

Abstract
High-throughput direct sequencing techniques have recently opened the possibility to sequence genomes from Pleistocene organisms. Here we analyze DNA sequences determined from a Neandertal, a mammoth, and a cave bear. We show that purines are overrepresented at positions adjacent to the breaks in the ancient DNA, suggesting that depurination has contributed to its degradation. We furthermore show that substitutions resulting from miscoding cytosine residues are vastly overrepresented in the DNA sequences and drastically clustered in the ends of the molecules, whereas other substitutions are rare. We present a model where the observed substitution patterns are used to estimate the rate of deamination of cytosine residues in single- and double-stranded portions of the DNA, the length of single-stranded ends, and the frequency of nicks. The results suggest that reliable genome sequences can be obtained from Pleistocene organisms.
Keywords: 454, deamination, depurination, paleogenomics
The retrieval of DNA sequences from long-dead organisms offers a unique perspective on genetic history by making information from extinct organisms and past populations available. However, three main technical challenges affect such studies. First, when DNA is preserved in ancient specimens, it is invariably degraded to a small average size (1). Second, chemical damage is present in ancient DNA (2) that may cause incorrect DNA sequences to be determined (3). Third, because ancient DNA is present in low amounts or absent in many specimens, traces of modern DNA from extraneous sources may cause modern DNA sequences to be mistaken for endogenous ancient DNA sequences (4–6). Recently, a DNA sequencing method based on highly parallel pyrosequencing of DNA templates generated by the PCR has been developed by 454 Life Sciences (454) (7). This method allows several hundred thousand DNA sequences of length 100 or 250 nt to be determined in a short time. It has been used to determine DNA sequences from the remains of three Pleistocene species: mammoths (8, 9), a cave bear (9), and a Neandertal (10). In all cases, the majority of DNA sequences retrieved are from microorganisms that have colonized the tissues after the death of the organisms. However, a fraction stem from the ancient organisms. In fact, the throughput of this technology, as well as other sequencing technologies currently becoming available (11), makes it possible to contemplate sequencing the complete genomes of extinct Pleistocene species (8, 10).
Here, we analyze DNA sequences determined on the 454 platform from an ≈38,000-year-old Neandertal specimen found at Vindija Cave, Croatia (10, 12), with respect to two features of particular significance for genomic studies of ancient DNA. First, we investigate the DNA sequence context around strand breaks in ancient DNA. This has not been previously possible, because when PCR is used to retrieve ancient DNA sequences, primers that target particular DNA sequences are generally used and thus the ends of the ancient DNA molecules are not revealed. Second, we investigate the patterns of nucleotide misincorporations in the ancient DNA sequences as a function of their position in ancient DNA fragments. Although there is strong evidence that the majority of such misincorporations are due to deamination of cytosine residues to uracil residues (3), which code as thymine residues, it is unclear whether other miscoding lesions are present in any appreciable frequency in ancient DNA or how miscoding lesions are distributed along ancient DNA molecules. When relevant, we use comparable data from an ≈43,000-year-old mammoth bone (9) from the Bol'shaya Kolopatkaya river, Russia, an ≈42,000-year-old cave bear bone from Ochsenhalt Cave, Austria (13), a contemporary human, and DNA sequences of the Vindija Neandertal cloned in a plasmid vector (14) to ask whether the patterns seen are general features of Pleistocene DNA sequences or are caused by the 454 sequencing process. Finally, we develop a model that allows us to estimate features of ancient DNA preservation and discuss the implications of our findings for the determination of complete genome sequences from Pleistocene organisms.
Results and Discussion
The 454 Process.
Because aspects of the 454 sequencing process are of crucial importance for the analyses presented, we briefly review some of its essential features. In a first step, a double-stranded DNA extract is end-repaired and ligated to two different synthetic oligonucleotide adaptors termed A and B. From each successfully ligated molecule, one of the DNA strands is isolated and subjected to emulsion PCR, during which each template remains isolated from other templates on a Sepharose bead carrying oligonucleotides complementary to one of the adaptors, producing beads each coated with ≈10 million copies of one DNA molecule. Up to 800,000 such DNA-containing beads are then loaded onto a multiwell glass plate, and their sequences are determined by pyrosequencing (7).
The end repair of the template DNA and ligation of adapters, which are critical for the analyses in this paper, are described in more detail in Fig. 1. First, T4 DNA polymerase is used to remove single-stranded 3′-overhanging ends and to fill in 5′-overhanging ends (Fig. 1ii). Simultaneously, 5′-ends are phosphorylated by using T4 polynucleotide kinase. Thus, while the 5′-ends of the sequences eventually generated reflect the 5′-ends present in the ancient DNA fragments, the 3′-ends correspond to the terminal 5′-position on the opposite, nonsequenced strand and are not necessarily the original 3′-end of the sequenced strand. Adaptor ligation is achieved in two enzymatic steps. First, the two double-stranded adaptors, A and B, which are not phosphorylated to avoid formation of adaptor dimers, are ligated to the 5′-ends of the target molecules (Fig. 1iii). Ligation products carrying at least one B adaptor are captured and the strand-displacing Bst DNA polymerase is used to make the ligation products fully double-stranded, displacing the downstream adaptor strands (Fig. 1iv). Finally, by NaOH-mediated denaturation of the two DNA strands, the A-to-B strands are released, recovered and used as templates for emulsion PCR, whereas B-to-A strands remain immobilized on the beads (Fig. 1v).
Fig. 1.

The 454 library preparation process. Double-stranded DNA molecules (i) (yellow) are made blunt-ended by T4 DNA polymerase, 5′-phosphorylated (stars) by T4 polynucleotide kinase (ii) and ligated to one strand of nonphosphorylated double-stranded adaptors A (green) and B (blue) (iii). Ligation products carrying the biotinylated B adaptor are captured on Streptavidin beads (red), and the strand-displacing Bst DNA polymerase is used to extend the nicks between adaptors and template (iv). The DNA strands are then denatured, releasing the A-to-B strands (v), which are isolated and used as templates for emulsion PCR.
Ancient DNA Fragmentation.
To investigate whether fragmentation of ancient DNA occurs predominantly at certain bases or in certain sequence contexts, we analyzed the base composition close to the 5′ and 3′ ends of DNA sequences, i.e., near the sites of breaks in the template DNA. To avoid confounding of the results by sequencing errors or misincorporations close to the ends of the sequences (see below), and to allow the sequence context outside the sequenced fragments to be analyzed, we aligned each 454 sequence to a reference genome, extended the alignment in both directions to include the entire 454 sequence, and used the reference sequence to gauge the base composition on both sides of the ends of the ancient DNA template. To avoid 3′-ends that were limited by 454 sequencing length, we used only sequences where the 3′-ends could be identified by the presence of a B adaptor.
Fig. 2 shows the base composition of the human reference genome from 10 bases outside the terminal Neandertal base sequenced to 20 bases into the sequence for the 5′- and 3′-end, respectively. Across most of the Neandertal molecules, C and G are each present at ≈22% frequency and A and T at ≈28%. Because the average proportion of G and C in the human genome is 20.5% each (15), and this is reflected in the contemporary human DNA sequenced by 454 [supporting information (SI) Fig. 5], this suggests a slight overall bias toward GC-rich sequences in the ancient reads. Strikingly, at the -1 position of the 5′-ends, i.e., the first position upstream of the 5′-most base sequenced, the frequency of G is elevated from ≈22% seen across all Neandertal reads analyzed to 29% (Fisher's exact test, P < 2.2 × 10−16), and the frequency of A is elevated from ≈28% to 31% (P = 3.5 × 10−10), whereas C and T are depressed. Conversely, at the position + 1 downstream of 3′-ends, the frequency of C (P < 2.2 × 10−16) as well as T (P = 1.32 × 10−5) is elevated to ≈30%, whereas G and A are depressed. At the 5′-most sequenced positions, A is depressed to 23% (P < 2.2 × 10−16), whereas T is elevated to 31% (P = 4.7 × 10−13), whereas at the 3′-most sequenced position, A is elevated to 32% (P = 2.8 × 10−12) and T is depressed to 23% (P < 2.2 × 10−16).
Fig. 2.

Base composition at ends of Neandertal DNA sequences. The base composition of the human reference sequence is plotted as a function of distance from 5′- and 3′-ends of Neandertal sequences.
Although 5′-ends of 454 sequences represent the positions of 5′-breaks of the sequenced ancient template strand, the 3′-ends represent the positions of 5′-breaks on the complementary strand (Fig. 1). Therefore, the data show that immediately before a strand break, guanine residues as well as adenine residues are elevated relative to cytosine and thymine residues. When modern human DNA sequenced by the 454 process is analyzed in the same way, no elevation of purines adjacent to strand breaks are seen but instead a slight elevation of C and depression of A at −1 positions (Fig. 2). This suggests that the patterns seen in the Neandertal data are due to a fragmentation process that has affected the ancient DNA rather than a bias in what fragments are sequenced efficiently by the 454 process. That an increased occurrence of purines immediately 5′ to strand breaks is typical of the Neandertal DNA prepared from the Vindija specimen is supported by the fact that an excess of guanine residues adjacent to strand breaks is seen also in Neandertal DNA from the same specimen that was cloned in a plasmid vector and subsequently sequenced (14) (SI Fig. 5). Interestingly, the overall GC content of the cloned Neandertal sequences is ≈50% vs. 41% in the human genome (15), suggesting that some feature of the cloning process introduces a bias for GC-rich ancient sequences that is stronger than in the direct 454 sequencing.
In the mammoth and cave bear (SI Fig. 5) DNA directly sequenced on the 454 platform, an excess of G as well as A is similarly seen immediately adjacent to breaks. However, in the cave bear, A is increased more than G. These results suggest that purines (G and A) may be overrepresented immediately 5′ to strand breaks in many or most ancient specimens. A mechanism that is likely to be responsible for this is depurination, i.e., the hydrolysis of purine bases from the deoxyribose-phosphate backbone of DNA. After depurination events, the sugar phosphate backbone is susceptible to hydrolysis 3′ to the depurinated site (16). DNA is affected by depurination under many conditions (17), and baseless sites have been shown to occur in ancient DNA (1). It should be noticed, however, that this appears to explain only in the order of 10% of all strand breaks in the directly sequenced Neandertal sample.
It should also be noted that, in addition to an elevation of purines adjacent to breaks, other base compositional aberrations close to ends of molecules are seen in some specimens. In the mammoth, there is an excess of T and a decreased amount of G at the second position upstream of the strand break. This is also seen in a permafrost-preserved mastodon sample (unpublished observation), indicating that this may be related to the permafrost environment. Further analyses of several ancient specimens are necessary to elucidate how frequently processes in addition to depurination are involved in strand breaks in ancient DNA samples from different preservation conditions.
Nucleotide Misincorporations.
Because each 454 sequence is derived from one single-stranded molecule, each of the 12 possible base differences to related genomes, e.g., C to G, can be distinguished from its complementary change, i.e., G to C (9, 18). Thus, the patterns and prevalence of each possible nucleotide misincorporation can be estimated. When this is done across large numbers of 454 sequence reads, the number of substitutions where any single nucleotide (e.g., C) changes to another particular nucleotide (e.g., T) should be equal to the number of substitutions where the complementary nucleotide (i.e., G) changes to the complementary nucleotide (i.e., A), unless nucleotide misincorporations occur (9). When such strand-equivalent reciprocal nucleotide substitutions are analyzed in DNA sequences from Pleistocene organisms, C to T changes are more frequent than G to A changes (9, 10, 18). Furthermore, in contrast to DNA sequences determined from modern DNA, the rates of both G to A changes and C to T changes are elevated above the rates of the other two transitions. Whereas there is ample evidence that deamination of cytosine residues to uracil (U) residues in ancient DNA is responsible for the excess C to T substitutions (3), the G to A substitutions are enigmatic. They could be caused by deamination of guanine residues to xanthine (X) residues, which are read by the DNA polymerase used in the 454 sequencing process as adenine residues, thus potentially causing G to A misincorporations (9). However, because the efficiency with which X is misread as A by the DNA polymerase is low, it is unclear whether this is enough to account for the effect observed.
We analyzed the frequency at which each of the 12 substitutions occur as a function of their distance from the 5′- and the 3′- ends (as defined by the presence of a B adaptor), respectively, of the Neandertal DNA sequences. Fig. 3 shows that in agreement with previous findings, C to T and G to A substitutions are drastically elevated, whereas other substitutions show similar and low rates. However, strikingly, C to T and G to A substitutions are unequally and differently distributed along the DNA molecules. The frequency of C to T substitutions are elevated at least 50-fold above other substitutions at the 5′-most nucleotide position of molecules, where ≈21% of all cytosine residues in the human reference sequence are read as thymine residues in the ancient sequences. C to T substitutions then decrease rapidly over the first ≈10 nucleotides of the molecules, after which they steadily decrease toward the 3′ ends, although they remain elevated relative to the other substitutions, except G to A. In stark contrast, G to A substitutions appear not to be elevated above other substitutions until ≈20 nucleotides into the molecules from the 5′ end when they increase steadily in frequency until the last ≈10 positions, where they increase to ≈60-fold above background at the 3′-most position of molecules. Other substitutions not only are much more rare but also do not appear to vary significantly as a function of position along DNA sequences, although the power to detect any such variation is obviously low because of their low frequency.
Fig. 3.

Misincorporation patterns in Neandertal DNA sequences. The frequencies of the 12 possible mismatches are plotted as a function of distance from 5′- and 3′-ends. At each position, the substitution frequency, e.g., C-T, is calculated as the proportion of human reference sequence positions carrying C where the 454 sequence is T. The 10 5′- and 10 3′-most nucleotides were removed from the 3′- and 5′-graphs, respectively.
In mammoth sequences similarly determined by the 454 technology (SI Fig. 6), higher numbers of all substitutions are seen across the reads because of the greater evolutionary distances between the mammoth and elephant genomes than between the Neandertal and human genomes. This makes misincorporations harder to identify. However, elevated C to T substitutions at 5′-ends and elevated G to A substitutions at 3′-ends are readily detectable. The same is true for direct sequences generated from a cave bear (SI Fig. 6). In the bacterial plasmid library prepared from the same Neandertal individual from which the 454 sequencing was performed (14), elevation of C to T substitutions at 5′-ends and of G to A substitutions at 3′-ends of inserts are similarly seen, although less dramatic than for the directly sequenced DNA (SI Fig. 6). In contrast, no such increase is seen in nebulized modern human DNA analyzed in a way identical to the Neandertal DNA (SI Fig. 6), showing this is a feature not of the 454 technology per se but of the ancient DNA.
Overhanging Ends and Nicks.
Because the 5′-ends produced by 454 sequencing represent the 5′-ends of the template molecules, the elevation of C to T substitutions at 5′-ends must stem from some process that results in cytosine residues being read as thymine residues. Deamination of cytosine to uracil has been shown to occur in ancient DNA (1, 3) and to cause nucleotide misincorporations (3). Therefore, deaminated cytosine residues in the ancient template strands sequenced are presumably responsible for the C to T substitutions seen in the 5′-ends of molecules. Taken at face value, the elevated G to A misincorporations at 3′-ends of molecules could be due to modified guanine residues in ancient templates. However, given that G to A substitutions at 3′-ends of molecules are similar in frequency and pattern to C to T substitutions at 5′-ends of molecules, and given that the 3′-ends of 454 template molecules may represent filled-in 5′-overhanging ends on the complementary strand (Fig. 1), we suggest that the elevated G to A substitutions at 3′-ends are the result of C to T substitutions on the complementary 5′-ends of the original template molecules. Indeed, although it has been previously suggested that all misincorporations seen by direct 454 sequencing reflect miscoding lesions on the sequenced strand (8–10), there are two steps in the 454 sequencing process where complementary changes on the strand to be sequenced could be created. First, if a miscoding lesion, e.g., a uracil residue, is present on a overhanging 5′-end (Fig. 4A), T4 DNA polymerase will insert a complementary base during end repair, i.e., an adenine residue, opposite the miscoding uracil residue. Subsequently, either the original damaged strand is sequenced, and a C to T substitution as a result of the uracil residue will be observed near the 5′-end of the sequence, or, alternatively, the nondamaged strand is sequenced, and a complementary G to A substitution will be observed near the 3′-end of the sequence. Second, when the strand-displacing Bst DNA polymerase is used to complete the adaptors, the enzyme can extend from any nick or gap in the template molecules, displacing the original strand downstream to the end of the template strand (Fig. 4B). If this is the case, miscoding lesions present downstream of the nick on the template strand will cause a misincorporation on the newly synthesized sequenced strand, for example, an adenine residue inserted opposite a uracil residue. Thus, downstream of a nick or gap, miscoding lesions present on the sequenced strand will be removed, and miscoding lesions on the opposite strand will be seen as misincorporations. 5′-overhanging ends as well as DNA nicks will therefore cause the rate of C to T substitutions to decrease and the rate of G to A substitutions to increase from the 5′- to the 3′-end of molecules.
Fig. 4.

Miscoding lesions and the 454 process. During preparation of templates for 454 sequencing, the ends of DNA fragments are first repaired by T4 DNA polymerase (A), and in a later step linkers are filled in by Bst DNA polymerase (B). During blunt-end repair by T4 DNA polymerase (A), miscoding lesions (black circles) on 3′-overhanging ends are removed, whereas miscoding lesions on 5′-overhangs result in complementary misincorporations (white circles) in the resultant 454 sequences. Similarly, extension by the strand-displacing Bst DNA polymerase (B) causes miscoding lesions in the template DNA downstream of nicks or gaps to result in complementary misincorporations in the sequences generated.
Fig. 3 shows that the frequency of C to T substitutions decreases steadily throughout the molecule toward 3′-ends even after the 20 first 5′-nucleotides, whereas G to A substitutions do not seem to be elevated to the very 5′-ends. This further supports the suggestion that the primary lesion underlying these patterns is one that affects cytosine residues and causes them to be read as thymine residues.
In summary, the patterns of C to T and G to A substitutions along ancient DNA molecules strongly suggest that the overwhelming majority of misincorporations in ancient DNA are due to deamination of cytosine residues. As a corollary, the previously proposed modified guanine residues that produce G to A substitutions (9, 18) either do not exist or are rare in comparison with deaminated cytosine residues.
Overhanging Ends and Deamination.
The high frequency of C to T misincorporations at the 5′-ends of ancient DNA sequences and the correspondingly high frequency of G to A misincorporations at the 3′-ends imply that deamination of cytosine residues is significantly elevated at the 5′-ends of ancient DNA molecules. This could be caused either by a tendency of cytosine residues at the ends of molecules to undergo deamination or a tendency of strand breaks to occur near deaminated cytosine residues. In the latter case, one would expect to see an elevation of cytosine residues in aligned reference sequences around strand breaks. However, this is not the case (Fig. 2). Therefore, we propose that cytosine residues close to the ends of ancient DNA molecules are more susceptible to deamination than cytosine residues more internal in the molecule.
One possible mechanism underlying this is the presence of single-stranded overhanging ends in the ancient DNA, because the rate of cytosine deamination is ≈2 orders of magnitude higher in single- than in double-stranded DNA (17). An alternative and not mutually exclusive mechanism is “DNA breathing” in the ends of molecules, which could cause them to be partially single-stranded and thus more susceptible to deamination. The former mechanism is supported by the fact that the elevation of G to A substitutions at 3′-ends is similar in magnitude to that of C to T substitutions at 5′-ends. This is expected if the effect is mainly due to single-stranded overhangs, which are filled in by T4 DNA polymerase during end-repair, because this will produce G to A substitutions at every 3′-end extended into a 5′-overhanging end with a deaminated cytosine residue. By contrast, if the end effects stemmed from elevated damage to double-stranded DNA, modified cytosine residues would be complemented only during the nick extension step, and therefore (unless all molecules carried nicks) the elevation of C to T substitutions in 5′-ends would be greater than the elevation of G to A substitutions in 3′-ends.
A Model of Ancient DNA Damage.
Given the data presented above, we conclude that cytosine deamination is the major factor causing nucleotide misincorporations in ancient DNA, and that ancient DNA contains single-stranded ends as well as nicks, which lead to apparent G to A substitutions in 454 sequence data. We furthermore suggest that cytosine deamination is more prevalent in single-stranded ends of molecules than in the interior of molecules. By formalizing these findings in a statistical model, we can estimate several parameters relevant to the extent of degradation of the Neandertal DNA.
In this model, we estimate the following four parameters: the frequency of nicks, which we model as occurring with uniform probability per base (ν); the average length of single-stranded overhanging ends, which we take to follow a geometric distribution with the parameter λ; the frequency of deaminated cytosine residues in double-stranded DNA (δ); and the frequency of deaminated cytosine residues in single-stranded DNA (δss). Note that this model explicitly incorporates two points that influence the output of 454 sequencing. First, when nicks are found on opposite strands and downstream of each other, the fragment is lost during the nick repair stage of 454 template preparation, because the molecule will split when the replication forks meet. This causes the distribution of first nicks in the sequenced fragments to be uniform rather than geometric. Second, the model accounts for the fact that the end repair step in the 454 protocol (Fig. 1) eliminates all 3′-overhanging ends and preserves only 5′-overhanging ends.
Given this model (details in SI Text), we use maximum likelihood to estimate the four parameters given the Neandertal data (Table 1). The estimated fraction of deaminated cytosine residues in single-stranded DNA is 68% (95% confidence interval (C.I.), 65–71%) and in double-stranded DNA 0.97% (C.I., 0.87–1.1%]. This is in keeping with previous work (17), which has shown the deamination rate of cytosine residues to be ≈2 orders of magnitude higher in single- than in double-stranded DNA. The average length of single-stranded overhanging ends is estimated to be 1.6–1.8 nucleotides and the frequency of single-stranded nicks 2.4% (C.I. 1.7–3.6%), i.e., about one nick or gap per 50 nucleotides. Note that while the C.I. for the lengths of overhanging ends is narrow the C.I. for nick frequency is a much larger fraction of the estimate, indicating that our power to estimate the nick frequency is relatively low.
Table 1.
Maximum likelihood estimates (MLE) for four features of Neandertal DNA sequences
| Parameter | MLE | 95% C.I. |
|---|---|---|
| Deamination, double-stranded DNA (δ̂) | 0.0097 | (0.0087, 0.011) |
| Deamination, single-stranded DNA (δ̂ss) | 0.68 | (0.65, 0.71) |
| Nick frequency per base (ν̂) | 0.024 | (0.017, 0.036) |
| Length of single-stranded overhangs (λ̂) | 0.36 | (0.35, 0.38) |
If we simulate the expected C to T and G to A misincorporation frequencies along a hypothetical Neandertal sequence using the parameter estimates above, the results fit the observed data quite well, indicating that our assumptions are broadly consistent with the data (SI Fig. 7). The application of this model to future data sets will provide a framework for evaluating the error probability of any nucleotide position generated from ancient DNA by 454 sequencing and will reveal to what extent these parameters vary from specimen to specimen and with preservation conditions.
Considerations for Genome Sequencing.
An exciting possibility opened up by high-throughput direct sequencing of DNA is that entire genomes can in principle be determined from Pleistocene organisms such as mammoths (8) or Neandertals (10). However, two main potential problems need to be considered in such undertakings: first, errors in the DNA sequences caused by lesions in the ancient DNA and, second, contamination of extracts by contemporary DNA, in particular contamination of Neandertal extracts by contemporary human DNA. The findings presented have bearing on both of these issues.
To address the first point, we estimated the extent of errors for all 12 substitutions in the Neandertal sequences and the contemporary human sequences, respectively, determined on the 454 platform. To do this, we compare the substitutions assigned to the human lineage and the lineage leading to the DNA sequences determined on the 454 platform in alignments to the human and chimpanzee genome sequences and assume that any acceleration of the latter lineage is because of nucleotide misincorporations and sequencing errors (19). Our results show that except for C to T and G to A misincorporations, no other nucleotide misincorporations in the Neandertal sequences are elevated above the rate of approximately four errors per 10,000 bp we estimate for the contemporary human 454 sequences (SI Fig. 8). The sole exception is G-T misincorporations, which appear slightly elevated in the Neandertal sequences but still <1 in 1,000. This could represent small levels of 8-hydroxyguanine, an oxidation product of guanine, which has previously been detected in ancient DNA (2) and is known to cause G-T transversions (20, 21). Thus, except for C to T, G to A, and perhaps G to T substitutions, nucleotide substitutions observed in Neandertals relative to humans and chimpanzees are as reliable as if they had been determined from contemporary DNA. For C to T and G to A substitutions, their reliability depends greatly on their positions in the sequencing reads. Although at the first or last positions of reads they are >50-fold increased above background levels of Neandertal–human changes (Fig. 3), at position 20 from 5′-ends C to T substitutions are only ≈3-fold increased while at position 20 from 3′-ends G to A substitutions are ≈2-fold increased. Using the model presented, the reliability of C to T and G to A substitutions can be estimated as a function of their positions in sequencing reads and incorporated into genome sequencing pipelines. In general, such substitutions located away from the ends of the molecules retrieved will be relatively reliable. Provided that eventually sufficient coverage of the Neandertal genome is achieved, nucleotide misincorporations should therefore not prevent a reliable Neandertal or mammoth genome sequence from being determined.
With respect to contamination of Neandertal DNA by modern human DNA, it has been argued that endogenous sequences are expected to differ from contaminating sequence by being of shorter length and by carrying more nucleotide misincorporations and thus that the length distribution and the extent of nucleotide misincorporations could be used to estimate the extent of contamination (14). However, the lengths of the endogenous DNA fragments differ from fossil to fossil and even among parts of a single fossil (unpublished observation). It is also impossible to exclude that contemporary DNA that contaminates fossils or laboratory reagents is degraded to a short average length either during cellular decay or after entering the fossil (4, 5). Furthermore, it has been shown that modern human DNA contaminating ancient bones may carry nucleotide misincorporations typical of ancient DNA sequences (6, 22). This suggests that neither fragment size nor misincorporations represent efficient ways to distinguish endogenous from contaminating DNA sequences.
The only way to positively identify contamination is by DNA sequences that distinguish the organism under study from potential contaminants. One such DNA sequence is the hypervariable region I (HVRI) of the mitochondrial DNA (mtDNA), which has been determined from 13 Neandertals (12, 23–31) and found to differ from contemporary humans by multiple substitutions. This can be exploited to estimate the relative amounts of endogenous mtDNA and contaminating human mtDNA in extracts prepared from Neandertal fossils (10). To control for contamination at subsequent stages of the 454 process, the DNA sequences produced from an extract can similarly be analyzed for mtDNA sequences. Thus, the mtDNA sequences identified from the Neandertal presented here fall outside the variation of modern humans (10) and all seven mtDNA HVR sequences that have subsequently been retrieved from this 454 library (SI Fig. 9) show sequence positions that match the mtDNA sequences previously determined from this specimen (12) and distinguish them from modern human mtDNAs (R.E.G., unpublished results). As more sequences become available also from other rapidly evolving regions of the Neandertal genome, e.g., the Y chromosome, it will be possible to arrive at even more accurate estimates of the contamination rate in the sequences produced by these approaches.
Although such assays allow Neandertal DNA extracts free of mtDNA contamination to be identified and the final sequences produced to be similarly assayed for contamination, two further experimental approaches are in our opinion crucial to minimize contamination. First, all steps up to the ligation of adaptors or plasmid vectors to the ancient DNA should be performed in a laboratory dedicated exclusively to work on ancient DNA extractions under conditions that minimize the risk of contamination. Second, adaptors or vectors that are specifically designed and exclusively used for a particular project should be used. This will allow contamination from DNA derived from other sources than the specimen as well as from other DNA libraries prepared in the same facilities to be detected. Although such adaptors have not been used in the generation of the Neandertal data analyzed here (10, 14), they are now used in the Neandertal genome project.
Given such precautions as well as the patterns of nucleotide misincorporations seen in Neandertal DNA, we are confident that it will be technically feasible to achieve a reliable Neandertal genome sequence.
Materials and Methods
DNA sequence reads from each run on the 454 machine as well as from the plasmid library (14) were aligned against each other to identify repeat reads that stem from a technical artifact related to low concentration DNA libraries (see SI Text for details). The sequence with the best match to the target species from each repeat cluster was aligned to reference genomes by using Megablast 2.2.12. This local alignment was then extended to encompass the entire 454 sequence read up to the end of the read or the B adaptor (see SI Text). The resulting alignments were used to analyze base composition in reference genomes at the ends of the alignments as well as nucleotide substitutions relative the reference genomes. For model parameter estimations and error rate estimations, 454 reads were aligned to the human (hg18) as well as the chimpanzee (panTro2) genomes.
Supplementary Material
Acknowledgments
We thank Graham Coop, Tom Evans, Laurent Excoffier, Christine Green, Michael Hofreiter, Nick Patterson, and Matthias Stiller for helpful discussions and the Max Planck Society for financial support. P.L.F.J was supported by National Institutes of Health Grant R01-GM40282 (to Montgomery Slatkin).
Abbreviations
- 454
454 Life Sciences
- mtDNA
mitochondrial DNA
- C.I.
confidence interval.
Note Added in Proof.
Similar conclusions with regard to C to T and G to A misincorporations have been independently achieved by using both novel experimental evidence and reanalyses of 454 sequencing data (32).
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequences reported in this paper have been deposited as follows. Directly sequenced Neandertal and mammoth sequences have been deposited in the European Molecular Biology Laboratory database (Neandertal accession nos. CAAN02000001-CAAN02470991, mammoth accession nos. CAAM02000001–CAAM02064265) and in the National Center for Biotechnology Information trace archive under GenomeProject IDs 18313 (Neandertal) and 17621 (mammoth). Cave bear and contemporary human sequences have been deposited in the National Center for Biotechnology Information trace archive under GenomeProject IDs 19671 (cave bear) and 19675 (human).
This article contains supporting information online at www.pnas.org/cgi/content/full/0704665104/DC1.
References
- 1.Pääbo S. Proc Natl Acad Sci USA. 1989;86:1939–1943. doi: 10.1073/pnas.86.6.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Höss M, Jaruga P, Zastawny TH, Dizdaroglu M, Pääbo S. Nucleic Acids Res. 1996;24:1304–1307. doi: 10.1093/nar/24.7.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hofreiter M, Jaenicke V, Serre D, Haeseler Av A, Pääbo S. Nucleic Acids Res. 2001;29:4793–4799. doi: 10.1093/nar/29.23.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hofreiter M, Serre D, Poinar HN, Kuch M, Pääbo S. Nat Rev Genet. 2001;2:353–359. doi: 10.1038/35072071. [DOI] [PubMed] [Google Scholar]
- 5.Pääbo S, Poinar H, Serre D, Jaenicke-Despres V, Hebler J, Rohland N, Kuch M, Krause J, Vigilant L, Hofreiter M. Annu Rev Genet. 2004;38:645–679. doi: 10.1146/annurev.genet.37.110801.143214. [DOI] [PubMed] [Google Scholar]
- 6.Malmström H, Stora J, Dalen L, Holmlund G, Götherström A. Mol Biol Evol. 2005;22:2040–2047. doi: 10.1093/molbev/msi195. [DOI] [PubMed] [Google Scholar]
- 7.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poinar HN, Schwarz C, Qi J, Shapiro B, Macphee RD, Buigues B, Tikhonov A, Huson DH, Tomsho LP, Auch A, et al. Science. 2006;311:392–394. doi: 10.1126/science.1123360. [DOI] [PubMed] [Google Scholar]
- 9.Stiller M, Green RE, Ronan M, Simons JF, Du L, He W, Egholm M, Rothberg JM, Keats SG, Ovodov ND, et al. Proc Natl Acad Sci USA. 2006;103:13578–13584. doi: 10.1073/pnas.0605327103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green RE, Krause J, Ptak SE, Briggs AW, Ronan MT, Simons JF, Du L, Egholm M, Rothberg JM, Paunovic M, Pääbo S. Nature. 2006;444:330–336. doi: 10.1038/nature05336. [DOI] [PubMed] [Google Scholar]
- 11.Bentley DR. Curr Opin Genet Dev. 2006;16:545–552. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 12.Serre D, Langaney A, Chech M, Teschler-Nicola M, Paunovic M, Mennecier P, Hofreiter M, Possnert G, Pääbo S. PLoS Biol. 2004;2:313–317. doi: 10.1371/journal.pbio.0020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noonan JP, Hofreiter M, Smith D, Priest JR, Rohland N, Rabeder G, Krause J, Detter JC, Pääbo S, Rubin EM. Science. 2005;309:597–599. doi: 10.1126/science.1113485. [DOI] [PubMed] [Google Scholar]
- 14.Noonan JP, Coop G, Kudaravalli S, Smith D, Krause J, Alessi J, Chen F, Platt D, Pääbo S, Pritchard JK, et al. Science. 2006;314:1113–1118. doi: 10.1126/science.1131412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 16.Lindahl T, Andersson A. Biochemistry. 1972;11:3618–3623. doi: 10.1021/bi00769a019. [DOI] [PubMed] [Google Scholar]
- 17.Lindahl T. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 18.Gilbert MT, Binladen J, Miller W, Wiuf C, Willerslev E, Poinar H, Carlson JE, Leebens-Mack JH, Schuster SC. Nucleic Acids Res. 2007;35:1–10. doi: 10.1093/nar/gkl483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sankoff D, Cedergren RJ. In: Time Warps, String Edits, and Macromolecules: The Theory and Practice of Sequence Comparison. Sankoff D, Kruskal JB, editors. New York: Addison-Wesley; 1983. [Google Scholar]
- 20.Moriya M. Proc Natl Acad Sci USA. 1993;90:1122–1126. doi: 10.1073/pnas.90.3.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakabeppu Y, Sakumi K, Sakamoto K, Tsuchimoto D, Tsuzuki T, Nakatsu Y. Biol Chem. 2006;387:373–379. doi: 10.1515/BC.2006.050. [DOI] [PubMed] [Google Scholar]
- 22.Sampietro ML, Gilbert MT, Lao O, Caramelli D, Lari M, Bertranpetit J, Lalueza-Fox C. Mol Biol Evol. 2006;23:1801–1807. doi: 10.1093/molbev/msl047. [DOI] [PubMed] [Google Scholar]
- 23.Krings M, Stone A, Schmitz RW, Krainitzki H, Stoneking M, Pääbo S. Cell. 1997;90:19–30. doi: 10.1016/s0092-8674(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 24.Krings M, Capelli C, Tschentscher F, Geisert H, Meyer S, von Haeseler A, Grossschmidt K, Possnert G, Paunovic M, Pääbo S. Nat Genet. 2000;26:144–146. doi: 10.1038/79855. [DOI] [PubMed] [Google Scholar]
- 25.Ovchinnikov IV, Gotherstrom A, Romanova GP, Kharitonov VM, Liden K, Goodwin W. Nature. 2000;404:490–493. doi: 10.1038/35006625. [DOI] [PubMed] [Google Scholar]
- 26.Schmitz RW, Serre D, Bonani G, Feine S, Hillgruber F, Krainitzki H, Pääbo S, Smith FH. Proc Natl Acad Sci USA. 2002;99:13342–13347. doi: 10.1073/pnas.192464099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beauval C, Maureille B, Lacrampe-Cuyaubere F, Serre D, Peressinotto D, Bordes JG, Cochard D, Couchoud I, Dubrasquet D, Laroulandie V, et al. Proc Natl Acad Sci USA. 2005;102:7085–7090. doi: 10.1073/pnas.0502656102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lalueza-Fox C, Sampietro ML, Caramelli D, Puder Y, Lari M, Calafell F, Martinez-Maza C, Bastir M, Fortea J, de la Rasilla M, et al. Mol Biol Evol. 2005;22:1077–1081. doi: 10.1093/molbev/msi094. [DOI] [PubMed] [Google Scholar]
- 29.Caramelli D, Lalueza-Fox C, Condemi S, Longo L, Milani L, Manfredini A, de Saint Pierre M, Adoni F, Lari M, Giunti P, et al. Curr Biol. 2006;16:R630–R632. doi: 10.1016/j.cub.2006.07.043. [DOI] [PubMed] [Google Scholar]
- 30.Lalueza-Fox C, Krause J, Caramelli D, Catalano G, Milani L, Sampietro ML, Calafell F, Martinez-Maza C, Bastir M, Garcia-Tabernero A, et al. Curr Biol. 2006;16:R629–30. doi: 10.1016/j.cub.2006.07.044. [DOI] [PubMed] [Google Scholar]
- 31.Orlando L, Darlu P, Toussaint M, Bonjean D, Otte M, Hanni C. Curr Biol. 2006;16:R400–R402. doi: 10.1016/j.cub.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 32.Brotherton P, Endicott P, Sanchez JJ, Beaumont M, Barnett R, Austin J, Cooper A. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
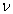{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
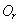{kind=link}
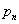{kind=link}
{kind=link}
{kind=link}
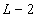{kind=link}
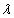{kind=link}
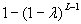{kind=link}
{kind=link}
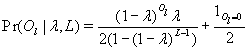{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
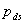{kind=link}
{kind=link}
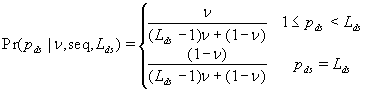{kind=link}
{kind=link}
{kind=link}
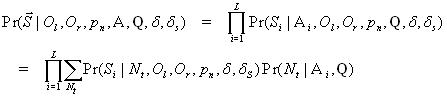{kind=link}
{kind=link}
{kind=link}
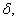{kind=link}
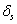{kind=link}
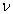{kind=link}
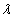{kind=link}
{kind=link}
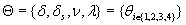{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}