

Abstract
A common feature of many human neurodegenerative diseases is the accumulation of insoluble ubiquitin-containing protein aggregates in the CNS. Although Drosophila has been helpful in understanding several human neurodegenerative disorders, a loss-of-function mutation has not been identified that leads to insoluble CNS protein aggregates. The study ofDrosophila mutations may identify unique components that are associated with human degenerative diseases. The Drosophila blue cheese (bchs) gene defines such a novel degenerative pathway. bchs mutants have a reduced adult life span with the age-dependent formation of protein aggregates throughout the neuropil of the CNS. These inclusions contain insoluble ubiquitinated proteins and amyloid precursor-like protein. Progressive loss of CNS size and morphology along with extensive neuronal apoptosis occurs in aged bchs mutants. BCHS protein is widely expressed in the cytoplasm of CNS neurons and is present over the entire length of axonal projections. BCHS is nearly 3500 amino acids in size, with the last 1000 amino acids consisting of three functional protein motifs implicated in vesicle transport and protein processing. This region along with previously unidentified proteins encoded in the human, mouse, and nematode genomes shows striking homology along the full length of the BCHS protein. The high degree of conservation between Drosophila and human bchssuggests that study of the functional pathway of BCHS and associated mutant phenotype may provide useful insights into human neurodegenerative disorders.
Keywords: neurodegeneration, ubiquitin, APPL, protein aggregates, apoptosis, Drosophila
Introduction
With an increase in the number of older adults, age-associated neural degenerative disorders have become an area of growing public concern, medical attention, and scientific study. Although significant progress has been made in characterizing many aspects of human neurodegeneration, the multifactorial nature and the long period over which these diseases develop have hampered informative research. Considering these difficulties, recent work has highlighted the value of studying disease-associated genes in model organisms and has led to the genetic study of neurodegeneration inDrosophila (Mutsuddi and Nambu, 1998; Chan and Bonini, 2000;Muqit and Feany, 2002).
Two main approaches have been productive: (1) the study of mutations in endogenous Drosophila genes and their neurodegenerative phenotypes and (2) the study of transgenic flies expressing disease-associated variants of human proteins. Among the endogenous genes and proteins that have been studied are homologs of human proteins, including the amyloid precursor protein, presenilins, and superoxide dismutase (Phillips et al., 1989; Mutsuddi and Nambu, 1998;Reiter et al., 2001), as well as genes involved in retinitis pigmentosa (i.e., rhodopsin and arrestin) (Davidson and Steller, 1998; Alloway et al., 2000; Kiselev et al., 2000). Work on these genes, their corresponding proteins, and interacting partners in flies has led to unique insights into normal function and consequences associated with human degenerative diseases.
Expression of disease-associated proteins in flies has included studies of polyglutamine expansion proteins (Warrick et al., 1999;Fernandez-Funez et al., 2000; Kazemi-Esfarjani and Benzer, 2000;Steffan et al., 2001; Kazantsev et al., 2002) as well as mutant forms of α-synuclein (Feany and Bender, 2000; Auluck et al., 2002) and tau (Wittmann et al., 2001). For most of these studies the animals develop hallmark features associated with the human disease counterpart. These include the formation of protein aggregates marked with ubiquitin, neuronal apoptosis, and the loss of neural tissues. This work underscores the conserved nature of cellular pathology and degenerative cascades. These transgenic models have allowed genetic screens to identify modifiers of degenerative phenotypes (enhancers or suppressors) that would be difficult to characterize in other systems (Warrick et al., 1999; Fernandez-Funez et al., 2000; Kazemi-Esfarjani and Benzer, 2000; Steffan et al., 2001; Kazantsev et al., 2002).
Given the success of studying Drosophila neurodegenerative loci and misexpressed human disease genes, it is somewhat surprising that endogenous Drosophila mutations have not been identified with certain key features associated with common late-onset human neurodegenerative disorders, notably the formation of protein aggregates containing ubiquitinated proteins and amyloid precursor-like protein (APPL) (Bence et al., 2001; Sherman and Goldberg, 2001; Ding et al., 2002). This study closes this gap by identification and characterization of a new Drosophila gene, blue cheese (bchs), which defines a novel degenerative pathway displaying this pathology. Inspiring the mutant name, we find the formation of protein inclusions throughout the CNS ofbchs mutants. These inclusions contain insoluble ubiquitinated protein aggregates and the Drosophila APPL and stain positive with the amyloid dye thioflavine S. Along with abnormal retinal and CNS morphology and size, aged mutant brains show extensive neuronal apoptosis. Loss-of-function mutations in bchs also result in a significantly shortened adult life span. The BCHS protein is highly conserved from nematodes to humans and contains motifs implicated in membrane vesicle transport and protein processing. Given the substantial level of homology, it is reasonable to propose functional and pathological consequences as a result of the loss of BCHS in other species.
Materials and Methods
Drosophila culture and mutagenesis. Stocks and crosses were grown on standard Drosophila medium at 25°C until eclosion. For aging studies and protein and confocal analysis, newly emerged adults were placed in fresh vials containing 25 individuals and were aged at 29°C for the times indicated. Flies were transferred to fresh food every 2–3 d. For analysis of life span the number of dead individuals for each genotype was counted during transfer, and the percentage remaining alive was calculated. Life span is given as the mean time of death. Aging profiles were plotted with CA-Cricket Graph III program, and p value comparisons were prepared by using Microsoft Excel and the online program GraphPad (http://www.graphpad.com/quickcalcs/ttest2.cfm).
New mutations in bchs were generated by using established P-element mutagenesis techniques and EP(2)2299 transposable element (Rio, 1991). Initially, all lines containing mobilization events were examined for viability and health over Df(2L)clot7 and as homozygotes and were tested for female fertility (dsf+). Of the 80 mobilized lines meeting these criteria, nearly one-half demonstrated precise removal of the P-element as determined via genomic Southern blot, PCR, and/or sequencing analysis in and around the original insertion site (see Fig.1B). The remaining lines contain small deletions or rearrangements to the 5′ region of bchs and were grouped into 13 molecular classifications for aging analysis.
Fig. 1.

The bchs region, mutations, protein structure, and species alignment. A, The location and extent of deletions uncovering bchs are illustrated. The map of the bchs genomic region and deletion boundaries is as described previously [Finley et al. (1998); see flybase website in Materials and Methods]. The location and arrangement of thebchs and dsf genes are indicated, as are the 402Cy P-element (w+;darkgray) and P-elements generatingbchs mutations (bchs3and bchs4; light gray). The 402Cy P-element is the parent of the Df(2L)dsf3 and Df(2L)dsf4 deletions and the bchs3 insertion. It is carried on the CyO chromosome used as a wild-type control in aging experiments. B, The intron/exon structure ofbchs was determined by comparison and alignment of cDNA and genomic sequences. The locations of BEACH, WD40, and FYVE protein motifs and potential phosphorylation sites are indicated. Sites of P-element insertion are indicated. The extent of thebchs5 internal deletion (determined by DNA sequencing) is indicated by the hatched bar. A precise excision derivative, thebchsrev1 allele, was determined by DNA sequencing and is indicated by arrows. C,Drosophila, vertebrate, and nematode BCHS protein alignments were generated by comparison of the inferred amino acid sequences. The overall size, protein domains, and amino acid sequence of the different BCHS proteins are conserved, including the first 2500 amino acids possibly identifying functional motifs within this region. The identified domains within the C-terminal region have the highest amino acid identity over divergent phyla, reaching >80% over extended regions of the protein.
Molecular techniques. Cloning of the bchs genomic region and the deletion boundaries have been described previously (Finley et al., 1998) (http://flybase.bio.indiana.edu/). blue cheese cDNAs were isolated from libraries (Tulle Hazelrigg, Columbia University, New York, NY), using standard techniques and genomic restriction fragment probes. Subcloned cDNA fragments were sequenced with vector primers or primers designed to match previously sequenced DNA regions on an ABI automated sequencer. Contiguous sequences were assembled with Sequencher software (Gene Codes, Ann Arbor, MI). BLAST, ProfileScan [run remotely, National Center for Biotechnology Information (NCBI), Bethesda, MD], or DNA STAR programs were used for data base searches, sequence comparisons, and alignments. For Northern blots total RNA was isolated (RNeasy Total RNA Kit; Qiagen, Chatsworth, CA), and RNA from 20 heads was loaded per lane. Transferred blots were hybridized with 32P-labeled random primed probe frombchs cDNA clones via established techniques.
Mammalian and nematode homologs of BCHS were identified by tblastn searches run remotely at NCBI, using all or part of the BCHS sequence as query. The majority of human and mouse BCHS sequences (with the exception of short 3′ cDNA contig, KIAA0993) was assembled by linking of previously unidentified and unconnected exons from human or mouse genomic sequence. Regions of weak homology toDrosophila were checked further for splice site and reading frame alignment and by cross comparison between the human and mouse sequences.
In situ hybridization. Pharate adults were embedded in Tissue Freezing Medium (Triangle Biomedical Sciences, Durham, NC) and cryostat sectioned (20 μm). In situhybridization followed described procedures (Finley et al., 1998). Riboprobes were synthesized by using T3 or T7 polymerase with a linearized pBluescript vector containing 5′ bchs cDNA templates and digoxygenin-coupled nucleotides (DIG RNA Labeling Kit, Boehringer Mannheim, Indianapolis, IN).
Plastic section histology. Heads, from flies of a particular age, were dissected from the body and proboscis and fixed overnight in 3% glutaraldehyde, 3% paraformaldehyde in 0.1 mcacodylate buffer, pH 7.4, at 4°C. Heads were washed after primary fixation in 0.1 m cacodylate buffer and postfixed on ice in 1% OsO4/0.1 m cacodylate buffer for 2 hr. After two washes in water the tissues were dehydrated in a standard acetone series and infiltrated with Epon 812 (EMS grade). Heads were oriented relative to the head cuticle before sectioning. Semithick sections (1.0 μm) were stained warmed for 2 min with 0.5% methylene blue, 0.5% borax, and 0.5% AzureII and rinsed with water. Sections were dehydrated before mounting with Permount (EMS grade, Fisher Scientific, Houston, TX).
BCHS antibody production, protein preparation, and immunoblot analysis. The BCHS C terminus (183 amino acids) was subcloned into the pGEX-KG vector (Amersham Biosciences, Piscataway, NJ). Guinea pigs were immunized with purified BCHS-fusion protein (glutathione beads, Sigma, St. Louis, MO) by following standard techniques. Antibodies were affinity purified by using membrane-bound fusion protein (Immobilon-P, Millipore, Bedford, MA) and standard techniques.
Approximately 30 flies for a particular age and genotype were placed in individual 15 ml conical centrifuge tubes, flash-frozen in liquid nitrogen, and vortexed several times. Severed heads were separated from thoraxes and abdomens by using a standard tea sieve. Heads were homogenized in PBS, 1% Triton X-100, and protease inhibitors at 4°C and centrifuged at 15,000 × g (Johnston et al., 1998;Mohtashami et al., 2001). Supernatants were collected as the Triton X-100 soluble fraction, and the remaining pellet was resuspended in 50 mm Tris, pH 7.5, and 2% SDS with protease inhibitors and then sonicated (Johnston et al., 1998). After centrifugation the supernatant was collected as the Triton X-100 insoluble fraction. For each sample 30 μg of protein (Bio-Rad, Hercules, CA) was resolved by 4–20% SDS-PAGE (Invitrogen, Carlsbad, CA) and electroblotted. Western blots were probed sequentially with anti-ubiquitin (1:1000 dilution;Zymed Laboratories, San Francisco, CA), anti-histone 2B (1:500 dilution; James Kadonaga, University of California, San Diego, CA), and anti-BCHS (1:200 dilution) antibodies with the use of standard techniques. Specific antibody binding was detected with standard ECL reagents.
Fluorescence confocal microscopy. For aggregate formation studies the flies were aged at 29°C for the indicated times. Under anesthesia the heads were removed and placed in PBS; the surrounding cuticle was dissected and the brains fixed for 45 min at 25°C in PBT (1× PBS with 0.1% Triton X-100) with 3.5% paraformaldehyde. After three PBT washes the brains were incubated in ubiquitin monoclonal antibody (1:100 dilution; Zymed Laboratories) and/or APPL antibody (preabsorbed, 1:2000 dilution; Kalpana White, Brandeis University, Waltham, MA) in PBT with 3% normal goat serum (NGS) as blocking agent overnight at 4°C. After three washes with PBT the samples were incubated in the indicated secondary antibodies (1:200 dilution, PBT with 3% NGS; Jackson ImmunoResearch, West Grove, PA) for 1 hr. For actin detection a 1:250 dilution of phalloidin-TRITC (100 μg/ml stock; Sigma) was added to the secondary antibody incubation. To label protein aggregates, we added a 1:100 dilution of thioflavine S (direct yellow 7, 1 mg/ml stock; Sigma) to the secondary incubation. Samples were washed three times (in PBT), mounted, and imaged with anOlympus 1× 70 inverted confocal microscope and Fluoview digital imaging software.
To detect the BCHS protein, we dissected staged larval and adult tissues, fixed them (PBS, 3.5% paraformaldehyde) for 25 min at room temperature, and washed them three times in PBT. Samples were incubated in BCHS antibody (1:50 dilution; affinity-purified) and with either ELAV antibody (1:1000 dilution; Developmental Studies Hybridoma Bank, Iowa City, IA) or REPO antibody (preabsorbed fixed embryos, 1:1000 dilution; Gerd Technau, University of Mainz, Mainz, Germany) overnight at 4°C in PBT containing 3% NGS. Tissues were washed three times in PBT and incubated in the indicated secondary antibodies (1:200 dilution, PBT with 3% NGS; Jackson ImmunoResearch) for 1 hr. Washed samples were confocal imaged as described.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling analysis. Aged brains were dissected and fixed (PBS, 4% paraformaldehyde) for 20 min at room temperature. Tissues were washed twice in PBS, once in PBS plus 0.1% Triton X-100 and 0.1% sodium citrate for 5 min, and then twice in PBS. The final wash was removed carefully with 3 mm paper strips. TUNEL analysis was performed by following the manufacturer's instructions (Boehringer Mannheim). Direct fluorescence confocal microscopy was used to detect fluorescein-labeled DNA strand breaks. For each CNS image a single compressed confocal stack was made from a series of 10 optical sections (2.0 μm each).
Results
Loss of bchs results in premature adult death
Initial work on the bchs gene began during our characterization of the 26A salivary chromosome region and the adjacentdissatisfaction gene (dsf) (Fig.1A) (Finley et al., 1998). Since this initial study we have generated or identified multiple bchs alleles and deletions of the surrounding genomic region. These include Df(2L)dsf3 (bchs1) and Df(2L)dsf4 (bchs2) chromosomes, which remove ∼60 kb of sequence containing both bchs and dsf genes, as well as the Df(2L)clot7 and Df(2L)w3 deletions (Fig.1A) (Finley et al., 1998). P-element insertion alleles bchs3 (isolated in our lab) andbchs4 [isolated by Rorth as EP(2L)2299] (Rorth, 1996; Kraut et al., 2001) interrupt the first intron and substantially lower BCHS protein expression (Fig. 1B; also see below).
Starting with the bchs4 allele, we generated several internal deletions by P-element mobilization (Rio, 1991; Finley et al., 1998). These substantially alter or remove intron 1 and portions of the bchs coding sequence. For example, thebchs5 allele deletes most of the first intron, second exon, and much of the third exon, removing the translation start site and the first ∼200 amino acids of the protein (Fig. 1B). Precise excisions of thebchs4 P-element, such asbchsrev1, restore the wild-type sequence and gene structure (Fig. 1B,arrows). All mutant alleles of bchs, either as homozygotes or in conjunction with larger regional deletions [Df(2L)clot7], generated normal Mendelian frequencies of adults that display appropriate motor, feeding, and grooming behaviors. This includes the Df(2L)dsf3 (bchs1) and Df(2L)dsf4 (bchs2) deletion alleles. Allbchs mutations generate males that are fertile, and genetic combinations that are dsf+ produce fertile females (i.e., bchs3,bchs4,bchs5).
In contrast to the normal survival found during larval and pupal development, bchs mutant adults demonstrate a significant decrease in life span (Fig. 2, Table1). When compared with wild-type controls, the mean life span is decreased between 40 and 45% to an average of between18 and 23 d when grown at 29°C (Table 1). This decrease in longevity includes all P-element misexcision alleles (Fig.2A), P-element insertions (Fig.2B), and any combination of deletions, which remove or alter both copies of the bchs gene (Fig.3C). Rescue of the phenotype and a return to normal life span is seen in those lines with precise P-element excisions that restore a wild-type sequence and gene structure (i.e.,bchsrev1) (Fig. 2A, Table 1). Nor is premature death associated only with higher temperatures. When maintained at 25°C,bchs3/Df(2L)clot7 flies have an average life span of 22.25 d (n = 219), whereas wild-type controls (Canton S) have an average of 36.8 d (n = 99). This is a similar 40% reduction in adult life span and indicates that early death of bchs mutant flies is not a temperature-dependent phenotype. The lack of pronounced developmental defects, normal behaviors in young adults, and premature death all indicate that the bchs gene is essential for normal adult survival and longevity.
Fig. 2.
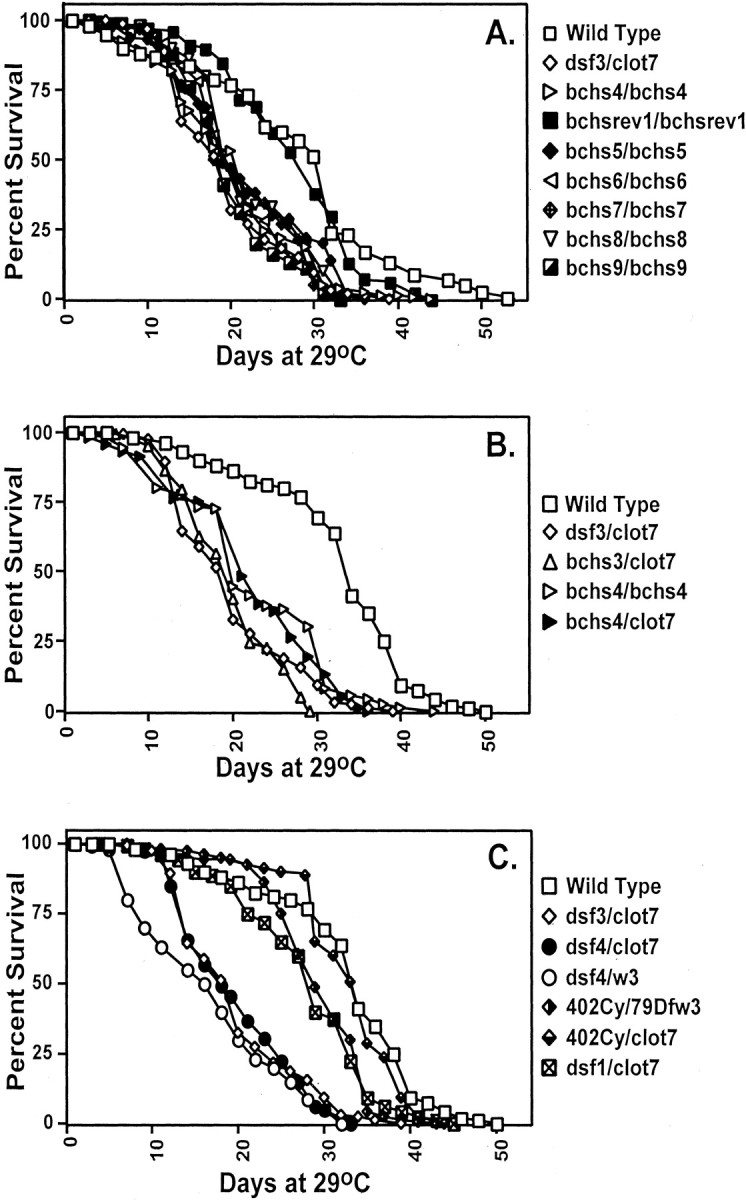
Premature death of bchs mutants. Newly eclosed males were collected and aged at 29°C for the duration of the experiments. A, Imprecise excisions of thebchs4 P-element, generating alterations in the 5′ region of the bchs gene, result in a shortened average life span, as shown by thebchs5/bchs5,bchs6/bchs6,bchs7/bchs7,bchs8/bchs8, andbchs9/bchs9fly lines. Rescue of premature death occurs when the samebchs4 P-element is removed precisely from its bchs location, as demonstrated by thebchsrev1/bchsrev1line. B, P-element insertions in the first intron ofbchs reduce average life span, as seen inbchs3/Df(2L)clot7,bchs4/bchs4, and bchs4/Df(2L)clot7 genetic combinations. C, Flies containing one or more wild-type copies of bchs, Canton S, 402Cy/Df(2L)w3, or 402Cy/Df(2L)clot7 have an average life span between 29.0 and 32.5 d. Adults with both copies of the bchs genomic region removed, Df(2L)dsf3/Df(2L)clot7, Df(2L)dsf4/Df(2L)w3, or Df(2L)dsf4/Df(2L)clot7 flies, show a 40–45% reduction in average life span when compared with controls. Mutating the dsf gene in the case of dsf1/Df(2L)clot7 flies does not alter average adult longevity significantly. Mean lifespan, SDs, and p values for a selection of genotypes are summarized in Table 1.
Table 1.
Summary of bchs alleles and phenotypes
| Genotype | Mean life span | SD | pvalue with WT | BCHS expression | Increase in IUCP* | Ubiquitin aggregates | APPL aggregates |
|---|---|---|---|---|---|---|---|
| Df(dsf3)/Df(clot7)bchs1 | 18.2 n = 190 | 6.3 | <0.0001 | 0 | 22.8 | + | + |
| Df(dsf4)/Df(clot7)bchs2 | 19.6 n = 136 | 6.5 | <0.0001 | 0 | nd | + | + |
| bchs3402Cy011/Df(clot7) | 19.1 n = 209 | 5.6 | <0.0001 | 0 | 16 | + | + |
| bchs4EP(2)2299/Df(clot7) | 21.7 n = 161 | 7.9 | <0.0001 | ++/0 | 6.9 | + | + |
| bchs4EP(2)2299/EP(2)2299 | 22.9 n = 135 | 8.3 | <0.0001 | ++/0 | nd | + | + |
| bchs5/bchs5 | 21.5 n = 99 | 8.5 | 0.00021-159 | nd | nd | + | nd |
| bchsrev1/bchsrev1 | 27.2 n = 81 | 7.7 | 0.811-159 | nd | nd | 0 | nd |
| Wild-type (Canton S) | 32 n = 123 | 9.1 | 1 | ++++ | 0 | 0 | 0 |
| 402CyO/Df(clot7) genotype control | 32.5 n = 151 | 5.8 | 0.67 | ++++ | nd | 0 | 0 |
| dsf1/Df(clot7) regional control | 31.5 n = 84 | 8.3 | 0.62 | +++ | nd | 0 | 0 |
Insoluble ubiquitin conjugated proteins.
The mean life span for each genotype is indicated, as are SDs andp value (Student t Test). Comparisons with wild-type (Canton S) and
F1-159: note that pvalues were calculated from wild-type controls aged at the same time and under the same conditions (average = 27.6, SD = 11.9, andn = 73). The + and 0 symbols used for BCHS expression summarize the Northern and Western analyses of the message and/or protein. The +/0 symbols used forbchs4 allele reflect the presence of the protein in young adults and its absence in aged individuals. The relative levels of ubiquitin and APPL aggregate formation detected by confocal (starting at 10 d of age) are summarized by + (presence) or 0 (absence) symbols. Of the three independent experiments done to examine levels of insoluble ubiquitin-conjugated proteins (IUCP), the representative Western blot in Figure 6 was used to calculate the fold increase after correcting for load (histone 2B) and normalization to wild-type controls.
Fig. 3.
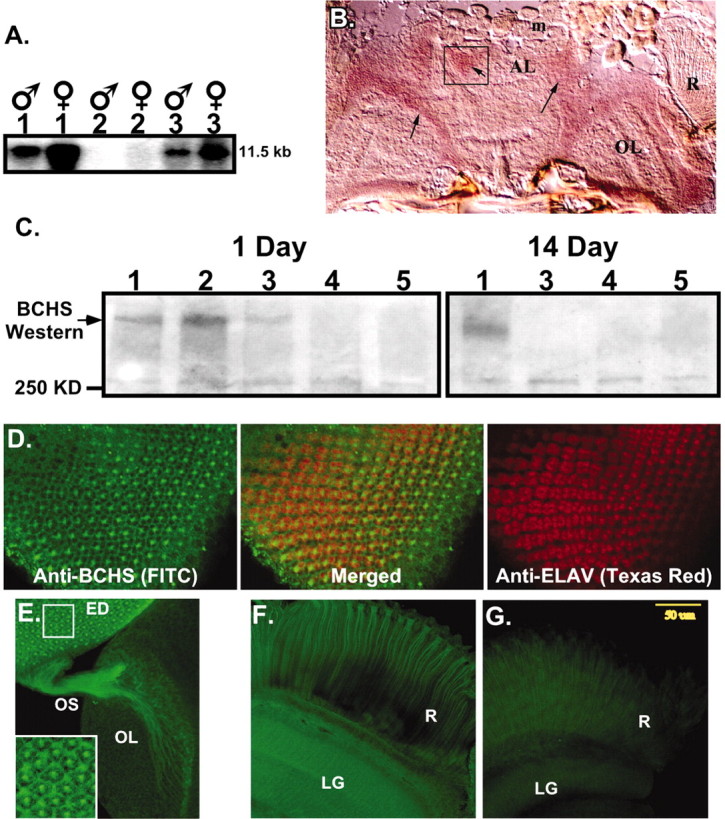
BCHS mRNA and protein expression patterns.A, Northern blot analysis of total RNA from 20 adult male and female heads. Wild-type (1) anddsf1/Df(2L)clot7 (3) mutant brains express a single, nearly 12.0 kb mRNA that is absent from Df(2L)dsf3/Df(2L)clot7 flies (2). B, Frozen sections from wild-type pharate adults (Finley et al., 1998) were used for in situ hybridization to bchs mRNA. Specific labeling for bchs is detected in a horizontal section through the head at the level of the antennal lobes. Labeling is seen in cortical regions containing neuronal cell bodies and is absent from neuropil areas. Arrows point to labeled cortical cells; the antenna lobe (AL), optic lobe (OL), and retina (R) are indicated. mRNA was not detected in muscles of the head (m) or other tissues. Sections through adult thorax also show uniformbchs labeling in thoracic and abdominal ganglia neurons and not in other tissues (data not shown). C, Western blot analysis of BCHS shows a protein running well above the largest molecular weight marker (250 kDa) in head extracts from 1-d-oldCanton S (1) and 402CyO/Df(2L)clot7 (2) wild-type controls. At this age bchs4/Df(2L)clot7 flies (3) express some BCHS, whereas BCHS is absent from bchs3/Df(2L)clot7 (4) and Df(2L)dsf3/Df(2L)clot7 (5) flies. At 2 weeks only wild-type flies (1) have detectable levels of the BCHS protein. Protein no longer is detected inbchs4/Df(2L)clot7 flies (3). D–F, Subcellular location of the BCHS protein. D, Immunofluorescence staining of a third instar larval eye disc double-labeled for BCHS (basal section;FITC) and neuronal-specific marker ELAV (apical section;Texas Red). The two proteins demonstrate similar temporal expression patterns in neurons, but ELAV is located primarily within the nucleus while BCHS is found in cytoplasm (center; merged panel).E, Eye disc (ED), optic stalk (OS), and attached larval optic lobe (OL) are stained for BCHS (FITC). The protein is found along the entire length of photoreceptor axons. The inset is an enhanced intensity image showing BCHS extending as far as growth cones. This pattern is visible only because of the lack of BCHS expression in surrounding undifferentiated neuroblasts at this developmental time point. F, Mature BCHS expression pattern in a retina (R) and laminal ganglion (LG) optical section of a 2-week-olddsf1/Df(2L)clot7 adult (n = 9). BCHS protein levels are still high in older adults (consistent with Western blots). The protein remains located in the cytoplasm of retinal axons and in laminal ganglia.G, Enhanced image of an identically prepared retina and attached CNS from an age-matched bchs mutant [Df(2L)dsf3/Df(2L)clot7; n = 11]. This level of staining is similar to secondary antibody alone and is substantially below the staining of bchs+animals.
Characterization of the bchs gene and protein
Genomic DNA from the bchs region was used to probe adult cDNA libraries. Thirty-four cDNAs were identified and fully or partially sequenced. Analysis and alignment of these sequences yielded a single continuous inferred mRNA for the bchs gene. The bchs mRNA consists of 11,140 nucleotides with a single open reading frame of 10,476 nucleotides. The mRNA encodes a predicted protein of 3492 amino acids with a molecular weight of 389,755 Da. We verified the mRNA sequence and mapped intron–exon junctions by direct sequencing of the 15 kb bchs genomic region (Fig.1B) and later verified these conclusions by using sequence data from the Drosophila genome project, which identified the gene as CG9011 and CG14001. Database screens and sequence homology comparisons reveal two distinct regions within the protein, a novel N-terminal portion (nearly 2500 amino acids) and a motif-rich C terminus containing three identifiable domains.
The first of these domains, termed BEACH, originally was classified in the lyst family of genes (lysosomal trafficking regulator) (Barbosa et al., 1996; Nagle et al., 1996; Tchernev et al., 2002). Recessive mutations in lyst cause the beige phenotype in mice and the Chediak–Higashi syndrome in humans. Both disorders are associated with lysosomal metabolism, trafficking, and protein-sorting defects. Humans and mice demonstrate pigmentation defects; however, humans have the additional pathology of immune disorders and mental retardation (Nagle et al., 1996; Faigle et al., 1998). As with other BEACH-containing proteins, BCHS also has a series of WD40 repeats that are thought to mediate protein–protein interactions (Neer et al., 1994).
The BCHS C terminus ends in a cysteine-rich FYVE finger domain (Fig.1B) (Burd and Emr, 1998; Corvera et al., 1999;Stenmark and Aasland, 1999; Wurmser et al., 1999; Cormont et al., 2001). This motif is found in an independent set of proteins with vesicle transport and protein-processing functions. Recent work indicates that FYVE fingers function as phosphatidyl inositol 3-phosphate [PtdIns(3)P] binding modules and downstream effectors of Rab protein signaling cascade (Kutateladze et al., 1999; Cormont et al., 2001). BCHS is the first identified protein to contain both BEACH and FYVE domains (Stenmark and Aasland, 1999).
Of the several hundred amino acids in BCHS capable of being phosphorylated, seven residues have surrounding sequence consensus motifs consistent with potential kinase phosphorylation sites (Fig.1B; 1 tyrosine, 4 serines, and 2 threonines). These residues may act as phosphorylation and interaction sites for a range of adapter and regulatory proteins functioning through SH2 or PTB domains (Shoelson, 1997).
Aside from potential phosphorylation sites, the first 2500 amino acids of BCHS lack significant homology to any previously identified protein or functional domain. However, comparison of EST and genomic sequences from different species reveals strong conservation of this region in novel proteins from nematodes (NP502420, NP502421, NP502422) to humans (KIAA0993) and mice (X144441). Indeed, using the Drosophila sequence as a scaffold, we have assembled the nearly complete amino acid sequence for worm, mouse, and human BCHS. The degree of amino acid identity among these different proteins is illustrated in Figure 1C. For identifiable motifs, like the BEACH domain, identity between Drosophilaand human BCHS can be >80% over 400 amino acids. In addition, the overall protein size and motif organization are also highly conserved between divergent species. The continuous homology and the multiple regions of high identity across the length of different BCHS proteins strongly suggest that they are conserved orthologs and are likely to perform similar cellular functions.
BCHS expression pattern and subcellular location
As a step toward characterizing bchs function, we examined the temporal and tissue expression patterns of thebchs RNA. Northern blot analysis reveals a nearly 12.0 kb transcript from adult head tissues of both wild-type anddsf1 mutant flies that is absent frombchs deletion alleles [Df(2L)dsf3] (Fig. 3A).In situ hybridization analysis of frozen sections reveals uniform levels of the bchs mRNA throughout the young adult CNS, but none in surrounding adipose or muscle tissues (Fig.3B). The signal is strongest in outer cortical regions of the CNS, areas dense in neuronal cell bodies (Fig. 3,arrows). The mRNA is not detected in regions of neuropil (axonal projections). A similar pattern of bchs neuronal expression is found in the cortical layer of the adult ventral ganglia (data not shown).
Immunoblot analysis with BCHS-specific antibodies identifies a large protein in neuronal tissue from young control animals andbchs4 mutants, which is absent in Df(2L)dsf3/Df(2L)clot7 andbchs3/Df(2L)clot7 flies (Fig.3C). At 2 weeks the BCHS protein is not detected in head preparations from any mutant strains, includingbchs4 animals. At this time wild-type BCHS exhibits a slightly faster electrophoretic mobility. These data indicate that the null and P-element bchs alleles substantially eliminate or lower expression of the protein (Table1).
To determine whether BCHS could mediate some aspects of vesicle and/or protein trafficking like other BEACH- or FYVE-containing proteins, we asked whether the BCHS protein is cytoplasmic and present in neuronal projections. We took advantage of the well characterized developmental profile and stereotypic organization of the Drosophilavisual system to examine BCHS localization within neurons and axons (Torroja et al., 1996; Hay et al., 1997). In the late larval eye disc we find BCHS expressed in photoreceptor cells after neuronal differentiation (Fig. 3D, FITC and merged image). The cytoplasmic localization of BCHS is best illustrated by comparison with the nuclear staining pattern of the ELAV protein (Texas Red). ELAV is localized primarily in photoreceptor nuclei, as illustrated by its staining in confocal sections from apical regions of the eye disc (Robinow and White, 1988). BCHS appears to be excluded from nucleus and instead generates a lattice-like staining pattern in confocal sections from basal regions of the disc. This indicates that BCHS is cytoplasmic within the eight neurons making up an ommatidial cluster. The bright central staining in each ommatidium marks BCHS in the confluence of photoreceptor axons before their projection toward the optic lobes of the CNS.
An examination of larval eye discs that remain attached to the brain reveals the presence of the BCHS protein in neuronal projections (Fig.3E). Most notably, the protein can be followed in axons projecting down the optic stalk and into the developing optic lobes. At this developmental time point BCHS reaches as far as growth cones and is visible only because of the lack of its expression in the surrounding neuroblasts. Later this staining pattern would be obscured by ubiquitous BCHS expression within neurons of this region. The enlarged inset image highlights the presence of BCHS within the photoreceptor cytoplasm.
We do not detect BCHS in glia or in most adult motor neuron projections (data not shown). Consistent with results from in situ and immunoblot analysis of adult brains, BCHS expression remains high in neuronal cytoplasm and projections, as shown in the 2-week-old adult retina and laminal ganglia (Fig. 3F). BCHS is absent from retinal or other neuronal tissue from age-matched null controls, as seen in an enhanced image (fivefold enhancement) with staining equivalent to secondary antibody alone (Fig. 3G). Together these data show that BCHS is present in neuronal cytoplasm and is present in CNS projections, which is consistent with a function in intracellular vesicle transport or protein trafficking.
bchs mutants demonstrate extensive neurodegeneration
Extensive expression of BCHS in the CNS and neural projections and its continued presence in older adults suggested that premature death is the result of progressive deterioration in neural tissues. Initial light microscopy and phase-contrast images of fixed tissues frombchs mutants showed a general loss of CNS size and morphology. The progressive nature of the bchs neural degenerative phenotype is best illustrated in the well defined and ordered structure of the adult retina.
Plastic-embedded tangential sections from 1-d-old wild-type andbchs3/Df(2L)clot7 adults show normal development of retinal structures. Ommatidia have the correct number of cells, and rhabdomeres are in the appropriate orientation (Fig. 4A,B). At 10 d the wild-type controls have normal mature ommatidial structures (Fig.4C), whereas thebchs3/Df(2L)clot7 adults show signs of degeneration (Fig. 4C). We find the beginnings of architectural disorganization as well as the development of vacuoles between clusters of ommatidia (Fig. 4D, indicated byarrows). At 14 d the structure of wild-type retinas still appears robust, with only an occasional vacuole being detected (Fig. 4E, arrow). At this time retinal structures from bchs3/Df(2L)clot7 mutants show increased degeneration (Fig. 4F) (Alloway et al., 2000). There is substantial loss of ommatidial morphology, a condensation of individual rhabdomeres, and the development of large vacuoles between ommatidia (Fig.4F, arrows), all tissue-specific features of a progressive degenerative process (Alloway et al., 2000).
Fig. 4.
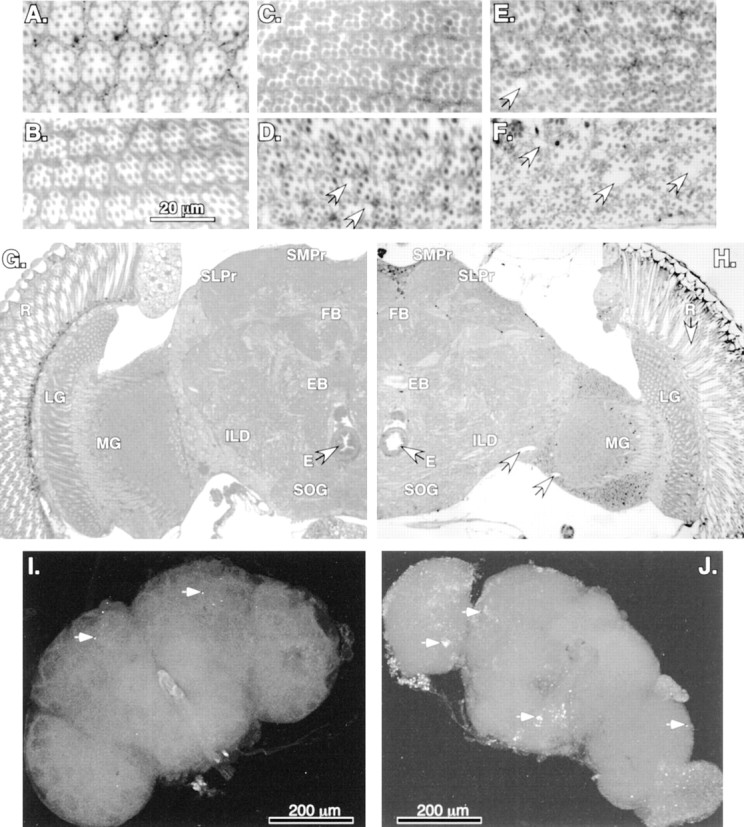
Neural degeneration and apoptosis inbchs mutants. A–F, Plastic-embedded tangential sections (1 μm) of adult retina stained with methylene blue. Minor differences in early ommatidial morphology are attributable to the depth of each individual section and position within a given eye. A, Retinal morphology of Canton Scontrols at 1 d of age is very similar to that ofbchs3/Df(2L)clot7 (B) individuals of the same age.C, At 10 d wild-type controls demonstrate the mature architecture and cellular morphology ofDrosophila retina, whereas age-matchedbchs3/Df(2L)clot7 flies (D) show the first signs of degeneration (vacuoles noted by arrows). E, At 14 d of age the wild-type retinas appear healthy and intact, with only the rare development of small vacuoles (arrow).F, At 14 d the degeneration detected earlier inbchs3/Df(2L)clot7 mutant flies has progressed further. There is a substantial loss of ommatidial architecture and an enlargement of vacuoles (arrows).G, H, Frontal sections (1 μm) from 10-d-old adults taken at the same depth within the head, near the central complex, as noted by the esophagus (E, arrows) and fan-shaped (FB) and ellipsoid bodies (EB). G, A section taken from 10-d-old controls demonstrates the characteristic organization and size of mature Drosophila neural structures. H, Age-matched section from abchs3/Df(2L)clot7 fly shows the presence of retinal degeneration (arrow) as well as atrophy of laminal (LG) and medullary (MG) ganglia, inferior lateral deutocerebrum (ILD), superior lateral (SLPr) and medial (SMPr) protocerebrum, and subesophageal ganglia (SOG) structures of the CNS. Nuclei and background appear darker in this image because of slight image enhancement.I, J, In situ TUNEL analysis of 2-week-old brains. In wild-type brains (I) the arrows indicate the limited number of cells undergoing apoptosis [402CyO/Df(2L)clot7; n = 10]. bchs null brains (J) have extensive TUNEL labeling, indicating significant numbers of cells undergoing apoptosis in most cortical regions of the CNS [Df(2L)dsf3/Df(2L)clot7; n = 7].
The neural degeneration associated with loss of bchs is not limited to the retina. In comparing plastic-embedded frontal sections from the same depth within the head from aged wild-type (n = 7 from 10 to 14 d) (Fig. 4G) andbchs3/Df(2L)clot7 (n = 10 from 10 to 14 d) (Fig. 4H) individuals, we consistently find atrophy of neural tissues in mutant animals. Using the esophagus (Fig. 4E, arrows) as a further positional landmark and starting at the periphery and working toward the central regions of the CNS, we find the retinal degeneration as demonstrated in Figure 4, D and F(arrow), clearly visible down the length of individual ommatidia. Loss of tissue size and morphology is also apparent in the laminal and medullary ganglia, inferior lateral deutocerebrum, superior lateral and medial protocerebrum, subesophageal ganglia, and fan-shaped and ellipsoid body regions of the CNS. Sections taken at 14 d demonstrate an increase in atrophy and fragility for bchsmutant brains. At later times mutant deterioration is significant enough to make the production of quality, complete sections to examine CNS morphology extremely difficult. Nevertheless, regions of sections from such older adults indicate a continued decrease in brain volume. In addition, dissected brains of multiple genotypes used for confocal microscopy (Figs. 4I,J,5A,B) show an approximately one-sixth decrease in linear dimension, indicating an ∼40% decrease in volume. Unlike in other characterized Drosophilamutations, which show CNS-wide neural degeneration and early adult death, we do not detect extensive vacuolization of bchsmutant brains. Occasional vacuoles that do develop are indicated (Fig.4H, arrows).
Fig. 5.
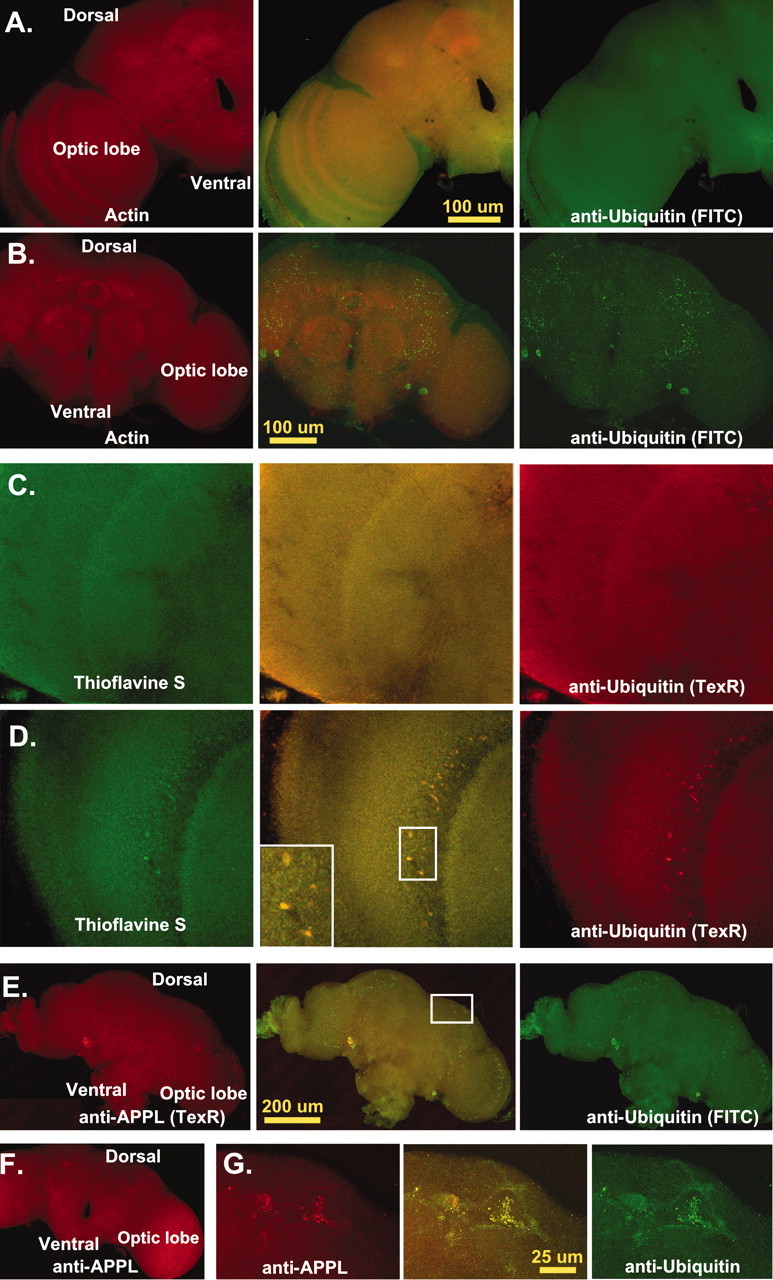
CNS protein aggregate formation.A–G, Single 0.2 μm transverse optical sections taken at least 5 μm into adult CNS. A, Optical section from an 11-d-old wild-type adult brain (n = 27) stained for actin (phalloidin-TRITC, Texas Red) and ubiquitin (FITC). Normal flies have a consistent pattern of actin and ubiquitin proteins within the CNS. Actin is enriched in neural projections and regions of synaptic innervation, whereas ubiquitin is stained uniformly throughout the entire CNS. B, Age-matched bchs null brains (n = 53) have deposits of ubiquitin in the CNS, mainly in regions of neuropil. C, Confocal images of 10-d-old wild-type controls (n = 20) stained for thioflavine S (green) and anti-ubiquitin (TexR).D, Identical images from age-matchedbchs3/Df(2L)clot7 flies (n = 15) stained with thioflavine S and for ubiquitin aggregates. E, Confocal sections from 11-d-old Df(2L)dsf3/Df(2L)clot7 adult brain (n = 22) stained for APPL (TexR) and ubiquitin (FITC).Drosophila APPL colocalizes with ubiquitin deposits in the CNS. F, APPL aggregates do not form in 2-week-old wild-type controls (n = 12). G,Inset enlargement (through portions of the mushroom body) shows the close association of ubiquitin and APPL in protein aggregates.
With the use of a range of light microscopy staining techniques for sectioned tissues, typically used to characterize the loss of neural morphology or development of neural tangles (including silver staining), we failed to detect obvious differences between the brains from aged bchs mutants and their wild-type counterparts (data not shown). However, we do find the development of dark inclusions throughout the CNS of older bchs flies, using phase-contrast light microscopy on fixed whole-mount preparations. This indicated the possible development of neural inclusions, which may be associated with the degeneration and premature death found withbchs mutants.
To characterize the temporal development and composition of these CNS inclusions better, we coupled chemical and immunohistochemical techniques used in the detection of protein aggregates with confocal microscopy of dissected, fixed Drosophila CNS preparations from a range of genotypes and ages. We began with specific proteins known to be included in pathological neural aggregates and focused on the protein degradation peptide signal, ubiquitin. This was in part attributable to its extensive association with a wide range of neural-toxic protein inclusions, mechanistic conservation, and the cross-reaction of commercial antibodies with the Drosophilaprotein (Warrick et al., 1999; Lai et al., 2001; Sherman and Goldberg, 2001).
The 10-d-old wild-type and mutant brains were immunostained for ubiquitin (FITC) and counterstained for actin (Texas Red). Confocal CNS sections from wild-type flies revealed a robust and characteristic staining pattern for both proteins (Fig. 5A). Actin primarily highlights neural projections (neuropil), whereas ubiquitin is distributed uniformly throughout the CNS. In optical sections from identically prepared and age-matched bchs mutants, only minor global changes were detected for actin. Unlike other studies ofin vivo aggregate formation, we did not observe incorporation of actin into inclusion-like structures (Fig.5B) (Suhr et al., 2001). This is in sharp contrast to ubiquitin. In 10-d-old bchs mutants we find immunoreactive ubiquitin concentrated in aggregate-like structures throughout the CNS (Fig. 5B). Extracellular deposits are even more pronounced at later ages (data not shown). Consistent with the progressive nature of the bchs phenotype, youngbchs mutant brains do not contain these ubiquitinated structures and are indistinguishable from their age-matched sibling controls (data not shown).
To determine whether bchs ubiquitin aggregates demonstrate structural similarities associated with other pathological conditions, we coupled ubiquitin staining with the fluorescent dye thioflavine S (Bacskai et al., 2001). Used to detect protein aggregates, thioflavine S binds to proteins that have taken on a β-pleated sheet conformation. It highlights abnormal in vivo and in vitro protein structures (often associated with neural pathology), including those containing Aβ peptide and mutant α-synuclein (Ostrerova-Golts et al., 2000; Bacskai et al., 2001). At 10 d we do not detect staining or colocalization of ubiquitin with thioflavine S within the CNS of wild-type controls (Fig. 5C), nor do we detect specific staining or aggregate formation in young bchs mutants or in age-matched controls (data not shown). We do find a close association of thioflavine S staining with ubiquitin containing inclusions in brains from 10-d-oldbchs mutants (Fig. 5D, inset). This indicates that a loss of bchs results in the formation of protein aggregates with structural similarities to aggregates associated with other progressive pathological conditions.
Other proteins also associated with human neurodegenerative disorders and CNS aggregate formations are conserved in Drosophila. This includes the amyloid precursor protein (APP) linked to Alzheimer's disease (Reiter et al., 2001). The Drosophilahomolog (APPL) is expressed abundantly in older adult neuronal tissues (Torroja et al., 1996). In 2-week-old bchs null brains we find that APPL (Texas Red) colocalizes with the CNS ubiquitinated inclusions (FITC) (Fig. 5E). Once again, these structures are genotype- and age-dependent and are not found in youngbchs mutant animals (data not shown) nor in age-matched controls containing a functional copy of bchs gene (Fig.5F). At higher magnification we find colocalization of ubiquitin and APPL in CNS inclusions (Fig. 5G). To varying degrees all tested bchs mutant alleles show similar ubiquitin aggregate formation over time, with the weakerbchs4 allele being the least affected at 10 d of age (Table 1).
Protein aggregate formation is often toxic to neurons and can initiate the apoptotic cell death pathway. We performed TUNEL analysis (direct labeling of DNA strand breaks) to determine whether aggregate formation in bchs mutant brains is associated with an increase in programmed cell death of neurons (Davidson and Steller, 1998; Abrams, 1999; Alloway et al., 2000; Kiselev et al., 2000). In 2-week-old wild-type control brains we find an average of 10–20 cells undergoing apoptosis (Fig. 4I, arrows). In contrast, age-matched bchs mutants show extensive CNS-wide apoptosis (Fig. 4J). Colocalization of dying cells with neuronal-specific (ELAV) (Robinow and White, 1988) or glial-specific (REPO) (Xiong and Montell, 1995) markers was not possible at this late developmental and degenerative time (data not shown). However, the cortical placement (location of neuronal cell bodies) and extensive number of cells highlighted by TUNEL labeling indicate that the majority of affected cells is neurons. Collectively, these results show that premature death of bchs adults is associated with age-dependent accumulation of neuronal abnormalities. This progressive condition is consistent with the formation of protein inclusions containing ubiquitin and APPL and with protein aggregates having β-pleated sheet properties. Also associated with aggregate formation is an increased neuronal apoptotic death for bchsmutants.
Immunoblot analysis of ubiquitin conjugates
Along with extensive ubiquitination and β-sheet structure, an additional feature of pathological protein aggregates is their tendency to be insoluble (Sherman and Goldberg, 2001). There is evidence that, along with removal of problematic proteins, sequestering cytotoxic peptides in insoluble aggregates may act as an intermediate mechanism to protect the cell from harmful secondary effects (Johnston et al., 1998; Marsh et al., 2000; Bence et al., 2001). To determine whether bchs mutants demonstrate an alteration in the ubiquitination pathway and whether older mutants develop substantial levels of insoluble proteins, we performed immunoblot analysis of CNS proteins that were obtained after sequential protein extractions (Fig. 6) (Johnston et al., 1998; Mohtashami et al., 2001). In the soluble protein fraction (Triton X-100 extraction) we do not detect a genotype- or age-specific change in levels of free ubiquitin or in the amounts of ubiquitin-conjugated proteins (Fig. 6A). This indicates that overall levels of ubiquitin and the steady-state level of soluble ubiquitin conjugates are not altered significantly for any age or genotype that was examined.
Fig. 6.

Immunoblot analysis of ubiquitin-conjugated proteins from adult Drosophila heads. Sequential protein extracts from heads of (1) wild-type, (2)bchs4/Df(2L)clot7, (3)bchs3/Df(2L)clot7, and (4) Df(2L)dsf3/Df(2L)clot7 flies were examined by Western blot analysis for free ubiquitin and conjugated forms of the protein. A, In the Triton X-100 fraction (readily soluble proteins) the levels of free ubiquitin and high molecular weight ubiquitin-conjugated proteins were not altered significantly for any genotype or age that was examined (1 or 14 d). Significant differences in less soluble ubiquitin-conjugated proteins also were not detected in young animals after more stringent extractions (SDS fraction). B, At 14 d there is a substantial increase in the amount of less soluble high molecular weight ubiquitinated conjugates in the SDS fraction for all three bchs mutant combinations, but not for wild-type controls. The bchs4 allele (2) has a nearly ninefold increase, whereas the Df(2L)dsf3/Df(2L)clot7 deletion genotype (4) has the highest level of accumulation (22.8-fold increase) when compared with wild-type controls. Results are summarized in Table 1.
Consistent with the absence of CNS aggregates in 1-d-oldbchs mutant adults, levels of ubiquitin-conjugated proteins from the insoluble protein fraction (SDS) are not significantly different for any particular genotype (genotype-independent) (Fig. 6B). This reinforces the observation that loss of bchs does not result in gross developmental defects associated with protein processing or turnover. However, in the insoluble protein fraction olderbchs mutants do demonstrate a sharp increase in levels of high molecular weight ubiquitinated proteins (genotype-dependent) (Fig.6B). When normalized to histone 2B (load control) and compared with age-matched wild-type controls, bchs mutant brains show between a ninefold (bchs4) and a 23-fold [Df(2L)dsf3/Df(2L)clot7] increase in levels of insoluble ubiquitinated proteins (Table 1). Along with the formation of age-dependent CNS protein inclusions (Fig. 5), these data indicate that a significant fraction of ubiquitinated, aggregated proteins is in an insoluble conformation. Given the potential role of BCHS in vesicle transport, these data suggest that the lack of BCHS may lead to alterations in the transport of proteins that are to be degraded.
Discussion
Loss of bchs identifies a novel degenerative pathway in Drosophila
In this paper we demonstrate that a loss of theDrosophila blue cheese gene results in premature adult death. Elimination of the protein by gene deletion, interruption of transcription with transposable P-elements, or removal of the translational start site all result in the same premature death phenotype. Of the 17 mutant bchs alleles tested for average life span, all were found to die early and behave as recessive loss-of-function mutations. For the alleles that were examined in detail, we find a tight correlation between lack of adult BCHS expression, premature death, and protein aggregate formation in the CNS. These data, along with phenotype rescue in those lines with clean P-element excisions and the absence of similar phenotype for other mutations in nearby genes (i.e., dsf), clearly indicate that removal of BCHS significantly reduces adult life span.
The decrease in longevity for bchs mutants is closely associated with the progressive formation of CNS-wide protein aggregates, which stain with thioflavine S. Although direct demonstration of ubiquitin/APPL coaggregates will require immunoelectron microscopy, at the level of confocal microscopy these inclusions at least partly consist of ubiquitinated proteins with APPL. Consistent with the thioflavine S staining of aggregates, Western blot analysis reveals that a substantial fraction of ubiquitinated proteins is located in the Triton X-100 insoluble protein fraction. This suggests that a sequestering mechanism may be functioning to buffer the cytotoxic effects of protein aggregate formation within the mutant CNS. The progressive formation of protein aggregates in bchsadults could be the precipitating factor initiating a degenerative cascade. The downstream consequences are the loss of retinal and CNS size and morphology, apoptosis of neurons, and premature death of olderbchs mutant individuals. These phenotypes define a novel mechanism of progressive CNS-wide neural degeneration forDrosophila.
Although there may be subtle developmental defects associated with a loss of bchs, several mechanistic functions for the protein can be eliminated because of a notable lack of phenotypes involving gross failures in nervous system development or synaptic function. Unlike other genes with major roles in neuronal differentiation or axonal path finding, bchs mutant animals emerge as adults at normal genetic ratios and breed with apparently normal efficiency. This also implies that many aspects of protein trafficking associated with earlier stages of development are substantially correct. The survival of BCHS mutants to adulthood also suggests that the protein is not essential for more global aspects of protein trafficking or vesicle transport. Therefore, it does not perform similar functions to those attributed to the Drosophila hrs gene (encoding a Snap 25 binding protein) (Lloyd et al., 2002). Similarly, youngbchs mutant adults do not demonstrate behavioral deficits expected of defective synaptic vesicle release, recycling, or refilling, such as those shown by the temperature-sensitive allele of the GTPase dynamin encoded by the shibere gene (Seugnet et al., 1997; Kiselev et al., 2000).
The neuropathology seen in bchs mutants is unlike that of other characterized Drosophila degenerative mutations. Recessive loss-of-function mutations in bubblegum,drop-dead, swiss cheese, spongecake,or eggroll often are associated with defects in either lipid metabolism or glial maintenance of adult neuronal tissues (Buchanan and Benzer, 1993; Xiong and Montell, 1995; Kretzschmar et al., 1997; Min and Benzer, 1997, 1999; Nakano et al., 2001). When compared withbchs, these mutations tend to have an even shorter adult life span, serious behavioral defects, and different neurodegenerative pathologies, including substantial neuropil vacuolization of the CNS. In contrast, bchs mutants have a less reduced adult life span, show few early behavioral defects, and do not develop a significant number of CNS vacuoles. The presence of BCHS in mature neurons and the degenerative CNS phenotype associated with its loss suggest that BCHS has a direct role in maintaining those neurons in which it is expressed.
A possible role in protein or vesicle trafficking for BCHS is suggested by the functional motifs identified in the C terminus of BCHS, most notably the BEACH and FYVE domains. Mutations in proteins containing either the BEACH or FYVE domain result in maturation and/or trafficking defects of membrane organelles, mainly involving the endosome and lysosome (Stenmark and Aasland, 1999; Lloyd et al., 2002; Tchernev et al., 2002). Consistent with this function and bchs mutant phenotypes, it is worth noting that during a dominant misexpression screen the overexpression of BCHS in developing larval motor neurons led to changes in synaptic structures, suggesting a role in importing or removing proteins from axons (Kraut et al., 2001).
The accumulation of ubiquitin-containing protein aggregates further supports a role for BCHS in trafficking or degradation of proteins. Although we do not find major alterations in the level of free ubiquitin or soluble ubiquitin protein conjugates, we do detect age-dependent accumulation of insoluble, ubiquitinated protein aggregates and specific proteins, including APPL. This suggests that ubiquitination still occurs but that there is a defect in transport or degradation of targeted proteins (Hicke, 2001; Pickart, 2001). It is possible that even a minor fraction of proteins with altered processing or trafficking could generate the progressive accumulation of insoluble ubiquitinated protein aggregates seen in bchs mutants.
Conserved degenerative pathways
A comparison of the mutant phenotypes of bchs to the phenotypes of other Drosophila neurodegeneration models strengthens the concept of the conserved nature of degenerative pathways between divergent phyla. Of interest are the direct comparisons that can be made between human and fly dominant missense mutations in the major opsin gene and retinitis pigmentosa. These mutations alter rhodopsin association with regulatory proteins, changing endocytotic clearance of rhodopsin from rhabdomeres. As a result, ubiquitinated protein aggregates form, photoreceptor neurons undergo apoptotic death, and there is progressive retinal degeneration. By the removal of components involved in this process (i.e., rhodopsin or arrestin) or by the block of protein transport or apoptosis, retinal degeneration is prevented in Drosophila (Davidson and Steller, 1998; Jacobson, 1998; Alloway et al., 2000; Kiselev et al., 2000). This process has notable similarities to bchs-induced degeneration: the slow development of neural degeneration in photoreceptor neurons, the formation of ubiquitinated protein aggregates in certain mutant conditions, and apoptotic death of neurons. There are several pronounced differences between thebchs phenotype and the mutant rhodopsin-induced degeneration. The bchs phenotype results from loss of a protein, not expression of an abnormal protein, and its loss affects the entire CNS, not just retinal neurons. The similarities and differences between the two systems indicate a CNS-wide role for BCHS in trafficking, accumulation, or turnover of proteins. This further suggests potential connections of BCHS with a number of disorders involving the accumulation of potentially toxic protein aggregates.
In many human neurodegenerative disorders defects in membrane vesicle transport and/or protein degradation are thought to play a key role in the inability of neurons to eliminate toxic insoluble peptides effectively. Wild-type α-synuclein and huntingtin proteins associate with membrane vesicles and are thought to be involved in synaptic plasticity or trafficking of subcellular vesicles (Jo et al., 2000;Bence et al., 2001; Sharon et al., 2001; Waelter et al., 2001). Dominant gain-of-function mutations in either gene result in the accumulation of these proteins along with ubiquitinated insoluble aggregates. There is growing evidence that this is mediated in part via a direct alteration in proteasome function. Similarly, an increase in cytotoxic amyloid Aβ1-42 peptide production and Alzheimer's disease may be associated with aberrant transport of APP, resulting in altered peptide production or degradation (Cataldo et al., 1996; Gandy and Petanceska, 2000;Gunawardena and Goldstein, 2001). Thus there are clear precedents in humans for changes in protein or vesicle trafficking that lead tobchs-like neurodegenerative phenotypes.
The similarity of bchs phenotypes to aspects of human neural degenerative disease and the high degree of similarity betweenDrosophila BCHS and the human BCHS raise important questions concerning the role of human BCHS in neural maintenance and neuropathology. Does human BCHS share conserved functions withDrosophila BCHS? This seems likely given the functional and phenotypic similarities between other Drosophilaneurodegenerative mutations and their human counterparts and the similar phenotypic consequences when human degeneration-inducing proteins or protein motifs are introduced into Drosophila. Interestingly, the human bchs locus, KIAA0993, Locus Link 23001 (previously identified from partial 3′ cDNAs) maps to a region associated with several cases of familial neurodegenerative disorders. In support of a role for mammalian BCHS in the brain, Northern blot analysis that uses mouse bchs cDNA probes reveals a 12 kbbchs transcript in murine neuronal cell lines and brain tissue (data not shown). Although human neuronal tissues have not been tested for BCHS expression, the possibility remains that the human counterpart is required for long-term function and survival of the nervous system, especially with regard to protein trafficking and processing. Ultimately, the identification of a novel degenerative pathway in a genetically tractable system opens up new areas of study involving ubiquitination, age-dependent protein aggregate formation, and their role concerning the health and survival of the nervous system.
Note added in proof. After the analysis for Figure 1C was completed, a full-length sequence for human BCHS appeared in GenBank under the name ALFY, NP_055806.1. This results from unpublished work of Simonsen, Birkeland, and Stenmark. Homology begins at BCHS amino acid 48 and extends to amino acid 3486. Identity based on BLAST alignment is 47% (63% positive).
Footnotes
This work was supported by National Institutes of Health Grants R01 MH57460 and R03 AG19614(M.M.), R01 GM56920A (B.J.T.), and R01 NS09658 (David Schubert). K.D.F. was supported by a National Institutes of Health Postdoctoral Training Grant (Salk Institute), National Institute of Neurological Disorders and Stroke Postdoctoral Fellowship, and the Pioneer Foundation. M.M. was a member of the National Cancer Institute-supported Cancer Center at the Salk Institute. We thank David Schubert for his support and advice regarding the area of neurodegeneration. We also thank Laura Knittel, David Acevedo, Michael Blower, Carlos Arias, and Charles Peto for technical assistance and Elizabeth Callan-Grabowski and Catherine Browning with help in manuscript preparation. Finally, we thank Dr. Kuo-Fen Lee for use of the confocal microscope facilities.
Correspondence should be addressed to either of the following: Kim D. Finley, Cellular Neurobiology Laboratory, The Salk Institute for Biological Studies, La Jolla, CA 92037-1099, E-mail: finley@salk.edu; or Michael McKeown, Molecular Biology, Cell Biology, and Biochemistry, GJ-363, Brown University, 69 Brown Street, Providence, RI 02912, E-mail: Michael_McKeown@Brown.edu.
References
- 1.Abrams JM. An emerging blueprint for apoptosis in Drosophila. Trends Cell Biol. 1999;9:435–440. doi: 10.1016/s0962-8924(99)01646-3. [DOI] [PubMed] [Google Scholar]
- 2.Alloway PG, Howard L, Dolph PJ. The formation of stable rhodopsin–arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron. 2000;28:129–138. doi: 10.1016/s0896-6273(00)00091-x. [DOI] [PubMed] [Google Scholar]
- 3.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 4.Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT. Imaging of amyloid-β deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–372. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- 5.Barbosa MD, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, Brandt SJ, Chotai D, Hodgman C, Solari RC, Lovett M, Kingsmore SF. Identification of the homologous beige and Chediak–Higashi syndrome genes. Nature. 1996;382:262–265. doi: 10.1038/382262a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin–proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 7.Buchanan RL, Benzer S. Defective glia in the Drosophila brain degeneration mutant drop-dead. Neuron. 1993;10:839–850. doi: 10.1016/0896-6273(93)90200-b. [DOI] [PubMed] [Google Scholar]
- 8.Burd CG, Emr SD. Phosphatidylinositol 3-phosphate signaling mediated by specific binding to RING FYVE domains. Mol Cell. 1998;2:157–162. doi: 10.1016/s1097-2765(00)80125-2. [DOI] [PubMed] [Google Scholar]
- 9.Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA. Properties of the endosomal–lysosomal system in the human CNS: disturbances mark most neurons in populations at risk to degenerate in Alzheimer's disease. J Neurosci. 1996;16:186–199. doi: 10.1523/JNEUROSCI.16-01-00186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan HY, Bonini NM. Drosophila models of human neurodegenerative disease. Cell Death Differ. 2000;7:1075–1080. doi: 10.1038/sj.cdd.4400757. [DOI] [PubMed] [Google Scholar]
- 11.Cormont M, Mari M, Galmiche A, Hofman P, Le Marchand-Brustel Y. A FYVE-finger-containing protein, Rabip4, is a Rab4 effector involved in early endosomal traffic. Proc Natl Acad Sci USA. 2001;98:1637–1642. doi: 10.1073/pnas.031586998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corvera S, D'Arrigo A, Stenmark H. Phosphoinositides in membrane traffic. Curr Opin Cell Biol. 1999;11:460–465. doi: 10.1016/S0955-0674(99)80066-0. [DOI] [PubMed] [Google Scholar]
- 13.Davidson FF, Steller H. Blocking apoptosis prevents blindness in Drosophila retinal degeneration mutants. Nature. 1998;391:587–591. doi: 10.1038/35385. [DOI] [PubMed] [Google Scholar]
- 14.Ding Q, Lewis JJ, Strum KM, Dimayuga E, Bruce-Keller AJ, Dunn JC, Keller JN. Polyglutamine expansion, protein aggregation, proteasome activity, and neural survival. J Biol Chem. 2002;277:13935–13942. doi: 10.1074/jbc.M107706200. [DOI] [PubMed] [Google Scholar]
- 15.Faigle W, Raposo G, Tenza D, Pinet V, Vogt AB, Kropshofer H, Fischer A, de Saint-Basile G, Amigorena S. Deficient peptide loading and MHC class II endosomal sorting in a human genetic immunodeficiency disease: the Chediak–Higashi syndrome. J Cell Biol. 1998;141:1121–1134. doi: 10.1083/jcb.141.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feany MB, Bender W. A Drosophila model of Parkinson's disease. Nature. 2000;204:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 17.Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, McCall A, Canal I, Orr HT, Zoghbi HY, Botas J. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- 18.Finley KD, Edeen PT, Foss M, Gross E, Ghbeish N, Palmer RH, Taylor BJ, McKeown M. dissatisfaction encodes a Tailless-like nuclear receptor expressed in a subset of CNS neurons controlling Drosophila sexual behavior. Neuron. 1998;21:1363–1374. doi: 10.1016/s0896-6273(00)80655-8. [DOI] [PubMed] [Google Scholar]
- 19.Gandy S, Petanceska S. Regulation of Alzheimer β-amyloid precursor trafficking and metabolism. Biochim Biophys Acta. 2000;1502:44–52. doi: 10.1016/s0925-4439(00)00031-4. [DOI] [PubMed] [Google Scholar]
- 20.Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32:389–401. doi: 10.1016/s0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- 21.Hay BA, Maile R, Rubin GM. P-element insertion-dependent gene activation in the Drosophila eye. Proc Natl Acad Sci USA. 1997;94:5195–5200. doi: 10.1073/pnas.94.10.5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hicke L. A new ticket for entry into budding vesicles—ubiquitin. Cell. 2001;106:527–530. doi: 10.1016/s0092-8674(01)00485-8. [DOI] [PubMed] [Google Scholar]
- 23.Jacobson MD. Anti-apoptosis therapy: a way of treating neural degeneration? Curr Biol. 1998;8:R418–R421. doi: 10.1016/s0960-9822(98)70267-2. [DOI] [PubMed] [Google Scholar]
- 24.Jo E, McLaurin J, Yip CM, St. George-Hyslop P, Fraser PE. α-Synuclein membrane interactions and lipid specificity. J Biol Chem. 2000;275:34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 25.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kazantsev A, Walker HA, Slepko N, Bear JE, Preisinger E, Steffan JS, Zhu YZ, Gertler FB, Housman DE, Marsh JL, Thompson LM. A bivalent huntingtin binding peptide suppresses polyglutamine aggregation and pathogenesis in Drosophila. Nat Genet. 2002;30:367–376. doi: 10.1038/ng864. [DOI] [PubMed] [Google Scholar]
- 27.Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science. 2000;287:1837–1840. doi: 10.1126/science.287.5459.1837. [DOI] [PubMed] [Google Scholar]
- 28.Kiselev A, Socolich M, Vinos J, Hardy RW, Zuker CS, Ranganathan R. A molecular pathway for light-dependent photoreceptor apoptosis in Drosophila. Neuron. 2000;28:139–152. doi: 10.1016/s0896-6273(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 29.Kraut R, Menon K, Zinn K. A gain-of-function screen for genes controlling motor axon guidance and synaptogenesis in Drosophila. Curr Biol. 2001;11:417–430. doi: 10.1016/s0960-9822(01)00124-5. [DOI] [PubMed] [Google Scholar]
- 30.Kretzschmar D, Hasan G, Sharma S, Heisenberg M, Benzer S. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J Neurosci. 1997;17:7425–7432. doi: 10.1523/JNEUROSCI.17-19-07425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kutateladze TG, Ogburn KD, Watson WT, de Beer T, Emr SD, Burd CG, Overduin M. Phosphatidylinositol 3-phosphate recognition by the FYVE domain. Mol Cell. 1999;3:805–811. doi: 10.1016/s1097-2765(01)80013-7. [DOI] [PubMed] [Google Scholar]
- 32.Lai EC, Deblandre GA, Kintner C, Rubin GM. Drosophila neuralized is a ubiquitin ligase that promotes the internalization and degradation of delta. Dev Cell. 2001;1:783–794. doi: 10.1016/s1534-5807(01)00092-2. [DOI] [PubMed] [Google Scholar]
- 33.Lloyd TE, Atkinson R, Wu MN, Zhou Y, Pennetta G, Bellen HJ. Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell. 2002;108:261–269. doi: 10.1016/s0092-8674(02)00611-6. [DOI] [PubMed] [Google Scholar]
- 34.Marsh JL, Walker H, Theisen H, Zhu YZ, Fielder T, Purcell J, Thompson LM. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Hum Mol Genet. 2000;9:13–25. doi: 10.1093/hmg/9.1.13. [DOI] [PubMed] [Google Scholar]
- 35.Min KT, Benzer S. spongecake and eggroll: two hereditary diseases in Drosophila resemble patterns of human brain degeneration. Curr Biol. 1997;7:885–888. doi: 10.1016/s0960-9822(06)00378-2. [DOI] [PubMed] [Google Scholar]
- 36.Min KT, Benzer S. Preventing neurodegeneration in the Drosophila mutant bubblegum. Science. 1999;284:1985–1988. doi: 10.1126/science.284.5422.1985. [DOI] [PubMed] [Google Scholar]
- 37.Mohtashami M, Stewart BA, Boulianne GL, Trimble WS. Analysis of the mutant Drosophila N-ethylmaleimide sensitive fusion-1 protein in comatose reveals molecular correlates of the behavioural paralysis. J Neurochem. 2001;77:1407–1417. doi: 10.1046/j.1471-4159.2001.00363.x. [DOI] [PubMed] [Google Scholar]
- 38.Muqit MM, Feany MB. Modeling neurodegenerative diseases in Drosophila: a fruitful approach? Nat Rev Neurosci. 2002;3:237–243. doi: 10.1038/nrn751. [DOI] [PubMed] [Google Scholar]
- 39.Mutsuddi M, Nambu JR. Neural disease: Drosophila degenerates for a good cause. Curr Biol. 1998;8:R809–R811. doi: 10.1016/s0960-9822(07)00506-4. [DOI] [PubMed] [Google Scholar]
- 40.Nagle DL, Karim MA, Woolf EA, Holmgren L, Bork P, Misumi DJ, McGrail SH, Dussault BJ, Jr, Perou CM, Boissy RE, Duyk GM, Spritz RA, Moore KJ. Identification and mutation analysis of the complete gene for Chediak–Higashi syndrome. Nat Genet. 1996;14:307–311. doi: 10.1038/ng1196-307. [DOI] [PubMed] [Google Scholar]
- 41.Nakano Y, Fujitani K, Kurihara J, Ragan J, Usui-Aoki K, Shimoda L, Lukacsovich T, Suzuki K, Sezaki M, Sano Y, Ueda R, Awano W, Kaneda M, Umeda M, Yamamoto D. Mutations in the novel membrane protein Spinster interfere with programmed cell death and cause neural degeneration in Drosophila melanogaster. Mol Cell Biol. 2001;21:3775–3788. doi: 10.1128/MCB.21.11.3775-3788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neer EJ, Schmidt CJ, Nambudripad R, Smith TF. The ancient regulatory-protein family of WD-repeat proteins. Nature. 1994;371:297–300. doi: 10.1038/371297a0. [DOI] [PubMed] [Google Scholar]
- 43.Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B. The A53T α-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci. 2000;20:6048–6054. doi: 10.1523/JNEUROSCI.20-16-06048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phillips JP, Campbell SD, Michaud D, Charbonneau M, Hilliker AJ. Null mutation of copper/zinc superoxide dismutase in Drosophila confers hypersensitivity to paraquat and reduced longevity. Proc Natl Acad Sci USA. 1989;86:2761–2765. doi: 10.1073/pnas.86.8.2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pickart CM. Ubiquitin enters the new millennium. Mol Cell. 2001;8:499–504. doi: 10.1016/s1097-2765(01)00347-1. [DOI] [PubMed] [Google Scholar]
- 46.Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001;11:1114–1125. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rio DC. Regulation of Drosophila P-element transposition. Trends Genet. 1991;7:282–287. doi: 10.1016/0168-9525(91)90309-E. [DOI] [PubMed] [Google Scholar]
- 48.Robinow S, White K. The locus elav of Drosophila melanogaster is expressed in neurons at all developmental stages. Dev Biol. 1988;126:294–303. doi: 10.1016/0012-1606(88)90139-x. [DOI] [PubMed] [Google Scholar]
- 49.Rorth P. A modular misexpression screen in Drosophila detecting tissue-specific phenotypes. Proc Natl Acad Sci USA. 1996;93:12418–12422. doi: 10.1073/pnas.93.22.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seugnet L, Simpson P, Haenlin M. Requirement for dynamin during Notch signaling in Drosophila neurogenesis. Dev Biol. 1997;192:585–598. doi: 10.1006/dbio.1997.8723. [DOI] [PubMed] [Google Scholar]
- 51.Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ. α-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci USA. 2001;98:9110–9115. doi: 10.1073/pnas.171300598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 53.Shoelson SE. SH2 and PTB domain interactions in tyrosine kinase signal transduction. Curr Opin Chem Biol. 1997;1:227–234. doi: 10.1016/s1367-5931(97)80014-2. [DOI] [PubMed] [Google Scholar]
- 54.Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 55.Stenmark H, Aasland R. FYVE-finger proteins—effectors of an inositol lipid. J Cell Sci. 1999;112:4175–4183. doi: 10.1242/jcs.112.23.4175. [DOI] [PubMed] [Google Scholar]
- 56.Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol. 2001;153:283–294. doi: 10.1083/jcb.153.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tchernev VT, Mansfield TA, Giot L, Kumar AM, Nandabalan K, Li Y, Mishra VS, Detter JC, Rothberg JM, Wallace MR, Southwick FS, Kingsmore SF. The Chediak–Higashi protein interacts with SNARE complex and signal transduction proteins. Mol Med. 2002;8:56–64. [PMC free article] [PubMed] [Google Scholar]
- 58.Torroja L, Luo L, White K. APPL, the Drosophila member of the APP family, exhibits differential trafficking and processing in CNS neurons. J Neurosci. 1996;16:4638–4650. doi: 10.1523/JNEUROSCI.16-15-04638.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waelter S, Scherzinger E, Hasenbank R, Nordhoff E, Lurz R, Goehler H, Gauss C, Sathasivam K, Bates GP, Lehrach H, Wanker EE. The huntingtin interacting protein HIP1 is a clathrin and α-adaptin-binding protein involved in receptor-mediated endocytosis. Hum Mol Genet. 2001;10:1807–1817. doi: 10.1093/hmg/10.17.1807. [DOI] [PubMed] [Google Scholar]
- 60.Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 61.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 62.Wurmser AE, Gary JD, Emr SD. Phosphoinositide 3-kinases and their FYVE domain-containing effectors as regulators of vacuolar/lysosomal membrane trafficking pathways. J Biol Chem. 1999;274:9129–9132. doi: 10.1074/jbc.274.14.9129. [DOI] [PubMed] [Google Scholar]
- 63.Xiong WC, Montell C. Defective glia induce neuronal apoptosis in the repo visual system of Drosophila. Neuron. 1995;14:581–590. doi: 10.1016/0896-6273(95)90314-3. [DOI] [PubMed] [Google Scholar]