a spontaneous redistribution between membranes because of changes of lipid environment in caveolae/lipid rafts.21
Decreases in the sphingomyelin content have also been reported in lipid rafts isolated from T lymphocytes and the colons of mice fed a diet enriched in n3-PUFA.5,71,72 Treating cells with sphingomyelinase, which cleaves the phosphorylcholine head group of sphingomyelin, leading to ceramide production, causes a significant displacement of cholesterol from lipid raft membranes and a modification of lipid raft signaling.81-83 Because DHA22:6,n3 was shown to activate neutral sphingomyelinase in cultured MDA-MB-231 breast cancer cells,84 the activation of sphingomyelinase followed by ceramide production could represent a potential mechanism for the DHA22:6,n3-induced cholesterol depletion from caveolae/lipid raft reported here. Sphingomyelinase activity and sphingomyelin and ceramide content were not analyzed in this article and will be the focus of future study.
As reported in Cos-1 cells, Fyn can be acylated by fatty acids other than myristate and palmitate.7 Acylation of Fyn by unsaturated fatty acids caused the displacement of Fyn from membrane rafts in Cos-1 cells.7 Determining whether SFKs can be acylated by DHA22:6,n3, which would provide an additional mechanism for selective displacement of Fyn and c-Yes from caveolae/lipid rafts in endothelial cells, is beyond the scope of this study.
In conclusion, we have characterized the involvement of caveolae/lipid rafts in mediating cytokine-induced proinflammatory signaling in primary cultures of retinal vascular endothelial cells. The modification of phospholipids residing in the caveolae/lipid rafts and the depletion of caveolae/lipid raft-specific cholesterol provide a compelling model to explain the molecular mechanism of DHA22:6,n3-induced displacement of Src family kinases and thus the immunomodulatory effect of DHA22:6,n3 on endothelial cells.
Acknowledgments
The authors thank Leslie A. Dybas and Todd A. Lydic for technical assistance.
Supported by National Eye Institute Grant EY016077, Juvenile Diabetes Research Foundation Grant 2−2005−97 (JVB), National Institute of Diabetes and Digestive and Kidney Diseases Grant DK43220, U.S. Department of Agriculture Grant 2003−5200−3400, and the Michigan Agriculture Experimental Station (DBJ).
Footnotes
Disclosure: W. Chen, None; D.B. Jump, None; W.J. Esselman, None; J.V. Busik, None
References
- 1.Lee TH, Hoover RL, Williams JD, et al. Effect of dietary enrichment with eicosapentaenoic and docosahexaenoic acids on in vitro neutrophil and monocyte leukotriene generation and neutrophil function. N Engl J Med. 1985;312:1217–1224. doi: 10.1056/NEJM198505093121903. [DOI] [PubMed] [Google Scholar]
- 2.Trebble T, Arden NK, Stroud MA, et al. Inhibition of tumour necrosis factor-alpha and interleukin 6 production by mononuclear cells following dietary fish-oil supplementation in healthy men and response to antioxidant co-supplementation. Br J Nutr. 2003;90:405–412. doi: 10.1079/bjn2003892. [DOI] [PubMed] [Google Scholar]
- 3.Diaz O, Berquand A, Dubois M, et al. The mechanism of docosahexaenoic acid induced phospholipase D activation in human lymphocytes involves exclusion of the enzyme from lipid rafts. J Biol Chem. 2002;277:39368–39378. doi: 10.1074/jbc.M202376200. [DOI] [PubMed] [Google Scholar]
- 4.Fan YY, Ly LH, Barhoumi R, McMurray DN, Chapkin RS. Dietary docosahexaenoic acid suppresses T cell protein kinase C-theta lipid raft recruitment and IL-2 production. J Immunol. 2004;173:6151–6160. doi: 10.4049/jimmunol.173.10.6151. [DOI] [PubMed] [Google Scholar]
- 5.Fan YY, McMurray DN, Ly LH, Chapkin RS. Dietary (n-3) polyunsaturated fatty acids remodel mouse T-cell lipid rafts. J Nutr. 2003;133:1913–1920. doi: 10.1093/jn/133.6.1913. [DOI] [PubMed] [Google Scholar]
- 6.Lee JY, Plakidas A, Lee WH, et al. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res. 2003;44:479–486. doi: 10.1194/jlr.M200361-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Liang X, Nazarian A, Erdjument-Bromage H, et al. Heterogeneous fatty acylation of Src family kinases with polyunsaturated fatty acids regulates raft localization and signal transduction. J Biol Chem. 2001;276:30987–30994. doi: 10.1074/jbc.M104018200. [DOI] [PubMed] [Google Scholar]
- 8.Stulnig TM. Immunomodulation by polyunsaturated fatty acids: mechanisms and effects. Int Arch Allergy Immunol. 2003;132:310–321. doi: 10.1159/000074898. [DOI] [PubMed] [Google Scholar]
- 9.Stulnig TM, Huber J, Leitinger N, et al. Polyunsaturated eicosapentaenoic acid displaces proteins from membrane rafts by altering raft lipid composition. J Biol Chem. 2001;276:37335–37340. doi: 10.1074/jbc.M106193200. [DOI] [PubMed] [Google Scholar]
- 10.Zeyda M, Staffler G, Horejsi V, Waldhausl W, Stulnig TM. LAT displacement from lipid rafts as a molecular mechanism for the inhibition of T cell signaling by polyunsaturated fatty acids. J Biol Chem. 2002;277:28418–28423. doi: 10.1074/jbc.M203343200. [DOI] [PubMed] [Google Scholar]
- 11.Zeyda M, Szekeres AB, Saemann MD, et al. Suppression of T cell signaling by polyunsaturated fatty acids: selectivity in inhibition of mitogen-activated protein kinase and nuclear factor activation. J Immunol. 2003;170:6033–6039. doi: 10.4049/jimmunol.170.12.6033. [DOI] [PubMed] [Google Scholar]
- 12.Chen W, Esselman WJ, Jump DB, Busik JV. Anti-inflammatory effect of docosahexaenoic acid on cytokine-induced adhesion molecule expression in human retinal vascular endothelial cells. Invest Ophthalmol Vis Sci. 2005;46:4342–4347. doi: 10.1167/iovs.05-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stulnig TM, Berger M, Sigmund T, et al. Polyunsaturated fatty acids inhibit T cell signal transduction by modification of detergent-insoluble membrane domains. J Cell Biol. 1998;143:637–644. doi: 10.1083/jcb.143.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–467. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 15.Anderson RG. Caveolae: where incoming and outgoing messengers meet. Proc Natl Acad Sci USA. 1993;90:10909–10913. doi: 10.1073/pnas.90.23.10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson RG, Jacobson K. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science. 2002;296:1821–1825. doi: 10.1126/science.1068886. [DOI] [PubMed] [Google Scholar]
- 17.Smart EJ, Graf GA, McNiven MA, et al. Caveolins, liquid-ordered domains, and signal transduction. Mol Cell Biol. 1999;19:7289–7304. doi: 10.1128/mcb.19.11.7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Almeida RF, Loura LM, Fedorov A, Prieto M. Lipid rafts have different sizes depending on membrane composition: a time-resolved fluorescence resonance energy transfer study. J Mol Biol. 2005;346:1109–1120. doi: 10.1016/j.jmb.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 19.Douglass AD, Vale RD. Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell. 2005;121:937–950. doi: 10.1016/j.cell.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaikh SR, Cherezov V, Caffrey M, Stillwell W, Wassall SR. Interaction of cholesterol with a docosahexaenoic acid-containing phosphatidylethanolamine: trigger for microdomain/raft formation? Biochemistry. 2003;42:12028–12037. doi: 10.1021/bi034931+. [DOI] [PubMed] [Google Scholar]
- 21.Silvius JR. Role of cholesterol in lipid raft formation: lessons from lipid model systems. Biochim Biophys Acta. 2003;1610:174–183. doi: 10.1016/s0005-2736(03)00016-6. [DOI] [PubMed] [Google Scholar]
- 22.Subczynski WK, Kusumi A. Dynamics of raft molecules in the cell and artificial membranes: approaches by pulse EPR spin labeling and single molecule optical microscopy. Biochim Biophys Acta. 2003;1610:231–243. doi: 10.1016/s0005-2736(03)00021-x. [DOI] [PubMed] [Google Scholar]
- 23.Edidin M. The state of lipid rafts: from model membranes to cells. Annu Rev Biophys Biomol Struct. 2003;32:257–283. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- 24.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 25.Viola A, Schroeder S, Sakakibara Y, Lanzavecchia A. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science. 1999;283:680–682. doi: 10.1126/science.283.5402.680. [DOI] [PubMed] [Google Scholar]
- 26.Simons K, Vaz WL. Model systems, lipid rafts, and cell membranes. Annu Rev Biophys Biomol Struct. 2004;33:269–295. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- 27.Cho CH, Lee CS, Chang MK, et al. Localization of VEGFR-2 and phospholipase D2 in endothelial caveolae is involved in VEGF-induced phosphorylation of MEK and ERK. Am J Physiol Heart Circ Physiol. 2004;286:H1881–H1888. doi: 10.1152/ajpheart.00786.2003. [DOI] [PubMed] [Google Scholar]
- 28.Feng Y, Venema VJ, Venema RC, et al. VEGF-induced permeability increase is mediated by caveolae. Invest Ophthalmol Vis Sci. 1999;40:157–167. [PubMed] [Google Scholar]
- 29.Foster LJ, De Hoog CL, Mann M. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc Natl Acad Sci USA. 2003;100:5813–5818. doi: 10.1073/pnas.0631608100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ko YG, Lee JS, Kang YS, Ahn JH, Seo JS. TNF-alpha–mediated apoptosis is initiated in caveolae-like domains. J Immunol. 1999;162:7217–7223. [PubMed] [Google Scholar]
- 31.Legler DF, Micheau O, Doucey MA, Tschopp J, Bron C. Recruitment of TNF-receptor 1 to lipid rafts is essential for TNF-alpha-mediated NF-kappaB activation. Immunity. 2003;18:655–664. doi: 10.1016/s1074-7613(03)00092-x. [DOI] [PubMed] [Google Scholar]
- 32.Shenoy-Scaria AM, Dietzen DJ, Kwong J, Link DC, Lublin DM. Cysteine3 of Src family protein tyrosine kinase determines palmitoylation and localization in caveolae. J Cell Biol. 1994;126:353–363. doi: 10.1083/jcb.126.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shenoy-Scaria AM, Gauen LK, Kwong J, Shaw AS, Lublin DM. Palmitylation of an amino-terminal cysteine motif of protein tyrosine kinases p56lck and p59fyn mediates interaction with glycosyl-phosphatidylinositol anchored proteins. Mol Cell Biol. 1993;13:6385–6392. doi: 10.1128/mcb.13.10.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xing L, Venegas AM, Chen A, et al. Genetic evidence for a role for Src family kinases in TNF- family receptor signaling and cell survival. Genes Dev. 2001;15:241–253. doi: 10.1101/gad.840301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Vliet C, Bukczynska PE, Puryer MA, et al. Selective regulation of tumor necrosis factor-induced Erk signaling by Src family kinases and the T cell protein tyrosine phosphatase. Nat Immunol. 2005;6:253–260. doi: 10.1038/ni1169. [DOI] [PubMed] [Google Scholar]
- 36.Viviani B, Bartesaghi S, Gardoni F, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.al-Ramadi BK, Welte T, Fernandez-Cabezudo MJ, et al. The Src-protein tyrosine kinase Lck is required for IL-1-mediated costimulatory signaling in Th2 cells. J Immunol. 2001;167:6827–6833. doi: 10.4049/jimmunol.167.12.6827. [DOI] [PubMed] [Google Scholar]
- 38.Chou MT, Wang J, Fujita DJ. Src kinase becomes preferentially associated with the VEGFR, KDR/Flk-1, following VEGF stimulation of vascular endothelial cells. BMC Biochem. 2002;3:32. doi: 10.1186/1471-2091-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Munshi N, Groopman JE, Gill PS, Ganju RK. c-Src mediates mitogenic signals and associates with cytoskeletal proteins upon vascular endothelial growth factor stimulation in Kaposi’s sarcoma cells. J Immunol. 2000;164:1169–1174. doi: 10.4049/jimmunol.164.3.1169. [DOI] [PubMed] [Google Scholar]
- 40.He H, Venema VJ, Gu X, et al. Vascular endothelial growth factor signals endothelial cell production of nitric oxide and prostacyclin through flk-1/KDR activation of c-Src. J Biol Chem. 1999;274:25130–25135. doi: 10.1074/jbc.274.35.25130. [DOI] [PubMed] [Google Scholar]
- 41.Weis S, Cui J, Barnes L, Cheresh D. Endothelial barrier disruption by VEGF mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol. 2004;167:223–229. doi: 10.1083/jcb.200408130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang YY, Walker JL, Huang A, et al. Expression of 5-lipoxygenase in pulmonary artery endothelial cells. Biochem J. 2002;361(pt 2):267–276. doi: 10.1042/0264-6021:3610267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen W, Jump DB, Grant MB, Esselman WJ, Busik JV. Dyslipidemia, but not hyperglycemia, induces inflammatory adhesion molecules in human retinal vascular endothelial cells. Invest Ophthalmol Vis Sci. 2003;44:5016–5022. doi: 10.1167/iovs.03-0418. [DOI] [PubMed] [Google Scholar]
- 44.Artwohl M, Roden M, Waldhausl W, Freudenthaler A, Baumgartner-Parzer SM. Free fatty acids trigger apoptosis and inhibit cell cycle progression in human vascular endothelial cells. FASEB J. 2004;18:146–148. doi: 10.1096/fj.03-0301fje. [DOI] [PubMed] [Google Scholar]
- 45.Busik JV, Olson LK, Grant MB, Henry DN. Glucose-induced activation of glucose uptake in cells from the inner and outer blood-retinal barrier. Invest Ophthalmol Vis Sci. 2002;43:2356–2363. [PubMed] [Google Scholar]
- 46.Lisanti MP, Sargiacomo M, Scherer PE. Purification of caveolae-derived membrane microdomains containing lipid-anchored signaling molecules, such as GPI-anchored proteins, H-Ras, Src-family tyrosine kinases, eNOS, and G-protein alpha-, beta-, and gamma-subunits. Methods Mol Biol. 1999;116:51–60. doi: 10.1385/1-59259-264-3:51. [DOI] [PubMed] [Google Scholar]
- 47.Kim HY, Salem N., Jr. Separation of lipid classes by solid phase extraction. J Lipid Res. 1990;31:2285–2289. [PubMed] [Google Scholar]
- 48.Chen W, Esselman WJ, Jump DB, Busik JV. Anti-inflammatory effect of docosahexaenoic acid (DHA) on cytokine induced adhesion molecule expression in human retinal vascular endothelial cells. Invest Ophthalmol Vis Sci. 2005;46:4342–4347. doi: 10.1167/iovs.05-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schroder S, Palinski W, Schmid-Schonbein GW. Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am J Pathol. 1991;139:81–100. [PMC free article] [PubMed] [Google Scholar]
- 50.Joussen AM, Poulaki V, Mitsiades N, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002;16:438–440. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 51.Joussen AM, Murata T, Tsujikawa A, et al. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158:147–152. doi: 10.1016/S0002-9440(10)63952-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McLeod DS, Lefer DJ, Merges C, Lutty GA. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. 1995;147:642–653. [PMC free article] [PubMed] [Google Scholar]
- 53.Miyamoto K, Khosrof S, Bursell SE, et al. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci USA. 1999;96:10836–10841. doi: 10.1073/pnas.96.19.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Joussen AM, Poulaki V, Qin W, et al. Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol. 2002;160:501–509. doi: 10.1016/S0002-9440(10)64869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adamis AP. Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol. 2002;86:363–365. doi: 10.1136/bjo.86.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53:2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]
- 57.Chaudhary A, Mishra A, Sethi S. Oxidized omega-3 fatty acids inhibit proinflammatory responses in glomerular endothelial cells. Nephron Exp Nephrol. 2004;97:e136–e145. doi: 10.1159/000079178. [DOI] [PubMed] [Google Scholar]
- 58.De Caterina R, Bernini W, Carluccio MA, Liao JK, Libby P. Structural requirements for inhibition of cytokine-induced endothelial activation by unsaturated fatty acids. J Lipid Res. 1998;39:1062–1070. [PubMed] [Google Scholar]
- 59.De Caterina R, Liao JK, Libby P. Fatty acid modulation of endothelial activation. Am J Clin Nutr. 2000;71(suppl):213S–223S. doi: 10.1093/ajcn/71.1.213S. [DOI] [PubMed] [Google Scholar]
- 60.Kim I, Moon SO, Kim SH, et al. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J Biol Chem. 2001;276:7614–7620. doi: 10.1074/jbc.M009705200. [DOI] [PubMed] [Google Scholar]
- 61.Mishra A, Chaudhary A, Sethi S. Oxidized omega-3 fatty acids inhibit NFkappaB activation via a PPARalpha-dependent pathway. Arterioscler Thromb Vasc Biol. 2004;24:1621–1627. doi: 10.1161/01.ATV.0000137191.02577.86. [DOI] [PubMed] [Google Scholar]
- 62.Gaide O, Favier B, Legler DF, et al. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat Immunol. 2002;3:836–843. doi: 10.1038/ni830. [DOI] [PubMed] [Google Scholar]
- 63.Kiely JM, Hu Y, Garcia-Cardena G, Gimbrone MA., Jr. Lipid raft localization of cell surface E-selectin is required for ligation-induced activation of phospholipase C gamma. J Immunol. 2003;171:3216–33224. doi: 10.4049/jimmunol.171.6.3216. [DOI] [PubMed] [Google Scholar]
- 64.Marmor MD, Julius M. Role for lipid rafts in regulating interleukin-2 receptor signaling. Blood. 2001;98:1489–1497. doi: 10.1182/blood.v98.5.1489. [DOI] [PubMed] [Google Scholar]
- 65.Monastyrskaya K, Hostettler A, Buergi S, Draeger A. The NK1 receptor localizes to the plasma membrane microdomains, and its activation is dependent on lipid raft integrity. J Biol Chem. 2005;280:7135–7146. doi: 10.1074/jbc.M405806200. [DOI] [PubMed] [Google Scholar]
- 66.Patel VP, Moran M, Low TA, Miceli MC. A molecular framework for two-step T cell signaling: Lck Src homology 3 mutations discriminate distinctly regulated lipid raft reorganization events. J Immunol. 2001;166:754–764. doi: 10.4049/jimmunol.166.2.754. [DOI] [PubMed] [Google Scholar]
- 67.Labrecque L, Royal I, Surprenant DS, et al. Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol Biol Cell. 2003;14:334–347. doi: 10.1091/mbc.E02-07-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vamosi G, Bodnar A, Vereb G, et al. IL-2 and IL-15 receptor alpha-subunits are coexpressed in a supramolecular receptor cluster in lipid rafts of T cells. Proc Natl Acad Sci USA. 2004;101:11082–11087. doi: 10.1073/pnas.0403916101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hillman N, Cox S, Noble AR, Gallagher PJ. Increased numbers of caveolae in retinal endothelium and pericytes in hypertensive diabetic rats. Eye. 2001;15(pt 3):319–325. doi: 10.1038/eye.2001.103. [DOI] [PubMed] [Google Scholar]
- 70.Bucci M, Roviezzo F, Brancaleone V, et al. Diabetic mouse angiopathy is linked to progressive sympathetic receptor deletion coupled to an enhanced caveolin-1 expression. Arterioscler Thromb Vasc Biol. 2004;24:721–726. doi: 10.1161/01.ATV.0000122362.44628.09. [DOI] [PubMed] [Google Scholar]
- 71.Ma DW, Seo J, Davidson LA, et al. n-3 PUFA alter caveolae lipid composition and resident protein localization in mouse colon. FASEB J. 2004;18:1040–1042. doi: 10.1096/fj.03-1430fje. [DOI] [PubMed] [Google Scholar]
- 72.Ma DW, Seo J, Switzer KC, et al. n-3 PUFA and membrane microdomains: a new frontier in bioactive lipid research. J Nutr Biochem. 2004;15:700–706. doi: 10.1016/j.jnutbio.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 73.Li Q, Tan L, Wang C, et al. Polyunsaturated eicosapentaenoic acid changes lipid composition in lipid rafts. Eur J Nutr. 2006;45:144–151. doi: 10.1007/s00394-005-0574-7. [DOI] [PubMed] [Google Scholar]
- 74.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 75.Hwang D. Fatty acids and immune responses—a new perspective in searching for clues to mechanism. Annu Rev Nutr. 2000;20:431–456. doi: 10.1146/annurev.nutr.20.1.431. [DOI] [PubMed] [Google Scholar]
- 76.Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276:36059–36062. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- 77.Brown DA, London E. Structure of detergent-resistant membrane domains: does phase separation occur in biological membranes? Biochem Biophys Res Commun. 1997;240:1–7. doi: 10.1006/bbrc.1997.7575. [DOI] [PubMed] [Google Scholar]
- 78.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 79.Gulbins E, Li PL. Physiological and pathophysiological aspects of ceramide. Am J Physiol Regul Integr Comp Physiol. 2006;290:R11–R26. doi: 10.1152/ajpregu.00416.2005. [DOI] [PubMed] [Google Scholar]
- 80.Schroeder R, London E, Brown D. Interactions between saturated acyl chains confer detergent resistance on lipids and glycosylphos-phatidylinositol (GPI)-anchored proteins: GPI-anchored proteins in liposomes and cells show similar behavior. Proc Natl Acad Sci USA. 1994;91:12130–12134. doi: 10.1073/pnas.91.25.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ito J, Nagayasu Y, Yokoyama S. Cholesterol-sphingomyelin interaction in membrane and apolipoprotein-mediated cellular cholesterol efflux. J Lipid Res. 2000;41:894–904. [PubMed] [Google Scholar]
- 82.Megha, London E. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): implications for lipid raft structure and function. J Biol Chem. 2004;279:9997–10004. doi: 10.1074/jbc.M309992200. [DOI] [PubMed] [Google Scholar]
- 83.Yu C, Alterman M, Dobrowsky RT. Ceramide displaces cholesterol from lipid rafts and decreases the association of the cholesterol binding protein caveolin-1. J Lipid Res. 2005;46:1678–1691. doi: 10.1194/jlr.M500060-JLR200. [DOI] [PubMed] [Google Scholar]
- 84.Wu M, Harvey KA, Ruzmetov N, et al. Omega-3 polyunsaturated fatty acids attenuate breast cancer growth through activation of a neutral sphingomyelinase-mediated pathway. Int J Cancer. 2005;117:340–348. doi: 10.1002/ijc.21238. [DOI] [PubMed] [Google Scholar]