

Abstract
The metabolic control of respiration is still poorly understood, due mainly to the lack of suitable approaches for studying it in vivo. Experiments on isolated mammalian mitochondria have indicated that a relatively small fraction of each of several components of the electron transport chain is sufficient to sustain a normal O2 consumption rate. These experiments, however, may not reflect accurately the in vivo situation, due to the lack in the mitochondrial fraction of essential cytosolic components and to the use of excess of substrates in the in vitro assays. An approach is described here whereby the control of respiration by cytochrome c oxidase (COX; EC 1.9.3.1) was analyzed in intact cultured human osteosarcoma 143B.TK− cells and other wild-type cells and in mitochondrial DNA mutation-carrying human cell lines. Surprisingly, in wild-type cells, only a slightly higher COX capacity was detected than required to support the endogenous respiration rate, pointing to a tighter in vivo control of respiration by COX than generally assumed. Cell lines carrying the MERRF mitochondrial tRNALys gene mutation, which causes a pronounced decrease in mitochondrial protein synthesis and respiration rates, revealed, in comparison, a significantly greater COX capacity relative to the residual endogenous respiration rate, and, correspondingly, a higher COX inhibition threshold above which the overall respiratory flux was affected. The observed relationship between COX respiratory threshold and relative COX capacity and the potential extension of the present analysis to other respiratory complexes have significant general implications for understanding the pathogenetic role of mutations in mtDNA-linked diseases and the tissue specificity of the mutation-associated phenotype.
Keywords: flux control; N,N,N′,N′-tetramethyl-p-phenylenediamine; polarography; MERRF mutation
The rapid accumulation of knowledge concerning mitochondrial diseases, especially those caused by mitochondrial DNA (mtDNA) mutations, has stimulated in recent years a strong interest in the metabolic control of oxidative phosphorylation (OXPHOS). In particular, the discovery of threshold effects in the capacity of a mtDNA mutation to produce an OXPHOS defect in the presence of varying amounts of wild-type mtDNA has called attention to the degree of control that a particular step exerts in the OXPHOS pathway. It has been emphasized that application of the metabolic control theory (1, 2) to the study of mitochondrial metabolism can be a valuable approach for determining the level of control exerted by different OXPHOS steps on the rate of mitochondrial respiration (3), and for identifying and quantifying enzymatic defects caused in the OXPHOS machinery by mitochondrial or nuclear DNA mutations (4–7). Recently, this approach has been applied to isolated rat tissue mitochondria, by using increasing concentrations of inhibitors of complex I (rotenone), complex III (myxothiazol or antimycin A), or complex IV (potassium cyanide) to mimic the effects of mutations affecting these complexes (4–7). These experiments have produced results suggesting that the activity of each of several components of the respiratory chain is in relatively large excess over the amount required to support the endogenous respiration rate. However, a limitation in the metabolic control analysis applied to isolated mitochondria is the significant alteration of the in vivo situation produced by the possible loss of essential metabolites, especially under nonrigorous isolation conditions, and by the disruption of the normal interactions of mitochondria with cytosolic components that may play a crucial role in channeling of respiratory substrates to the organelles. Furthermore, the in vitro use of saturating concentrations of a given respiratory substrate may alter the control exerted by the corresponding step in the pathway. To obviate these problems, in the present work, an approach has been developed for the analysis of overall electron flux to oxygen in intact cells and of the control by cytochrome c oxidase (COX; EC 1.9.3.1) of the respiratory flux. This approach has been applied to several types of wild-type cultured human cells and to mutant cell lines (8) carrying the mitochondrial tRNALys gene mutation associated with the MERRF encephalomyopathy (9). These experiments unexpectedly have revealed that the wild-type cell lines tested have only a slightly higher COX capacity than required to support the endogenous respiration rate. Furthermore, in the mutant cell lines, a significantly greater COX capacity relative to the residual endogenous respiration rate was detected. These observations and their potential extension to other respiratory complexes are relevant for understanding the mechanisms underlying the mtDNA mutation-associated phenotype in mitochondrial diseases and, in general, the metabolic control of respiration.
MATERIALS AND METHODS
Cell Lines and Media.
The pT1 and pT4 human cell lines, carrying in nearly homoplasmic form the A-to-G transition at position 8344 in the mitochondrial tRNALys gene that is associated with the MERRF syndrome (9), and the pT3 cell line, carrying in homoplasmic form the wild-type version of the tRNALys gene, previously were isolated by transfer into human mtDNA-less (ρ0) 206 cells of mitochondria from myoblasts of a MERRF patient (8). The transformants were grown in DMEM, supplemented with 10% fetal bovine serum (FBS) and 100 μg/ml bromodeoxyuridine (BrdUrd). The parental line of ρ0206, 143B.TK− (10), was grown in DMEM with 5% FBS and 100 μg/ml BrdUrd. The cell line 701.2.8c, which is a simian virus 40 T-antigen gene-transformed derivative (B. Wold, personal communication) of the human fibroblast strain GM701 (11), and HeLa cells (strain ATCC CCL2) were grown in DMEM with 10% FBS. The ρ0206 cell line was grown in DMEM with 5% FBS, 100 μg/ml BrdUrd, and 50 μg/ml uridine.
DNA Analysis.
Quantification of the MERRF mutation in the mitochondrial tRNALys gene was carried out by PCR amplification of a mtDNA segment encompassing that mutation and allele-specific termination of primer extension (12) using an appropriate 5′-32P-labeled primer.
Measurements of Endogenous and [Ascorbate + N,N,N′,N′-Tetramethyl-p-phenylenediamine (TMPD)]-Dependent Respiration in Intact Cells.
Exponentially growing 143B, HeLa, or 701.2.8c cells, fluid changed the day before the measurements, were collected by trypsinization and centrifugation, and resuspended at 3 to 4 × 106 cells per ml in 1.5 ml of DMEM lacking glucose, supplemented with 5% FBS, equilibrated at 37°C. Other cell samples were washed once in Tris-based, Mg2+-, Ca2+-deficient (TD) buffer (0.137 M NaCl/5 mM KCl/0.7 mM Na2HPO4/25 mM Tris·HCl, pH 7.4 at 25°C), and resuspended at 3 to 4 × 106 cells per ml in 1.5 ml of the same buffer at 37°C. Each sample was transferred into a 1.5-ml water-jacketed chamber containing a small magnetic bar (Gilson), and connected to a circulating water bath at 37°C and a YS model 5300 biological oxygen monitor (Yellow Springs Instruments), and recording of oxygen consumption was started. The cell samples resuspended in TD buffer were read directly and after addition of 2,4-dinitrophenol (DNP) to 25–30 μM, or of DNP and rotenone (Sigma) (100 nM), or of DNP and antimycin A (Sigma) (20 nM), or of DNP and potassium cyanide (KCN) (2 mM), or of DNP, antimycin A, TMPD (Fluka) (0.1 to 2.0 mM), and sodium ascorbate (10 mM).
KCN Titration of COX Activity in Intact Cells.
Exponentially growing cells, fluid changed the day before, were collected by trypsinization and centrifugation, and resuspended, at either 2 to 4 × 106 cells per ml (143B, pT3) or 1 to 1.5 × 107 cells per ml (pT1, pT4), in TD buffer containing 25–30 μM DNP for KCN titration of “integrated” COX activity, or in TD buffer containing 25–30 μM DNP/20 nM antimycin A/0.2 mM TMPD/10 mM sodium ascorbate, equilibrated at 37°C, for KCN titration of “isolated” COX activity. Two 1.5-ml portions of each cell suspension were transferred into chambers attached to the same circulating water bath and the same oxygen monitor. After measurement of the starting oxygen consumption rate, small amounts of a 10 mM KCN solution in H2O, adjusted to neutral pH with HCl, were sequentially introduced at appropriate intervals into one of the chambers near the bottom, using a Hamilton syringe fitted with a 2.5-inch (6.3-cm)-long needle (Hamilton). The intervals of addition were chosen to allow adequate time for constant slope measurements and to construct detailed inhibition curves. The KCN-inhibited activity was measured after each addition of KCN and expressed as a percentage of the uninhibited activity measured, in parallel, in the other chamber. For the measurement of (ascorbate + TMPD)-dependent isolated COX activity, the oxygen consumption rate was corrected for nonspecific KCN-insensitive oxidations, as recorded in a control experiment with mtDNA-less ρ0206 cells, which contain no cytochrome c oxidase and show no detectable KCN-sensitive (ascorbate + TMPD)-dependent respiration.
Oxygen Consumption Measurements in Digitonin-Permeabilized Cells.
Exponentially growing 143B.TK−, pT3, pT1, or pT4 cells were collected by trypsinization and centrifugation, resuspended at 4 to 8 × 106 cells per ml in 1.5 ml of measurement buffer (13) at 37°C and transferred into a 1.5-ml Gilson chamber. After equilibration for a few minutes, the coupled endogenous respiration rate was measured, and digitonin was added from a 10% solution in dimethyl sulfoxide to 15 μg per 106 cells. After another equilibration for 10–15 min, sodium malate and glutamate were added to 5 mM, and, after adequate time for accurate slope measurement, rotenone was added to 100 nM. After maximal inhibition of O2 consumption was reached, sodium succinate and glycerol 3-phosphate were added to 5 mM. After an appropriate time, antimycin A was introduced into the chamber to 20 nM, and, after maximal inhibition of respiration was obtained, sodium ascorbate was added to 10 mM and TMPD, to 0.2 mM (13). A parallel experiment was carried out with the same cell suspension in the absence of inhibitors.
RESULTS AND DISCUSSION
Measurements of Endogenous Respiration Rate.
Fig. 1a shows that the osteoblastoma-derived human cell line 143B.TK− (10) can respire in a Tris/NaCl/NaHPO4 buffer (TD buffer), using endogenous substrates, at a rate indistinguishable from that observed in DMEM lacking glucose, which often is used for O2 consumption measurements on intact cells (14). This rate corresponds to ≈4.5 fmol of O2 per min per cell. In both TD buffer and DMEM (lower-glucose) medium, the respiration rate remains practically constant for the duration of an oxygraphic run (not shown). Addition of DNP to the TD buffer increases the O2 consumption rate by ≈35% (Fig. 1a). In all the experiments described below, the endogenous respiration rate was measured in the presence of DNP, to avoid any influence of proton cycling and OXPHOS on the respiratory flux. Accordingly, in Fig. 1a and subsequent figures, the DNP-uncoupled endogenous respiration rate is set equal to 100%. As shown in Fig. 1a, the endogenous respiration rate of DNP-uncoupled 143B cells is nearly completely sensitive to 100 nM rotenone, 20 nM antimycin A, or 2 mM KCN, specific inhibitors, respectively, of NADH dehydrogenase (complex I), bc1 complex (complex III), and COX (complex IV).
Figure 1.

Measurements of endogenous respiration rate or (ascorbate + TMPD)-dependent respiration rate by intact 143B.TK− cells. (a) Relative oxygen consumption rate by cells in DMEM lacking glucose, or in TD buffer in the absence or presence of DNP, or in TD buffer in the presence of DNP and either rotenone (Rot) or antimycin A (Ant) or KCN, or in TD buffer in the presence of DNP, antimycin A, ascorbate, and 0.2 mM TMPD. The data represent the means ± SE obtained in two determinations made in DMEM (−glucose) or TD, 7 determinations in TD+DNP, one determination in TD+ DNP+rotenone, or in TD+DNP+antimycin A, or in TD+DNP+KCN, and 12 determinations in TD+DNP, antimycin A, ascorbate, and 0.2 mM TMPD. (b) Dependence on concentration of TMPD, in the presence of a constant concentration of ascorbate, of the initial oxygen consumption rate associated with isolated COX activity in DNP- and antimycin A-treated 143B.TK− cells (•), HeLa cells (□), and the fibroblast cell line 701.2.8c (▵), expressed as a percentage of the endogenous rate. The data shown represent the means ± SE obtained in 2–12 determinations on 143B cells tested at each of different concentrations of TMPD (the error bars that fall within the individual data symbols are not shown), or the results of single determinations on HeLa cells or 7012–8c cells.
Test of Isolated COX Activity in Intact Cells.
By using the above system, the isolated COX activity could be tested polarographically by supplying the cells with 20 nM antimycin A to suppress the electron flow in the upstream segment of the respiratory chain, 0.2 mM TMPD as membrane-permeant one-equivalent electron donor, and an excess (10 mM) of ascorbate as primary reducing agent. The exclusive addition of ascorbate to antimycin A-inhibited cells did not result in any significant respiratory activity (not shown). It appears from Fig. 1a that the (ascorbate + 0.2 mM TMPD)-supported O2 consumption rate in antimycin A-inhibited, DNP-uncoupled 143B cells was only slightly lower (−8%) than the uncoupled endogenous respiration rate (Fig. 1a). This observation indicated that 0.2 mM TMPD was able to reduce cytochrome c at a rate sufficient to maintain a near-normal respiratory flux. It also was observed that permeabilization with digitonin of antimycin A- and (ascorbate + TMPD)-treated cells did not significantly affect the O2 consumption rate supported by ascorbate and TMPD (data not shown). This observation argues against any significant role in the present experiments of insufficient plasma membrane permeability to TMPD.
Fig. 1b shows a more detailed analysis of the dependence on the concentration of TMPD (from 0 to 2 mM), in the presence of a constant concentration of ascorbate (10 mM), of the COX activity, measured as an isolated step, in DNP- and antimycin A-treated 143B cells, HeLa cells, and fibroblasts of the human cell line 701.2.8c. It appears that, in 143B cells, the TMPD concentration is near-saturating above 2 mM, and starts becoming limiting at ≈0.2 mM, as previously observed for antimycin A-inhibited beef heart isolated mitochondria (15). The two other human wild-type cell lines, genetically unrelated, gave a substantially identical titration curve for TMPD. In the present work, a concentration of TMPD of 0.2 mM was chosen, because it supports a rate of respiration very close to the endogenous one in wild-type cells and also minimizes a possible direct electron transfer from TMPD to COX (16, 17).
KCN Titration of COX Activity in Intact Cells as Isolated Step and as Integrated Step.
COX activity was titrated with the specific inhibitor KCN both as isolated step, using 0.2 mM TMPD and 10 mM ascorbate as electron donors, in the presence of antimycin A as upstream inhibitor, and as respiratory chain integrated step (endogenous respiration) in the absence of ascorbate and TMPD in non-antimycin A-treated cells. In this way, it was possible to determine, at each concentration of KCN used, the percentage of inhibition of the isolated COX activity and how this inhibition affects the respiratory flux (4, 5, 7). It had been shown by others that, in isolated mitochondria, direct spectrophotometric titration by KCN of the isolated COX activity supported by an excess of reduced cytochrome c gives the same curve as the polarographic KCN titration of O2 consumption in the presence of ascorbate and 0.2 mM TMPD (5). We verified that, in intact 143B cells, the sensitivities to KCN of the isolated COX activity promoted by 0.2 mM and by 1.0 mM TMPD, as measured versus increasing concentrations of the inhibitor, were substantially identical (within 10%) (data not shown). Furthermore, to exclude the possibility that an intracellular accumulation of the oxidized form of TMPD during the titration experiment would interfere with the KCN sensitivity measurements, the effect of each KCN concentration on the initial rate of (ascorbate + TMPD)-supported O2 consumption was analyzed. Again very similar results were obtained in the presence of 0.2 mM and 1.0 mM TMPD (data not shown).
Fig. 2 shows that the KCN titration curve, in the presence of ascorbate and TMPD, of antimycin A-inhibited 143B cells was quasilinear for nonsaturating concentrations of the inhibitor. On the other hand, the variation of the endogenous respiration rate over the same range of KCN concentrations produced a curve that was clearly sigmoidal in shape. Very similar results were obtained in experiments carried out in the absence of DNP (data not shown). The difference in the KCN titration curves of COX activity as isolated step and as respiratory chain integrated step was consistent with the occurrence in 143B cells of an excess of COX activity over that required to maintain a normal O2 consumption rate. However, this excess appeared to be significantly lower than previously reported for isolated rat tissue mitochondria (4, 5, 7), suggesting a tighter in vivo control of respiration by COX in 143B cells than previously assumed.
Figure 2.

Inhibition by KCN of endogenous respiration rate in TD buffer in the presence of DNP (○), or of (ascorbate + 0.2 mM TMPD)-dependent respiration rate in TD buffer in the presence of DNP and antimycin A (•), in intact 143B.TK− cells and transmitochondrial human cell lines carrying mtDNA with the mitochondrial tRNALys gene mutation (pT1, pT4) or wild-type mtDNA (pT3), derived from a patient affected by the MERRF encephalomyopathy (8). The data shown represent the means ± SE obtained in six or seven determinations on 143B cells, three determinations on pT1 cells, and four determinations on pT4 cells (the error bars that fall within the limits of the individual data symbols are not shown), or the results of a single determination on pT3 cells.
The availability of 143B.TK− cell derivatives carrying in their mtDNA various proportions of the tRNALys gene mutation associated with the MERRF encephalomyopathy produced by introducing mitochondria from myoblasts of a MERRF patient into mtDNA-less (ρ0) 143B.TK− cells (8), offered the opportunity for investigating how the respiratory flux control by COX was affected in cells containing different levels of residual COX activity spared by the mutation. The pT1 cell line virtually lacks wild-type mtDNA. The pT4 cell line contains only 6.6% wild-type mtDNA, which is very close to the genetic protective threshold (18), and the pT3 cell line is apparently free of mutant mtDNA (8). The endogenous respiration rate of pT1 cells was found to be ≈8%, and that of pT4 cells, ≈24% of the 143B rate, whereas pT3 cells exhibited a respiratory activity comparable to that of the parental wild-type cell line (8) (see also Table 1). As shown in Fig. 2, the KCN inhibition curves for the isolated COX activity of pT1 and pT4 cells were very similar to that observed for 143B cells, whereas, in pT3 cells, the isolated COX appeared to be less sensitive to the inhibitor. While it is not known whether this lower sensitivity of the isolated step in pT3 cells reflected a decreased permeability of the cells to the inhibitor or another nuclear gene-controlled change(s), occurring presumably in the original ρ0 recipient cell, it was expected that a lower sensitivity to the inhibitor also would be exhibited by the KCN titration curve of the endogenous respiration rate of pT3 cells. Indeed, as shown in Fig. 2, the relative sensitivities to KCN of the endogenous respiration rate and of the isolated enzyme activity in pT3 cells were very similar to those found in 143B cells, with the titration curve of the endogenous respiration rate exhibiting a near-sigmoidal shape. By contrast, the KCN titration curve of the endogenous respiration rate of pT1 cells indicated a pronounced decrease in sensitivity as compared with 143B cells, while the titration curve of pT4 cells exhibited an intermediate behavior (Fig. 2).
Table 1.
COX threshold, relative COX capacity (COSRmax), endogenous respiration, and activities of respiratory enzymes in 143B cells and their mutant derivatives and in partially rotenone- or antimycin A-inhibited 143B cells
| Exp. | Measurement | 143B | pT3 | pT4 | pT1 | 143B +rotenone | 143B +antimycin A |
|---|---|---|---|---|---|---|---|
| 1 | COX threshold, % | 28.2 | 21.3 | 51.2 | 82.2 | 75.2 | 63.1 |
| COXRmax | 1.40 | 1.27 | 2.00 | 5.20 | 3.96 | 2.69 | |
| 2 | Respiration rate ratio, (ascorbate + TMPD) to endogenous | 1.39 ± 0.03 | 1.20 ± 0.04 | 2.06 ± 0.06 | 5.58 ± 0.11 | 3.60 | 2.50 |
| 3 | Respiration rate, fmol O2/min/cell | ||||||
| Endogenous | 4.47 (4.54) | 4.70 (4.70) | 1.02 (1.06) | 0.34 | |||
| Substrate-dependent* | |||||||
| Glutamate + malate | 2.82 (3.06) | 1.88 (2.27) | 0.33 (0.37) | 0.06 | |||
| Succinate + G-3-P | 3.88 (4.99) | 2.83 (3.70) | 1.14 (1.38) | 0.50 | |||
| Ascorbate + TMPD | 4.24 (6.36) | 3.86 (5.13) | 1.63 (2.20) | 1.44 |
Experiment 1. Data taken from experiments of Fig. 3 and similar experiments carried out with pT3 cells and with 143B cells partially inhibited in their respiration by 3 nM rotenone or 2.7 nM antimycin A. Experiment 2. Data taken from experiment of Fig. 1b with 143B cells and similar experiments carried out with the pT3 wild-type mtDNA-carrying transformant, the pT1 and pT4 MERRF mutation-carrying transformants, and 143B cells partially inhibited by 3 nM rotenone or 2.7 nM antimycin A. The data represent the means ± SE obtained in three determinations on 143B or pT3 or pT1 or pT4 cells, and the results of single determinations on rotenone- or antimycin A-treated cells. TMPD was 0.4 mM. Experiment 3. The data shown represent the respiration rates supported by endogenous substrates or added substrates (expressed in fmol of O2 per min per cell), using rotenone at 100 nM or antimycin A at 20 nM to block respiration supported by glutamate + malate or succinate + glycerol 3-phosphate (G-3-P), respectively, or, in a parallel experiment, without inhibitors (in parentheses). TMPD was 0.2 mM.
Digitonin-permeabilized cells.
Determination of COX Respiratory Threshold.
The difference between the mutant cell lines pT1 and pT4 and the wild-type cell lines 143B and pT3 in the relative KCN sensitivities of the endogenous respiration rate and of the isolated COX activity are best illustrated by a threshold plot (5, 7, 19), i.e., a plot of the relative endogenous respiration rate of 143B cells and of the pT3, pT4, and pT1 ρ0 cell transformants against percent inhibition of isolated COX activity by the same KCN concentrations. In this plot, the endogenous respiration rate remains fairly constant with increasing inhibition of the isolated enzyme, up to the percent inhibition (threshold) at which a further decrease in enzyme activity has a marked inhibitory effect on the rate of endogenous respiration. It is clear from Fig. 3a that the thresholds of COX inhibition above which the respiratory flux starts decreasing are higher in the mutant cell lines pT1 and pT4 than in the wild-type cell lines 143B and pT3. In particular, the estimated values are ≈28% in 143B cells, ≈21% in pT3 cells, ≈51% in pT4 cells, and ≈82% in pT1 cells (indicated by arrows in the expanded plots of Fig. 3 b–d; see also Exp. 1 in Table 1).
Figure 3.

Percentage of endogenous respiration rate as a function of percent of COX inhibition in intact 143B.TK− cells (○) and pT1 (□), pT3 (⋄), and pT4 (▵) transmitochondrial cell lines (a), and maximum COX capacity in 143B.TK− (b), pT1 (c), and pT4 cells (d). (a) The values (either means ± SE obtained in several experiments with 143B.TK− cells, pT1, and pT4 cells shown in Fig. 2, or values obtained in a single experiment with pT3 cells) for percent of endogenous respiration rate and percent of COX inhibition at each KCN concentration are plotted against each other. (b–d) The percent rates of endogenous respiration in the experiments using 143B, pT1, and pT4 cells illustrated in a are plotted against percent of COX inhibition by KCN, and the least-square regression line through the symbols (filled) beyond the inflection point of each curve (arrow) is extended to zero COX inhibition. The equations describing these extrapolated lines and the probability of the data fitting are shown.
Determination of Relative COX Capacity in Intact Cells.
In the expanded plots of Fig. 3, the least-square regression lines through the filled symbols beyond the inflection point in each curve are extrapolated to zero COX inhibition. The intersections of these lines with the ordinate axis give estimates of the maximum COX capacity as integrated step, expressed as percentages of the uninhibited endogenous respiration rate (Fig. 3 b–d; see also Exp. 1 in Table 1). One can see that the maximum COX capacity is only 1.4 times and 1.27 times as high as the endogenous respiration rate in 143B cells and, respectively, pT3 cells (Table 1), whereas it is twice the endogenous respiration rate in pT4 cells and 5.2 times as high in pT1 cells. The equations describing these extrapolated lines conform to the general formula:
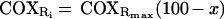 |
1 |
where x represents the percent inhibition of COX activity at [i] concentration of inhibitor, as detected by KCN titration of the isolated step (Fig. 2), COXRi represents the endogenous respiration rate at [i] concentration of inhibitor, expressed as percentage of the uninhibited endogenous respiration rate, and COXRmax represents the ratio of the maximum COX capacity to the uninhibited endogenous respiration rate. The threshold value of x at which COXRi = uninhibited endogenous respiration rate (=100) can be calculated from Eq. 1 as follows:
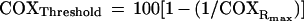 |
2 |
where COXRmax is defined above. Hereafter, COXRmax is defined as relative capacity of COX.
In the TMPD titration curve for 143B cells shown in Fig. 1b, the relative uncoupled respiration rate supported by 0.4 mM TMPD in the presence of antimycin A reached a value of 1.39 (indicated by a dashed line), which is almost identical to the COXRmax value previously determined by extrapolation of the KCN inhibition data in the threshold plot of Fig. 3b to zero COX inhibition (1.40). It is worth noticing that the relative uncoupled respiration rate of two other, genetically unrelated, human wild-type cell lines (HeLa cells and 701.2.8c cells) reached at 0.4 mM TMPD a very similar value (Fig. 1b). These observations suggested the possibility that titration of the relative uncoupled respiration rate at 0.4 mM TMPD in antimycin A-treated cells would provide a simplified approach for the estimation of the COXRmax and, therefore, of the COX respiratory threshold by Eq. 2 given above. This possibility was verified by determining the ratios of (ascorbate + 0.4 mM TMPD)-dependent respiration rate to endogenous respiration rate in intact antimycin A-treated pT1 and pT4 cells in TD + DNP. As shown in Table 1 (Experiments 1 and 2), the latter ratios were very similar, within 10%, to the values for COXRmax determined by extrapolation of the COX inhibition data in the threshold plots of Figs. 3 c and d. The observation that the relative uncoupled respiration rate supported by 0.4 mM TMPD in the presence of antimycin A reached in intact cells of several cell lines a value very close to the COXRmax value determined by extrapolation of the KCN inhibition data is in full agreement with the previous finding that, in beef heart submitochondrial particles, the steady-state level of reduction of cytochrome c reached a maximum at ≈0.4 mM TMPD (17). Presumably, any increase in O2 consumption rate occurring in the presence of concentrations of TMPD >0.4 mM reflects direct electron transfer (16, 17) to the cytochrome c oxidase.
Also shown in Table 1 are the results of experiments in which the value of COXRmax was experimentally modified in 143B cells by exposing them to a low concentration of rotenone (≈3 nM) or antimycin A (≈2.7 nM) to decrease the rate of their endogenous respiration. It is clear that both rotenone and antimycin A increase the COX respiratory threshold and the COXRmax to values intermediate between those observed in pT4 and pT1 cells. Furthermore, here too, the values for COXRmax determined by extrapolation to zero COX inhibition of the KCN inhibition data in the partially rotenone- or antimycin A-inhibited cells are very similar to the ratios of (ascorbate + 0.4 mM TMPD)-dependent respiration rate to endogenous respiration rate determined under the same conditions.
Relationship of COX Respiratory Threshold to Relative COX Capacity.
This relationship, described by Eq. 2, has important implications for the metabolic control of respiration. Fig. 4 shows a plot of the COX respiratory thresholds (derived from the KCN inhibition data) versus the independently determined ratios of (ascorbate + 0.4 mM TMPD)-dependent respiration rate to endogenous respiration rate, used as estimators of COXRmax, in intact 143B, pT1, and pT4 cells, as well as in 3 nM rotenone- or 2.7 nM antimycin A-treated 143B cells. The theoretical value of COX threshold (= 0) expected for COXRmax = 1 also is plotted. The curve shown corresponds to Eq. 2, and one can see that it fits very closely the independently determined experimental data. The observed relationship between COX threshold and relative COX capacity indicates that, in the low range of relative COX capacities (between 1 and 2), any physiological change or inhibitor action or mutation that affects, even moderately, the activity of a component of the respiratory chain upstream of COX or the activity of COX may produce dramatic changes in the COX threshold, and, therefore, in the control of respiration by COX. These findings and their potential extension to other respiratory complexes are significant for understanding the pathogenetic role of disease-causing mtDNA mutations that produce only a moderate decrease in the activity of a given component of the respiratory chain (20–23), and for elucidating the basis of the striking tissue specificity of the mutation-associated phenotype (24).
Figure 4.

Relationship of COX respiratory threshold (COXT) to relative COX capacity. The data for COX thresholds derived from the COX inhibition data in wild-type 143B and pT3 cells, mutant pT1 and pT4 cells (Fig. 3 and Table 1), and 3 nM rotenone- or 2.7 nM antimycin A-treated 143B cells (Table 1) are plotted against the ratios of (ascorbate + 0.4 mM TMPD)-dependent O2 consumption rate to endogenous respiration rate (TMPD 0.4 mM/endog. resp.).
Polarographic Analysis of Activity of Respiratory Complexes in Digitonin-Permeabilized Cells.
In the case of the cell lines carrying the MERRF mutation (pT1 and pT4), the significant increase in their COX thresholds, as compared with those of cell lines carrying the same nuclear background (143B and pT3), and even the same mtDNA apart from the mutation (pT3), clearly indicates that COX exerts only a low level of control on the respiratory chain of these mutant cells. To obtain evidence about the rate-limiting step in the respiration of these cells, a polarographic analysis of the activity of the respiratory complexes was carried out in cells permeabilized with digitonin. As shown in Table 1 (Exp. 3), in pT4 and pT1 cells, the rotenone-sensitive (glutamate + malate)-dependent O2 consumption rate was decreased, relative to 143B cells, by 88% and 98%, respectively, whereas the antimycin A-sensitive (succinate + glycerol 3-phosphate)-dependent respiration rate was decreased by 71% and 87%, and the KCN-sensitive (ascorbate + 0.2 mM TMPD)-dependent respiration rate, by 62% and 66%. Furthermore, in non-rotenone-inhibited pT4 cells, the O2 consumption rate in the presence of glutamate, malate, succinate, and glycerol 3-phosphate was significantly higher than the coupled endogenous respiration (1.38 vs. 1.06), indicating that complex III was not rate-limiting in these cells. It is, therefore, clear from these observations that, in MERRF mutation-carrying cells, the activity of complex I is the rate-limiting step. This agrees with the finding of an undetectable labeling of the largest mtDNA-encoded subunits of this complex, ND4 and ND5, in [35S]methionine pulse-labeled mutant cells (8, 25).
The data of Exp. 3 in Table 1 are also relevant to the general question of how closely O2 consumption measurements carried out on isolated mitochondria or digitonin-permeabilized cells reflect the in vivo situation. In particular, one can see that the state 3 (glutamate + malate)-dependent respiration rate measured in digitonin-permeabilized cells in two parallel experiments was only 63% to 67%, 40% to 48%, 32% to 35%, and 18% of the coupled endogenous respiration in 143B, pT3, pT4, and pT1 cells, respectively. It is particularly significant that, after permeabilization with digitonin, the (glutamate + malate)-dependent respiration rate in 143B cells was 63% to 67% of the coupled endogenous respiration rate, whereas the (ascorbate + 0.2 mM TMPD)-dependent respiration rate was practically unaffected, as previously mentioned. These observations indicate that, under the above conditions, which would be expected to affect the environment of mitochondria much less dramatically than the isolation of the organelles, the relative COX capacity of 143B and pT3 cells was already significantly increased, as compared with the values in intact cells. In fact, one can estimate values of relative COX capacity of ≈2.2 and ≈3.1 by dividing the O2 consumption rate expected, on the basis of the data in Fig. 1b, in digitonin-permeabilized, antimycin A-treated 143B and pT3 cells, respectively, if the TMPD concentration were increased from 0.2 mM to 0.4 mM, by the (malate + glutamate)-dependent respiration rate in the same cells. It also should be noticed that, in Exp. 3 in Table 1 carried out in the absence of inhibitors, in which the endogenous electron flux was anticipated to increase the level of cytochrome c reduction, the (ascorbate + 0.2 mM TMPD)-dependent respiration rate measured in digitonin-permeabilized 143B and pT3 cells was also more than double (2.1 and 2.3 times, respectively) the (glutamate + malate)-dependent respiration rate in the same cells. These observations, which agree with other reports on O2 consumption measurements in intact and digitonin-permeabilized cells (21, 26, 27), emphasize the value of in vivo measurements for better understanding the role of developmental, physiological, or pathological factors in the metabolic control of respiration.
Acknowledgments
G.V. thanks Dr. N. Capitanio for helpful discussions, advice, and encouragement throughout the project. We are also very grateful to Drs. Y. Hatefi, S. Papa, and A. Chomyn for their critical reading of the manuscript and their valuable comments, to Drs. J. A. Enriquez and J. Cabezas for their help in the primer extensions experiments, and to B. Keeley, A. Drew, and R. Zedan for technical assistance. These investigations were supported by National Institutes of Health Grant GM-11726 to G.A.
Footnotes
Abbreviations: mtDNA: mitochondrial DNA; COX: cytochrome c oxidase; TMPD: N,N,N′,N′-tetramethyl-p-phenylenediamine; OXPHOS, oxidative phosphorylation; FBS, fetal bovine serum; DNP, 2,4- dinitrophenol; TD, Tris-based, Mg2+-, Ca2+-deficient.
References
- 1.Kacser H, Burns J A. In: Rate Control of Biological Processes. Davies D D, editor. Cambridge, U.K.: Cambridge Univ. Press; 1973. pp. 65–104. [Google Scholar]
- 2.Heinrich R, Rapoport T A. Eur J Biochem. 1974;42:89–95. doi: 10.1111/j.1432-1033.1974.tb03318.x. [DOI] [PubMed] [Google Scholar]
- 3.Groen A K, Wanders R J A, Westerhoff H V, Van der Meer R, Tager J M. J Biol Chem. 1982;257:2754–2757. [PubMed] [Google Scholar]
- 4.Letellier T, Malgat M, Mazat J-P. Biochim Biophys Acta. 1993;1141:58–64. doi: 10.1016/0005-2728(93)90189-m. [DOI] [PubMed] [Google Scholar]
- 5.Letellier T, Heinrich R, Malgat M, Mazat J-P. Biochem J. 1994;302:171–174. doi: 10.1042/bj3020171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor R W, Birch-Machin M A, Bartlett K, Lowerson S A, Turnbull D M. J Biol Chem. 1994;269:3523–3528. [PubMed] [Google Scholar]
- 7.Davey G P, Clark J B. J Neurochem. 1996;66:1617–1624. doi: 10.1046/j.1471-4159.1996.66041617.x. [DOI] [PubMed] [Google Scholar]
- 8.Chomyn A, Meola G, Bresolin N, Lai S T, Scarlato G, Attardi G. Mol Cell Biol. 1991;11:2236–2244. doi: 10.1128/mcb.11.4.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shoffner J M, Lott M T, Lezza A M S, Seibel P, Ballinger S, Wallace D C. Cell. 1990;61:931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- 10.King M P, Attardi G. Cell. 1988;52:811–819. doi: 10.1016/0092-8674(88)90423-0. [DOI] [PubMed] [Google Scholar]
- 11.Fung C H, Khachadurian A K. J Biol Chem. 1980;255:676–680. [PubMed] [Google Scholar]
- 12.Hofhaus G, Attardi G. Mol Cell Biol. 1995;15:964–974. doi: 10.1128/mcb.15.2.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hofhaus G, Shakeley R M, Attardi G. Methods Enzymol. 1996;264:476–483. doi: 10.1016/s0076-6879(96)64043-9. [DOI] [PubMed] [Google Scholar]
- 14.King M P, Attardi G. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 15.Brand M D, Vallis B P S, Kesseler A. Eur J Biochem. 1994;226:819–829. doi: 10.1111/j.1432-1033.1994.00819.x. [DOI] [PubMed] [Google Scholar]
- 16.Crinson M, Nicholls P. Biochem Cell Biol. 1992;70:301–308. doi: 10.1139/o92-047. [DOI] [PubMed] [Google Scholar]
- 17.Sagi-Eisenberg R, Gutman M. Eur J Biochem. 1979;97:127–132. doi: 10.1111/j.1432-1033.1979.tb13093.x. [DOI] [PubMed] [Google Scholar]
- 18.Yoneda M, Miyatake T, Attardi G. Molec Cell Biol. 1994;14:2699–2712. doi: 10.1128/mcb.14.4.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korzeniewski B, Mazat J-P. Biochem J. 1996;319:143–148. doi: 10.1042/bj3190143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majander A, Finel M, Savontaus M-L, Nikoskelainen E, Wikstrom M. Eur J Biochem. 1996;239:201–207. doi: 10.1111/j.1432-1033.1996.0201u.x. [DOI] [PubMed] [Google Scholar]
- 21.Hofhaus G, Johns D R, Hurko O, Attardi G, Chomyn A. J Biol Chem. 1996;271:13155–13161. doi: 10.1074/jbc.271.22.13155. [DOI] [PubMed] [Google Scholar]
- 22.Jun A S, Trounce I A, Brown M D, Shoffner J M, Wallace D C. Mol Cell Biol. 1996;16:771–777. doi: 10.1128/mcb.16.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuznetsov A V, Clark J F, Winkler K, Kunz W S. J Biol Chem. 1996;271:283–288. doi: 10.1074/jbc.271.1.283. [DOI] [PubMed] [Google Scholar]
- 24.Wallace D C. Annu Rev Biochem. 1992;61:1175–1212. doi: 10.1146/annurev.bi.61.070192.005523. [DOI] [PubMed] [Google Scholar]
- 25.Enriquez J A, Chomyn A, Attardi G. Nat Genet. 1995;10:47–55. doi: 10.1038/ng0595-47. [DOI] [PubMed] [Google Scholar]
- 26.Guan M-X, Fischel-Ghodsian N, Attardi G. Hum Molec Genet. 1996;5:963–971. doi: 10.1093/hmg/5.7.963. [DOI] [PubMed] [Google Scholar]
- 27.Dunbar D R, Moonie P A, Zeviani M, Holt I J. Hum Molec Genet. 1996;5:123–129. doi: 10.1093/hmg/5.1.123. [DOI] [PubMed] [Google Scholar]