

Abstract
The CaaX proteases Rce1p and Ste24p can independently promote a proteolytic step required for the maturation of certain isoprenylated proteins. Although functionally related, Rce1p and Ste24p are unrelated in primary sequence. They have distinct enzymatic properties, which are reflected in part by their distinct inhibitor profiles. Moreover, Rce1p has an undefined catalytic mechanism, whereas Ste24p is an established zinc-dependent metalloprotease. This study demonstrates that both enzymes are inhibited by peptidyl (acyloxy)methyl ketones (AOMKs), making these compounds the first documented dual specificity inhibitors of the CaaX proteases. Further investigation of AOMK-mediated inhibition reveals that varying the peptidyl moiety can significantly alter the inhibitory properties of AOMKs toward Rce1p and Ste24p and that these enzymes display subtle differences in sensitivity to AOMKs. This observation suggests that this compound class could potentially be engineered to be selective for either of the CaaX proteases. We also demonstrate that the reported sensitivity of Rce1p to TPCK is substrate-dependent, which significantly alters the interpretation of certain reports having used TPCK sensitivity for mechanistic classification of Rce1p. Finally, we show that an AOMK inhibits the isoprenylcysteine carboxyl methyltransferase Ste14p. In sum, our observations raise important considerations regarding the specificity of agents targeting enzymes involved in the maturation of isoprenylated proteins, some of which are being developed as anti-cancer therapeutic agents.
Keywords: Ras, protease, CaaX protein, post-translational modification, isoprenylation, (acyloxy)methyl ketone
Introduction
Rce1p and Ste24p1 are proteases that mediate the maturation of certain lipid-modified proteins, specifically those whose precursors have a C-terminal tetrapeptide CaaX motif (C- cysteine; a-aliphatic; X-one of several amino acids) [1, 2]. Their substrates (i.e., CaaX proteins) typically undergo three ordered post-translational modifications: covalent attachment of an isoprenoid lipid to the cysteine, proteolytic removal of the aaX tripeptide, and carboxyl methyl esterification of the exposed isoprenylated cysteine [2, 3]. While the proteases share a common function and are both ER-localized integral membrane proteins that possess multiple transmembrane spans, they are otherwise unrelated by primary sequence [4].
Several substrates of Rce1p have been described. Many but not all Rce1p substrates are involved in signal transduction. Some have key roles in cellular transformation (e.g. Ras, RhoB). Thus, agents that inhibit the maturation of CaaX proteins are hypothesized to have chemotherapeutic potential [3, 5]. The testing of this hypothesis has led to the development of farnesyltransferase inhibitors that are being examined for their ability to moderate tumor growth [6-9]. The inhibition of Rce1p holds similar anti-cancer potential [3, 10, 11]. By contrast, few substrates have been described for Ste24p. One specific target is the lamin A precursor. Defects in lamin A maturation are associated with abnormal musculo-skeletal development, varied laminopathies, and progeroid syndromes [12-14]. The only other known target of Ste24p is the precursor of the yeast a-factor mating pheromone, which is also a target of Rce1p [1, 15]. For both of its targets, Ste24p appears to catalyze not only CaaX cleavage but also a second cleavage distal to the farnesylated cysteine [16, 17]. Other targets of Ste24p likely exist but have not yet been identified. The yeast a-factor precursor is thus far unique as a CaaX protein in being a substrate of both Ste24p and Rce1p [1]. Once processed by either Rce1p or Ste24p, CaaX proteins are obligatory substrates of the isoprenylcysteine carboxyl methyltransferase (ICMT) [18]. The minimum recognition determinant for this ER-localized membrane protein is a farnesyl cysteine [19, 20]. Both proteolysis and carboxyl methylation can significantly alter the function, localization, and other properties of CaaX proteins [1, 10, 21].
The modern classification system for proteases designates four categories of proteolytic mechanisms: serine/threonine, cysteine, aspartic, and metal-dependent. Ste24p is a zinc-dependent metalloprotease. As expected, Ste24p possesses a consensus zinc metalloprotease motif that is essential for its activity, requires zinc for optimal activity, and is inhibited by zinc chelating compounds such as 1,10-phenanthroline [16, 22]. By contrast, the mechanistic classification of Rce1p has eluded definition, primarily because it lacks a readily identifiable protease motif. Rce1p has also been refractory to purification, which has hindered detailed biochemical and structural analysis of this integral membrane protein.
Rce1p is reportedly sensitive to certain serine/cysteine protease inhibitors (e.g. TPCK), and this sensitivity has been used in part to support a proposed cysteine protease classification for Rce1p [23-26]. Nevertheless, TPCK-sensitivity should be viewed cautiously when used as an indicator of protease classification because TPCK covalently modifies the active site histidine residues of both serine and cysteine proteases (e.g. chymotrypsin and papain, respectively), and possibly other catalytic types. Moreover, Rce1p is insensitive to thiol-modifying agents such as NEM and iodoacetamide, which further counters a cysteine protease classification for this enzyme [24, 27]. Certain mutational studies are also inconsistent with a cysteine protease classification for Rce1p [28]. Supporting a proposed metalloprotease classification for Rce1p are the observations that it requires certain glutamate and histidine residues for activity and its inhibition by 1,10-phenanthroline [24, 28]. Nevertheless, the partial sensitivity of Rce1p to a non-chelating form of phenanthroline (i.e., 4,7-phenanthroline) suggests that the inhibition by this compound class may be unrelated to chelating effects. It is also formally possible that Rce1p utilizes a novel proteolytic mechanism. The recent identification of a carboxyl peptidase requiring a Glu-Gln catalytic dyad reveals that additional proteolytic mechanisms may yet be discovered [29].
Several inhibitors of Rce1p in addition to those discussed above have been described [23-27, 30-35]. These fall into two classes: general non-specific inhibitors and substrate mimetics. The first class includes chloromethyl ketones, organomercurials (e.g. MSA, PHMB, and PHMS), and certain metal ions (i.e., Cu2+ and Zn2+). Rce1p is reportedly insensitive to many broad-spectrum inhibitors, such as EDTA, EGTA, antipain, chymostatin, pepstatin A, leupeptin, E64, and is partially sensitive to MMTS. It should be noted that the sensitivity of Rce1p to the serine protease inhibitors PMSF and DFP and the alkylating agent NEM has been inconsistently reported [24, 27, 30, 31]. The second class of Rce1p inhibitors is represented by substrates with non-cleavable peptide bonds, isoprenoid-like compounds, and bisubstrate analogues (i.e., compounds containing both a farnesylmimetic and peptidomimetic). These compounds typically act competitively to inhibit Rce1p, and some have IC50 values in the nM range [27, 32, 34-36]. Despite the various studies probing the enzymology and inhibitor profile of Rce1p, the mechanistic classification of Rce1p remains undefined, with cysteine and metalloprotease categories having been independently proposed [24, 37].
In this study, we have compared the inhibitor profiles of Rce1p and Ste24p orthologs using two primary agents, chloromethyl ketones and peptidyl (acyloxy)methyl ketones (AOMKs). AOMKs were included in our analysis because a pilot screen of about 20 compounds yielded an AOMK as a candidate yeast Rce1p inhibitor. Compounds of this class reportedly inhibit the cysteine protease cathepsin B [38-41], but the impact of AOMKs on Rce1p activity had not previously been reported. We evaluated three distinct orthologs of each enzyme to assess whether enzymatic profiles are evolutionarily conserved within each group and investigated the inhibitor profiles of these enzymes using two distinct assays to independently confirm our observations. The orthologs were all expressed using the yeast system to aid comparative evaluations. We present evidence that the inhibition of Rce1p and Ste24p by chloromethyl ketones is substrate-specific, and we report for the first time that AOMKs can inhibit both of the CaaX proteases as well as the yeast ICMT Ste14p. Additionally, we demonstrate that the peptidyl portion of AOMKs can modulate the inhibitory properties of this class of agent. Besides providing a new reagent that may be useful for probing the enzymology of CaaX modifying enzymes, this study underscores the caution that must be taken when evaluating the inhibitor profiles of these enzymes.
Materials and Methods
Yeast Strains and Plasmids
The yeast strains used in this study were SM3614 (MATa trp1 leu2 ura3 his4 can1 ste24Δ::LEU2 rce1Δ::TRP1) and RC757 (MATα sst2-1) [15, 42]. Plasmid-bearing versions of SM3614 were generated by transformation with the indicated plasmids according to published methods [43]. Transformed strains were routinely grown at 30 °C on synthetic complete dropout (SC-) media, as previously described [44]. The Rce1p, Ste24p, and Ste14p-encoding plasmids used in this study have previously been described and are listed in Table 1.
Table 1.
Plasmids used in this study.
Substrate Reagents
The peptide substrates used in this study were made synthetically. The fluorogenic substrates ABZ-KSKTKC(farnesyl)QLIM, ABZ-KSKTKC(farnesyl)VQLM, and ABZ-KSKTKC(farnesyl)VIQL were initially obtained from Wyeth Research (Pearl River, NY) and subsequently purchased from AnaSpec (San Jose, CA). ABZ is aminobenzoic acid, and QL is lysine ε-dinitrophenyl. The a-factor-based substrate YIIKGVFWDPAC(farnesyl)VIA was purchased from California Peptide (Napa, CA).
Peptidyl (acyloxy)methyl ketones and other compounds
Several distinct AOMKs were evaluated in this study (Figure 1). Z-Phe-Lys-2,4,6-trimethylbenzoyloxymethyl ketone (FKBK(CH3)3) was purchased from Bachem (Torrance, CA). A Phe-Ala derivative (FABK(CH3)3) was a gift from Dr. Jan Potempa (Jagiellonian University, Poland), and was subsequently synthesized in house according to standard chemical methods that will be described elsewhere (Porter, Deshert, Breevoort, Hembree, Dore and Schmidt, in preparation). Phe-Arg (FRBK(CH3)2), Phe-Gly (FGBK(CH3)2), AcTyr-Phe-Arg (YFRBK(CH3)2), and AcTyr-Phe-Gly (YFGBK(CH3)2) derivatives were obtained from Dr. Matthew Bogyo (Stanford University). Additional amounts of FRBK(CH3)2 and FGBK(CH3)2 were synthesized in house according to standard chemical methods that will be described elsewhere (Porter, Dechert, Breevoort, Hembree, Dore and Schmidt, in preparation). All inhibitors were dissolved in DMSO, with the exception of TLCK, which was dissolved in H2O. Other chemical reagents were purchased from Sigma-Aldrich.
Fig. 1. Chemical structures of peptidyl (acyloxy)methyl ketones.

The general chemical structure of an AOMK is shown (top). The names of the compounds used in this study are listed along with the substitutions specific to the indicated compound.
In vitro fluorescence-based CaaX proteolysis assay
An established fluorescence-based assay was used to monitor Rce1p-dependent cleavage of a quenched fluorogenic peptide substrate [36]. By using a slightly different substrate, this assay was adapted to monitor Ste24p activity. In brief, the assay involves mixing an appropriate fluorogenic substrate with membranes derived from yeast over-expressing the appropriate CaaX protease. The membranes used as the source of activity were isolated as 1 mg/ml total protein stocks in Lysis Buffer (50 mM Tris, pH 7.5, 0.2 M sorbitol, 1 mM EDTA, 0.2% NaN3, protease inhibitors CLP, aprotinin, PMSF) according to our reported methods [22, 28]. Prior to use, the membranes were diluted to 0.5 mg/ml with Assay Buffer (100 mM HEPES, pH 7.5, 5 mM MgCl2) and preincubated with DMSO or inhibitors for 10 min at 30 °C, unless otherwise indicated. The substrate was typically diluted with Assay Buffer to 40 μM from a 1 mM stock; a range of concentration (0-200 μM) was used in instances where kinetic parameters were sought. Assays were initiated by mixing equal volumes (50 μl) of the membrane and substrate dilutions in a 96-well plate suitable for use in a microtiter plate fluorometer. The fluorescence in the samples was measured at 420 nm every 30 sec over a 35-60 min time course at 30 °C using either a SpectraMax Gemini EM fluorometer (Molecular Devices) or a Bio-Tek Synergy fluorometer equipped with a 320/420 nm excitation/emission filter set. The collected data were graphed and initial linear slopes determined using Microsoft Excel. These values were used to calculate % activities relative to the DMSO-treated enzyme, which was always included as a control in each reaction set.
Kinetic Analyses
Kinetic parameters were typically determined using nonlinear regression methods (Prism 4.0 GraphPad Software Inc.). The Kitz-Wilson approach was used to compare the time-dependent interactions of TPCK and FKBK(CH3)3 with Rce1p [45]. According to the associated theory, an irreversible active-site directed inhibitor of an enzyme should show a linear decrease in ln[Et/E0] with time of incubation, where Et is the activity at time t and E0 is the activity of the uninhibited enzyme at time zero; the slope of this line is defined as the apparent inactivation rate Kapp. TPCK was previously shown to be an irreversible active-site inhibitor of bovine Rce1p by this analysis [23].
In vitro a-factor-based CaaX proteolysis assay
An established assay was used for the in vitro production of bioactive a-factor from the farnesylated pentadecapeptide precursor YIIKGVFWDPAC(farnesyl)VIA [16]. In brief, the assay involves mixing membranes derived from yeast over-expressing the appropriate CaaX protease with the farnesylated substrate. The membranes were isolated and diluted for the assay as described above. The substrate was diluted from a 100 μM stock to 40 μM using Assay Buffer (see above). Assays were initiated by mixing equal volumes (10 μl each) of the substrate and membrane components in a 96-well plate suitable for use in a PCR thermocycler. After an 8 min incubation at 30 °C, the samples were heated to 95 °C for 1 min to inactivate enzymatic activity, cooled, and supplemented with S-adenosylmethionine (1.7 mM final) and yeast membranes containing the Ste14p ICMT (0.33 mg/ml final) to initiate carboxyl methylation of cleaved products. For reactions with limited S-adenosylmethionine and Ste14p, the concentrations were 160 μM and 0.1 mg/ml respectively. The Ste14p-membranes were derived from a CaaX protease-deficient strain as previously described [16]. After 60 min of incubation at 30 °C, the samples were supplemented with copper sulfate (1.2 mM final) to stop the methylation reaction. The a-factor activity in each sample was determined using a biological response assay in which yeast supersensitive to the a-factor mating pheromone (RC757) undergo growth arrest in the presence of 7 nM or greater concentrations of pheromone [46]. The activity observed for a two-fold dilution series of a sample was compared to that of other samples for an assessment of relative activity measurements.
Inhibitor and Chaotropic Agent Treatments
To assess the inhibition by the compounds described in this study, the above assay protocols were modified such that diluted membrane samples were pretreated with the appropriate compound or control (DMSO or H2O) for 10 min at 30 °C prior to use. The pre-treated sample was split into two portions for use in each assay when evaluating substrate-specific effects. For reversibility experiments, pre-treated membranes were recovered by centrifugation at 16,000g for 15 min, washed twice with 2-fold excess of Wash Buffer (a 1:1 ratio of Lysis Buffer and Assay Buffer) containing varying amounts of NaCl (0-1.5 M) or urea (0-0.125 M), and finally resuspended to the original input volume with Wash Buffer lacking added salt or urea. The resuspended membranes were used directly in the fluorescence-based assay along with an unprocessed control to confirm that inefficient membrane recovery was not the reason for observed decreases in activity.
Results
Peptidyl (acyloxy)methyl ketones inhibit Rce1p
The enzymes evaluated in this study were isolated from CaaX protease deficient yeast (MATa rce1Δ ste24Δ) engineered to over-express the human, plant (A. thaliana), or native yeast Rce1p enzyme. Because the yeast CaaX proteases localize to the endoplasmic reticulum (ER) compartment in yeast, enriched ER membranes were used as the source of enzyme activity for our experiments [4]. Using a previously described fluorescence-based activity assay designed for human Rce1p, we observed Rce1p dependent activity for all three orthologs. Despite the common expression system and activity assay, the specific activities for the isolated membranes were quite distinct, with yeast Rce1p-containing membranes having the highest value (3.48 nmol/min/mg of total membrane protein), which was followed by plant and human Rce1p-containing membranes (0.89 and 0.10 nmol/min/mg, respectively). The activity observed was dependent on Rce1p since membranes derived from yeast deficient in CaaX proteases had inconsequential activity in this assay (0.0014 nmol/min/mg). The reason for the varied specific activities is unknown but could be attributable to differential enzymatic properties, expression, localization, and/or substrate specificity. For example, the specific activity of plant Rce1p is two-fold higher at pH 6.0 than at pH 7.5 (the pH used in this study) while yeast Rce1p has optimal activity at pH 7.5 and 35% less activity at pH 6.0.
Using the fluorescence assay, we evaluated the inhibitor profile of the three Rce1p orthologs. To facilitate cross-species comparisons, the observed activities for each ortholog were normalized to an appropriate mock treated control. Our analysis revealed that the orthologs were all inhibited by TPCK and, to a lesser extent, by TLCK (Figure 2). Variations in sensitivity were observed. For example, yeast Rce1p appeared least sensitive to chloromethyl ketones by comparison to the other enzymes, while human Rce1p had an increased sensitivity to TLCK by comparison. When AOMKs were evaluated, we observed that FKBK(CH3)3 inhibited all three Rce1p orthologs. Variations in sensitivity to FKBK(CH3)3 were also observed. For example, the human enzyme was relatively more sensitive than either the yeast or plant enzyme. The related compound FABK(CH3)3 inhibited the three Rce1p orthologs to a much lesser degree, even at concentrations as high at 1 mM (86%, 87% and 74% activity for Sc, At and Hs Rce1p respectively). This suggests that the dipeptidyl portion of this class of compound impacts inhibitor specificity by some manner. Generally, the three Rce1p enzymes were found to be sensitive to TPCK and FKBK(CH3)3 and relatively less sensitive to TLCK and FABK(CH3)3.
Fig. 2. Rce1p orthologs have sensitivity to FKBK(CH3)3 and TPCK as measured using a fluorescence assay.

The inhibitory effect of various compounds on the activity of S. cerevisiae (Sc), A. thaliana (At), and human (Hs) Rce1p (A-C, respectively) was evaluated using a fluorescence-based assay. The assay monitors cleavage of a quenched fluorogenic farnesylated peptide that is based on the C-terminal sequence of the K-Ras4b precursor (ABZ-KSKTKC(farnesyl)QLIM). The indicated Rce1p enzyme was over-expressed using a plasmid-based system in yeast lacking the chromosomal copies of RCE1 and STE24. Membranes enriched for Rce1p were isolated and used as the source of enzymatic activity. The compounds evaluated were FKBK(CH3)3 (FKBK), FABK(CH3)3 (FABK), TPCK, TLCK, and DMSO as a control. All compounds were used at 250 μM to pretreat the yeast-derived membranes used as the source of enzymatic activity. The DMSO-treated sample was defined as having 100% activity for each membrane set to facilitate cross-species comparative analyses. Each value represents the average activity of three independent reactions with the positive standard deviation of the measurements shown.
Because of the observed inhibition of Rce1p by FKBK(CH3)3, we evaluated additional AOMKs using the Ras-based fluorescence assay. The dipeptidyl AOMK FRBK(CH3)2 and tripeptidyl AOMK YFRBK(CH3)2 are similar to FKBK(CH3)3 in that they have a charged amino acid in the last position of the peptidyl portion of the compound (Figure 1); they mainly differ in having distinct peptidyl moieties and two rather than three methyl substitutions on the benzoyl group. FRBK(CH3)2 inhibited Rce1p and was the most potent Rce1p inhibitor of the AOMKs evaluated, whereas YFRBK(CH3)2 was the least inhibitory (Table 2). The dipeptidyl AOMK FGBK(CH3)2 and tripeptidyl YFGBK(CH3)2 are similar to FABK(CH3)3 in that they have a small uncharged amino acid in the last position of the peptidyl portion. None of these compounds inhibited Rce1p activity. Our results suggest that a positively charged amino acid in the last position of the dipeptidyl moiety of the AOMK is required for the ability of this compound class to inhibit Rce1p.
Table 2.
Effect of chloromethyl ketones and peptidyl (acyloxy)methyl ketones on the in vitro activity of yeast Rce1p and Ste24p.
| % Activity Remaininga | ||
|---|---|---|
| Compound | Rce1p | Ste24p |
| FKBK(CH3)3 | 51.3 ± 6.0 | 29.4 ± 1.0 |
| FABK(CH3)3 | 95.3 ± 2.5 | 95.1 ± 2.4 |
| FRBK(CH3)2 | 41.0 ± 4.4 | 58.6 ± 0.3 |
| FGBK(CH3)2 | 99.0 ± 3.2 | 112.7 ± 2.1 |
| YFRBK(CH3)2 | 80.2 ± 0.9 | 96.4 ± 4.6 |
| YFGBK(CH3)2 | 97.7 ± 1.6 | 99.4 ± 2.7 |
| TPCK | 77.4 ± 1.3 | 92.3 ± 3.4 |
| TLCK | 101.0 ± 0.0 | 94.7 ± 0.5 |
Values are averages (n ≥ 3 replicates) of percent activity remaining from studies using 100 μM of the indicated compound relative to a DMSO-treated control.
The inhibition of yeast Rce1p by TPCK is substrate-specific
To independently confirm the inhibitory effects of TPCK and certain AOMKs, we evaluated these compounds using a coupled CaaX proteolysis-methylation assay that generates the bioactive a-factor mating pheromone [16]. This assay utilizes a distinct substrate and readout that essentially eliminates any interference arising from unanticipated autofluorescing or quenching properties associated with compounds being evaluated. Using the coupled assay, we surprisingly observed that TPCK did not significantly inhibit a-factor production (Figure 3). An aliquot of the same TPCK-treated sample demonstrated reduced Rce1p activity when evaluated using the fluorescence assay (Figure 3B, lower panel). Except for the nature of the substrate, the reaction conditions for the proteolytic step in both assays were identical (i.e., the same buffer and concentrations of substrate and membrane). The activity observed in the presence of TPCK in the a-factor assay cannot be attributed to an unaccounted a-factor-specific proteolytic activity because membranes lacking Rce1p and Ste24p have insignificant activity in this assay [16]. Unlike TPCK, FKBK(CH3)3 inhibited regardless of the assay used. Consistent with our previous observations, neither TLCK nor FABK(CH3)3 inhibited Rce1p to any significant extent when evaluated using the a-factor assay.
Fig. 3. The sensitivity of yeast Rce1p to TPCK is substrate-specific.

A) The inhibitory effect of various compounds on the activity of yeast Rce1p was evaluated using an assay that monitors in vitro formation of the bioactive a-factor mating pheromone. The reaction conditions were similar to those described for Figure 2, except that the compounds were used at 400 μM to pretreat membranes and the initial proteolysis step of the reaction was followed by an in vitro methylation step. A portion of the final sample and two-fold serial dilutions were spotted onto a lawn of MATα sst2-1 cells that had been spread as a thin lawn on a YEPD plate. This MATα background is supersensitive to the a-factor mating pheromone and undergoes a strong growth arrest in its presence, as indicated by a zone of no growth (spot) in the lawn after incubation of the plate at 30 °C for 24 hrs. Each experiment was performed multiple times, and one representative replicate is shown. B) Graphical representation of data from trials using the a-factor assay (top) or the fluorescence assay (bottom) with the values at the top of each bar representing the average amount of activity observed. The sample order in the two panels is identical.
Inhibition of Rce1p by FKBK(CH3)3 is not readily reversible
As a first step toward investigating the nature of interaction between AOMKs and Rce1p, we evaluated whether FKBK(CH3)3-mediated inhibition was readily reversible by washing inhibitor-treated membranes with buffers containing salt (0-1.5 M NaCl) or urea (0-0.125 M). The washes themselves did not significantly affect the activity of untreated Rce1p by comparison to an unwashed sample (data not shown). Using this approach, we observed that FKBK(CH3)3-treated Rce1p could not be reactivated by any of the wash conditions employed (Table 3).
Table 3.
The effect of salt and urea washes on the in vitro activity of inhibitor-treated yeast Rce1p and Ste24p.
| % Activity Remaininga | ||||
|---|---|---|---|---|
| Rce1p | Ste24p | |||
| Wash | DMSOa | FKBK(CH3)3 | DMSO | FKBK(CH3)3 |
| 0 mM NaCl | 100.0 ± 0.5 | 27.3 ± 1.0 | 100.0 ± 0.7 | 30.3 ± 3.5 |
| 125 mM NaCl | 92.3 ± 0.9 | 28.9 ± 1.1 | 97.4 ± 8.7 | 35.7 ± 2.2 |
| 1500 mM NaCl | 99.0 ± 2.5 | 31.6 ± 0.2 | 113.4 ± 1.3 | 30.3 ± 1.5 |
| 0 mM urea | 100.0 ± 0.6 | 37.9 ± 0.4 | 100.0 ± 5.5 | 20.4 ± 10.7 |
| 62.5 mM urea | 94.9 ± 0.4 | 37.1 ± 6.0 | 100.6 ± 2.4 | 20.0 ± 1.7 |
| 125 mM urea | 92.6 ± 2.1 | 39.3 ± 0.6 | 96.2 ± 3.5 | 19.4 ± 0.5 |
Values are averages (n ≥ 2 replicates) of percent activity remaining after treatment under the indicated condition. Values for DMSO-treated samples are relative to the 0 mM wash condition while values for FKBK(CH3)3-treated samples are relative to the appropriate washed DMSO-treated sample.
Membranes were pretreated with DMSO or 250 μM of FKBK(CH3)3 for 10 min at 30 °C before being washed.
To further resolve the inhibitory mechanism of FKBK(CH3)3, we examined the kinetics of enzyme inhibition. This analysis revealed that FKBK(CH3)3 reduced the Vmax of Rce1p in a dose-dependent manner (Figure 4A and B). TPCK-treatment at the same concentration also reduced the Vmax of Rce1p, but to a lesser extent. The observed Km values were essentially unchanged relative to the untreated control. Linear transformations of the kinetic data (e.g., Lineweaver-Burke analysis) revealed that inhibition by FKBK(CH3)3 could be attributable to either a reversible noncompetitive or irreversible mechanism. By point of comparison, TPCK is reportedly an irreversible active-site inhibitor of Rce1p [23]. One characteristic of this type of inhibitor is that the extent of inhibition depends on time of exposure. We found that FKBK(CH3)3 did not inhibit Rce1p in a time-dependent manner (Figure 4C) [23]. We should also note a complicating issue regarding our analysis of kinetic parameters. During the course of our investigations, we determined that FKBK(CH3)3 partitions onto yeast membranes in a non-selective manner (Figure 4D). This observation suggests that the amount of free inhibitor in solution is actually less than predicted in our assays. This property would necessarily lead to an overestimation of IC50 and could alter the kinetic parameters observed in the presence of this compound.
Fig. 4. Kinetic analysis of FKBK(CH3)3 and TPCK mediated inhibition of Rce1p.

A) FKBK(CH3)3 and TPCK both reduce the Vmax of Rce1p. Initial reaction rates were determined after pretreatment with DMSO, 100 μM FKBK(CH3)3 or 100 μM TPCK using the fluorescence-based assay described in Figure 2. Data points were plotted and a best-fit curve determined by non-linear regression analysis using Prism 4.0. B) Summary of kinetic parameters observed after pretreatment with inhibitors. Km and Vmax were determined by non-linear regression analysis. C) FKBK(CH3)3 is a fast-acting inhibitor of Rce1p. FKBK(CH3)3 and TPCK were each used at 250 μM to pretreat Rce1p for the indicated times. Initial reaction rates were determined at various time points (Et) and used to calculate enzyme activity relative to the reaction rate for a DMSO-treated control at t = 0 min (E0), the time at which inhibitor and substrate were added simultaneously to the enzyme. The natural log of the resultant Et/E0 values were plotted vs. time and a best-fit line determined for each dataset. D) FKBK(CH3)3 partitions with yeast membranes. Samples containing buffer alone (buffer) or yeast membranes devoid of CaaX protease activity (membranes) were incubated with DMSO or 100 μM FKBK(CH3)3 for 10 min at 30 °C. The samples were centrifuged to clear membrane material and the clarified supernatants were used to resuspend Rce1p-containing membrane pellets that were prepared in parallel. The inhibitor activity associated with each preconditioned sample was determined using the assay described in Figure 2. Values reported for FKBK(CH3)3 are relative to the DMSO control for each condition.
Ste24p can be inhibited by AOMKs
In order to further understand the specificity of chloromethyl ketones and AOMKs, we sought to determine the inhibitor profile of Ste24p in relationship to these agents. For this study, we developed a fluorescence-based assay to monitor Ste24p activity. Initially, we evaluated the Rce1p substrate (CQLIM) for its ability to be cleaved by yeast Ste24p and determined that it was a poor substrate (Figure 5); similar results were obtained for plant and human Ste24p (Porter and Schmidt, unpublished observation). We next evaluated isoprenylated substrates that differed only in the placement of the quenching group. This analysis revealed that yeast Ste24p had a marked preference for the quencher at either the a2 (CVQLM) or X position (CVIQL) of the CaaX motif relative to the a1 position (CQLIM), while yeast Rce1p had a marked preference for the quencher at the a1 position. Using the CVIQL fluorogenic substrate, we determined the specific activities of several Ste24p orthologs. These values varied as had been observed for the Rce1p orthologs. Yeast Ste24p had the highest specific activity, followed by the human and plant enzymes (9.41 nmol/min/mg, 1.43 nmol/min/mg, 1.01 nmol/min/mg of total membrane protein, respectively). To our knowledge, this is the first assay described that is amenable for kinetic analyses of Ste24p activity. Accordingly, we have determined that yeast Ste24p has a KM of 10.9 μM using the CVIQL fluorogenic substrate and nonlinear regression methods.
Fig. 5. Quencher position impacts the specificity of Rce1p and Ste24p.
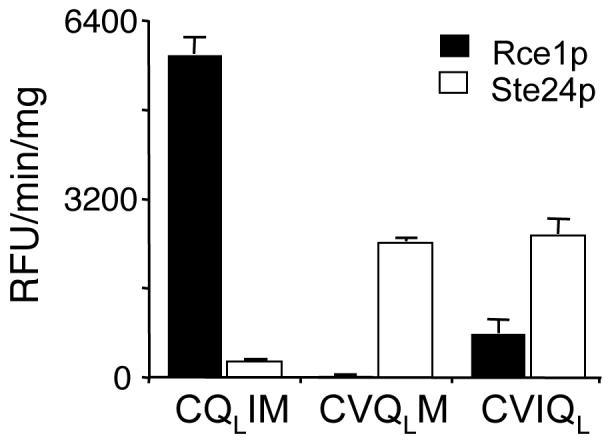
The cleavage of quenched fluorogenic peptides derived from K-Ras4b were evaluated using yeast membrane extracts containing either yeast Rce1p or Ste24p and the assay described in Figure 2. The substrates vary in the position of the quenching group, which is at either the a1 (CQLIM), a2 (CVQLM), or X position (CVIQL) of the CaaX motif. Otherwise, the samples are identical to the substrate described in Figure 2. Incorporation of the quenching group at the a2 or X position converts this substrate into a Ste24p reporter. The closed and open bars represent the activities of Rce1p and Ste24p, respectively.
Using the fluorescence-based assay, we evaluated the inhibitor profile of three Ste24p orthologs (Sc Ste24p, At Ste24p, and Hs Ste24p). FKBK(CH3)3consistently inhibited these enzymes (Figure 6). FABK(CH3)3 was a weak inhibitor at best, with human Ste24p appearing to be the most sensitive of the group. Neither TPCK nor TLCK had significant inhibitory activity. Yeast Ste24p was also determined to be inhibited by FRBK(CH3)2, but not by FGBK(CH3)2 or tripeptidyl AOMKs (Table 2). To investigate whether the observed inhibition profile of Ste24p was substrate-specific, we evaluated the effects of these compounds using the a-factor based assay. Paralleling our results with Rce1p, we observed that TPCK did not inhibit yeast Ste24p, whereas FKBK(CH3)3 remained a potent inhibitor by comparison (Figure 7). Like Rce1p, we also observed that the inhibition of Ste24p by FKBK(CH3)3 was not readily reversible (Table 3).
Fig. 6. Ste24p orthologs have comparable inhibitor profiles as measured using a fluorescence assay.

The inhibitory effect of the indicated compounds on the activity of S. cerevisiae (Sc), A. thaliana (At), and human (Hs) Ste24p (A-C, respectively) was evaluated as described in Figure 2, except that a different substrate (CVIQL) was used. All compounds were used at 250 μM to pretreat the yeast-derived membranes used as the source of enzymatic activity. The DMSO-treated sample was defined as having 100% activity for each membrane set to facilitate cross-species comparative analyses.
Fig. 7. The sensitivity of Ste24p to select inhibitors is not substrate-specific.

A) The inhibitory effect of various compounds on the activity of yeast Ste24p was evaluated using the a-factor assay and conditions described in Figure 3. B) Graphical representation of the data from all the trials (n > 3) with the values at the top of each bar representing the average amount of activity observed.
FKBK(CH3)3 inhibits the Ste14p ICMT
The ability of FKBK(CH3)3 to inhibit both Rce1p and Ste24p prompted us to examine whether the isoprenylcysteine carboxyl methyltransferase (ICMT) was a target of this compound. The integral membrane protein Ste14p is the yeast ICMT [20]. To investigate this issue, we took advantage of the fact that a-factor production relies on a coupled assay having distinct proteolytic and methylation steps (Steps 1 and 2, respectively). Thus, the inhibition of Ste14p can be evaluated by simply adding the agent after proteolysis has been completed and the protease heat-inactivated (Step 1). Under standard reaction conditions where Ste14p activity was added in excess, FKBK(CH3)3 had only a modest inhibitory effect on a-factor production when added during Step 2 (Figure 8A). The inhibition was less than that observed when added during Step 1, suggesting that FKBK(CH3)3 does indeed inhibit the CaaX proteases and that the loss of activity observed in the a-factor assay with this compound is due to synergistic inhibition of the CaaX protease and ICMT. The inhibitory effect of FKBK(CH3)3 was exaggerated when limiting amounts of Ste14p were present in the reaction (Figure 8B), demonstrating that the ICMT is indeed inhibited by FKBK(CH3)3.
Fig. 8. The Ste14p isoprenylcysteine carboxyl methyltransferase is inhibited by FKBK(CH3)3.

The a-factor assay was used to test for inhibition of the Ste14p ICMT. A) FKBK(CH3)3 was added immediately prior to the Rce1p mediated proteolysis step (Step 1) or the Ste14p mediated carboxyl methylation step (Step 2) but otherwise using the conditions described in Figure 3. A representative replicate data set (top panel) and a graph summarizing the results of the experiment (bottom panel) are shown. B) FKBK(CH3)3 was added to Step 1 or Step 2 as described in A, but in the presence of a limiting amount of Ste14p.
Discussion
This study reports several new findings. First, we document using a single expression system that Rce1p orthologs have a similar inhibitor profile to certain agents (Figure 2). This observation, when combined with the observation that Rce1p orthologs have conserved substrate specificity, strengthens the assertion that enzymatic studies of yeast Rce1p and other non-human orthologs will ultimately lead to a better understanding of the human enzyme, which is of biomedical importance [28]. Second, our study demonstrates that TPCK is an unreliable diagnostic inhibitor of Rce1p (Table 2 and Figure 3). This finding suggests the use of caution when interpreting previous studies where TPCK was used in efforts to define the mechanistic class of the enzyme [24, 27]. Lastly, we describe peptidyl AOMKs as novel agents that inhibit both Rce1p and Ste24p (Table 2), and in doing so we developed a new in vitro assay for monitoring Ste24p activity (Figure 5 and Figure 6). The AOMKs themselves represent new and potentially useful tools for investigating Rce1p and Ste24p enzymology that can perhaps serve as the basis for developing more potent inhibitors of these enzymes. This latter point is supported by the observation that varying the peptidyl moiety of AOMKs can yield an improved Rce1p inhibitor (i.e. FRBK(CH3)2). While no single AOMK evaluated in this study was absolutely specific for either Rce1p or Ste24p, our observation that Rce1p and Ste24p display differential sensitivity to certain AOMKs suggests that Rce1p or Ste24p specific inhibitors might be extractable from this class of compounds (Table 2).
Our observation that substrate context impacts the effectiveness of Rce1p inhibitors could explain the inconsistent effect of serine protease inhibitors and alkylating agents on Rce1p activity [24, 27, 30, 31]. We thus suggest that evaluating inhibitors of Rce1p in the context of multiple substrates is the best way to fully ascertain the inhibitory properties of a particular compound for this multi-substrate enzyme. Rce1p mutants should be similarly evaluated since we have observed that the activities of certain Rce1p mutants are substrate dependent [28]. We suspect that substrate context may also explain why yeast Rce1p C251A is reported to be both active and inactive [24, 28]. While we do not observe activity defects for Rce1p C251A when using any of our in vitro or in vivo assays, it remains a formal possibility that the initial report on this particular mutant utilized an enzyme/substrate combination that uncovers sensitivities that our combinations did not. The fact that most studies of Rce1p have been performed using a single enzyme/substrate combination certainly raises important concerns regarding the interpretation of reported data. We fully expect to continue evaluating Rce1p in the context of multiple substrates because of this issue.
Despite AOMKs being described as cysteine protease-specific inhibitors that are proposed to mediate their effects through covalent modification of an active site cysteine residue, it is unlikely that this mechanism applies in the context of Rce1p. First, we have observed that yeast Rce1p C251A is fully functional [28]; C251 is the only cysteine found among Rce1p orthologs that is invariably conserved. Moreover, we have observed that FKBK(CH3)3 inhibits Rce1p C251A to the same extent as the wildtype enzyme. Second, the fact that Ste24p and Ste14p, two enzymes reportedly lacking active site cysteine residues, are inhibited by FKBK(CH3)3 argues against a cysteine-directed modification. Lastly, mutational and bioinformatic analyses do not support Rce1p being a cysteine protease.
All together, our kinetic data suggest that FKBK(CH3)3 is a tightly bound but otherwise reversible noncompetitive inhibitor of Rce1p. We do not believe that FKBK(CH3)3 acts as an irreversible inhibitor because of a lack of time dependent behavior, but we cannot formally exclude that FKBK(CH3)3 acts faster than we can measure with our existing assays. Another caveat that must be considered is the propensity of FKBK(CH3)3 to partition with yeast membranes. This property necessarily alters the amount of free inhibitor available for interacting with Rce1p and could markedly alter our reported IC50 values and kinetic parameters. Whether other reported Rce1p inhibitors have the same property has not been addressed, but should be considered given our observations. Despite the above caveats, it is clear that FKBK(CH3)3 inhibits three distinct enzymes involved in CaaX protein maturation. Among several possible explanations for this observation, we suggest that FKBK(CH3)3 might be an allosteric inhibitor that binds to a common structural feature that is possessed by Rce1p, Ste24p, and Ste14p. Alternatively, FKBK(CH3)3 may perturb some aspect of membrane association or metal ion coordination, the latter being proposed as a shared property of these enzymes [16, 22, 37, 47].
As for TPCK, our data are consistent with it being an active-site irreversible inhibitor of yeast Rce1p, which is the reported mechanism described for this agent in the context of bovine Rce1p [23]. It is challenging to explain, however, the apparent substrate-specific inhibitory properties of TPCK. Among several possibilities, we suggest that the presence of TPCK at the active site interferes with the binding of low-affinity substrates (i.e. the highly charged K-Ras4b based fluorescent peptide) more so than high affinity substrates (i.e. the highly hydrophobic a-factor based peptide). It is also plausible that TPCK might bind Rce1p in a way that induces allosteric effects that alter substrate binding in a similar manner. Of note, TPCK is not the only substrate-specific inhibitor of Rce1p. In a recent screen aimed at identifying small molecule in vitro inhibitors of Rce1p, over 80% of hits found to inhibit cleavage of the fluorogenic K-Ras4b substrate were determined to be ineffective at preventing Rce1p-dependent a-factor production [48]. Clearly, determining the structure of Rce1p in complex with TPCK, an inhibitory AOMK, or any inhibitor is likely to resolve the mode of inhibition for these agents, but this is a daunting task given that Rce1p is an integral membrane protein possessing multiple membrane spans.
This study has provided new insight into the complex enzymology of Rce1p and new tools for future studies of the CaaX proteases. For example, our observation that dipeptidyl AOMKs can inhibit two presumably distinct proteases suggests that the mechanisms and/or active sites of the CaaX proteases Rce1p and Ste24p are more similar than previously perceived. Our findings also suggest that AOMKs could be modified into more potent and potentially specific inhibitors of either or both of the CaaX proteases. Finally, this study highlights the important need to evaluate enzyme inhibitors in the context of multiple substrates.
Acknowledgments
We are grateful to Drs. Claiborne Glover, Kelley Moremen, Jim Travis (all of the University of Georgia), and Jan Potempa (Jagiellonian University, Poland) for technical advice, critical discussions, and access to equipment, Wyeth Pharmaceuticals for providing initial samples of the fluorogenic K-Ras peptide substrates, Dr. Matthew Bogyo (Stanford University) for providing FR, YFR, FG, and YFG based AOMKs, and Andrea Lapham-Cardenas and Victoria Rogers for expert technical assistance. This work was supported by a Georgia Cancer Coalition Distinguished Cancer Clinician/Scientist Scholar award, a grant from the National Institutes of Health (to WKS), and an NIH research grant supplement to Promote Diversity in Health-Related Research (for SBP).
Abbreviations
- Z
benzyloxycarbonyl
- AOMK
(acyloxy)methyl ketone
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacetic acid
- E64
N-[N-(L-3-trans-carboxyoxiran-2-carbonyl)-L-leucyl]agmatine
- ICMT
isoprenylcysteine carboxyl methyltransferase
- TLCK
tosyl-L-lysyl-chloromethylketone
- TPCK
tosyl-L-phenylalanyl-chloromethylketone
- MMTS
methyl methanethiosulfonate
- MSA
mersalyl acid
- NEM
N-ethylmaleimide
- PHMB
p-hydroxymercuribenzoic acid
- PHMS
p-hydroxymercuriphenylsulfonic acid
- PMSF
phenylmethylsulfonylfluoride
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
There are various name designations for the Ras converting enzyme and the Sterile 24 protein - Rce1p and Ste24p/Afc1p (S. cerevisiae), Rce1/FACE-2 and Zmpste24/FACE-1 (human), AtRce1/AtFACE-2 and AtSte24 (A. thaliana), CeRce1p/CeFACE-2 and CeSte24p/CeFACE-1 (C. elegans), and the Type I (Ste24p) and Type II (Rce1p) CaaX proteases. In order to clearly convey the ortholog being described, and with apologies to others who study these enzymes, we have opted to use a standard naming convention for these enzymes - a species identifier followed by Rce1p or Ste24p.
References
- [1].Boyartchuk VL, Ashby MN, Rine J. Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science. 1997;275:1796–800. doi: 10.1126/science.275.5307.1796. [DOI] [PubMed] [Google Scholar]
- [2].Young SG, Ambroziak P, Kim E, Clarke S. Postisoprenylation protein processing: CXXX (CaaX) endoproteases and isoprenylcysteine carboxyl methyltransferase. In: Tamanoi F, Sigman DS, editors. The Enzymes. Academic Press; New York, NY: 2001. pp. 155–213. [Google Scholar]
- [3].Winter-Vann AM, Casey PJ. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat Rev Cancer. 2005;5:405–12. doi: 10.1038/nrc1612. [DOI] [PubMed] [Google Scholar]
- [4].Schmidt WK, Tam A, Fujimura-Kamada K, Michaelis S. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proc Natl Acad Sci USA. 1998;95:11175–80. doi: 10.1073/pnas.95.19.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gibbs J. Ras C-terminal Processing enzymes-New Drug Targets? Cell. 1991;65:1–4. doi: 10.1016/0092-8674(91)90352-y. [DOI] [PubMed] [Google Scholar]
- [6].Cox A, Der C. Farnesyltransferase inhibitors: promises and realities. Current Opinion in Pharmacology. 2002;2:388–393. doi: 10.1016/s1471-4892(02)00181-9. [DOI] [PubMed] [Google Scholar]
- [7].Baum C, Kirschmeier P. Preclinical and clinical evaluation of farnesyltransferase inhibitors. Curr Oncol Rep. 2003;5:99–107. doi: 10.1007/s11912-003-0096-5. [DOI] [PubMed] [Google Scholar]
- [8].Morgan MA, Ganser A, Reuter CW. Therapeutic efficacy of prenylation inhibitors in the treatment of myeloid leukemia. Leukemia. 2003;17:1482–98. doi: 10.1038/sj.leu.2403024. [DOI] [PubMed] [Google Scholar]
- [9].Zhu K, Hamilton AD, Sebti SM. Farnesyltransferase inhibitors as anticancer agents: current status. Curr Opin Investig Drugs. 2003;4:1428–35. [PubMed] [Google Scholar]
- [10].Kim E, Ambroziak P, Otto JC, Taylor B, Ashby M, Shannon K, Casey PJ, Young SG. Disruption of the mouse Rce1 gene results in defective Ras processing and mislocalization of Ras within cells. J. Biol. Chem. 1999;274:8383–8390. doi: 10.1074/jbc.274.13.8383. [DOI] [PubMed] [Google Scholar]
- [11].Bergo MO, Ambroziak P, Gregory C, George A, Otto JC, Kim E, Nagase H, Casey PJ, Balmain A, Young SG. Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol Cell Biol. 2002;22:171–81. doi: 10.1128/MCB.22.1.171-181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, Van Bruggen N, Carano RA, Michaelis S, Griffey SM, Young SG. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A. 2002;99:13049–54. doi: 10.1073/pnas.192460799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- [14].Young SG, Fong LG, Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria--new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res. 2005;46:2531–58. doi: 10.1194/jlr.R500011-JLR200. [DOI] [PubMed] [Google Scholar]
- [15].Tam A, Nouvet F, Fujimura-Kamada K, Slunt H, Sisodia SS, Michaelis S. Dual roles for Ste24p in yeast a-factor maturation: NH2-terminal proteolysis and COOH-terminal CAAX processing. J. Cell Biol. 1998;142:635–49. doi: 10.1083/jcb.142.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tam A, Schmidt WK, Michaelis S. The multispanning membrane protein Ste24p catalyzes CAAX proteolysis and NH2-terminal processing of the yeast a-factor precursor. J Biol Chem. 2001;276:46798–806. doi: 10.1074/jbc.M106150200. [DOI] [PubMed] [Google Scholar]
- [17].Corrigan DP, Kuszczak D, Rusinol AE, Thewke DP, Hrycyna CA, Michaelis S, Sinensky MS. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem J. 2005;387:129–38. doi: 10.1042/BJ20041359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Farh L, Mitchell D, Deschenes R. Farnesylation and Proteolysis are sequential, but distinct steps in the CaaX box modifcation pathway. Archives of Biochemistry and Biophysics. 1995;318:113–121. doi: 10.1006/abbi.1995.1211. [DOI] [PubMed] [Google Scholar]
- [19].Dai Q, Choy E, Chiu V, Romano J, Slivka SR, Steitz SA, Michaelis S, Philips MR. Human prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J. Biol. Chem. 1998;273:15030–15034. doi: 10.1074/jbc.273.24.15030. [DOI] [PubMed] [Google Scholar]
- [20].Romano JD, Schmidt WK, Michaelis S. The Saccharomyces cerevisiae prenylcysteine carboxyl methltransferase Ste14p is in the endoplasmic reticulum membrane. Mol. Biol. Cell. 1998;9:2231–47. doi: 10.1091/mbc.9.8.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bergo MO, Leung GK, Ambroziak P, Otto JC, Casey PJ, Young SG. Targeted inactivation of the isoprenylcysteine carboxyl methyltransferase gene causes mislocalization of K-Ras in mammalian cells. J. Biol. Chem. 2000;275:17605–10. doi: 10.1074/jbc.C000079200. [DOI] [PubMed] [Google Scholar]
- [22].Schmidt WK, Tam A, Michaelis S. Reconstitution of the Ste24p-dependent N-terminal proteolytic step in yeast a-factor biogenesis. J. Biol. Chem. 2000;275:6227–33. doi: 10.1074/jbc.275.9.6227. [DOI] [PubMed] [Google Scholar]
- [23].Chen Y, Ma YT, Rando RR. Solubilization, partial purification, and affinity labeling of the membrane-bound isoprenylated protein endoprotease. Biochemistry. 1996;35:3227–37. doi: 10.1021/bi952529s. [DOI] [PubMed] [Google Scholar]
- [24].Dolence JM, Steward LE, Dolence EK, Wong DH, Poulter CD. Studies with recombinant Saccharomyces cerevisiae CaaX prenyl protease Rce1p. Biochemistry. 2000;39:4096–104. doi: 10.1021/bi9923611. [DOI] [PubMed] [Google Scholar]
- [25].Cadiñanos J, Schmidt WK, Fueyo A, Varela I, Lopez-Otin C, Freije JM. Identification, functional expression and enzymic analysis of two distinct CaaX proteases from Caenorhabditis elegans. Biochem J. 2003;370:1047–54. doi: 10.1042/BJ20021514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cadiñanos J, Varela I, Mandel D, Schmidt WK, Díaz-Perales A, López-Otín C, JMP F. AtFACE-2, a prenylated-protein protease from Arabidopsis thaliana related to Ras converting enzymes. Journal of Biological Chemistry. 2003;278:42091–7. doi: 10.1074/jbc.M306700200. [DOI] [PubMed] [Google Scholar]
- [27].Ma Y-T, Gilbert B, Rando R. Inhibitors of the isoprenylated protein endoprotease. Biochemistry. 1993;32:2386–2393. doi: 10.1021/bi00060a033. [DOI] [PubMed] [Google Scholar]
- [28].Plummer LJ, Hildebrandt ER, Porter SB, Rogers VA, McCracken J, Schmidt WK. Mutational analysis of the ras converting enzyme reveals a requirement for glutamate and histidine residues. J Biol Chem. 2006;281:4596–605. doi: 10.1074/jbc.M506284200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fujinaga M, Cherney MM, Oyama H, Oda K, James MN. The molecular structure and catalytic mechanism of a novel carboxyl peptidase from Scytalidium lignicolum. Proc Natl Acad Sci U S A. 2004;101:3364–9. doi: 10.1073/pnas.0400246101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Akopyan TN, Couedel Y, Beaumont A, Fournie-Zaluski MC, Roques BP. Cleavage of farnesylated COOH-terminal heptapeptide of mouse N-ras by brain microsomal membranes: evidence for a carboxypeptidase which specifically removes the COOH-terminal methionine. Biochem Biophys Res Commun. 1992;187:1336–42. doi: 10.1016/0006-291x(92)90449-u. [DOI] [PubMed] [Google Scholar]
- [31].Georgopapadakou NH, Hall CC, Lambros T, Liu W, Watkins JD. A radiometric assay for Ras-processing peptidase using an enzymatically radiolabeled peptide. Anal Biochem. 1994;218:273–7. doi: 10.1006/abio.1994.1178. [DOI] [PubMed] [Google Scholar]
- [32].Hall CC, Watkins JD, Ferguson SB, Foley LH, Georgopapadakou NH. Inhibitors of farnesyltransferase and Ras processing peptidase. Biochem Biophys Res Commun. 1995;217:728–32. doi: 10.1006/bbrc.1995.2833. [DOI] [PubMed] [Google Scholar]
- [33].Dolence EK, Dolence JM, Poulter CD. Solid-phase synthesis of a farnesylated CaaX peptide library: inhibitors of the Ras CaaX endoprotease. J Comb Chem. 2000;2:522–36. doi: 10.1021/cc000026m. [DOI] [PubMed] [Google Scholar]
- [34].Schlitzer M, Winter-Vann A, Casey PJ. Non-peptidic, non-prenylic inhibitors of the prenyl protein-specific protease Rce1. Bioorg Med Chem Lett. 2001;11:425–7. doi: 10.1016/s0960-894x(00)00685-5. [DOI] [PubMed] [Google Scholar]
- [35].Craig KS, Williams DE, Hollander I, Frommer E, Mallon R, Collins K, Wojciechowicz D, Tahir A, van Soest R, Andersen RJ. Novel sesterterpenoid and norsesterterpenoid RCE-protease inhibitors isolated from the marine sponge Hippospongia sp. Tetrahedron Letters. 2002;43:4801–4804. [Google Scholar]
- [36].Hollander I, Frommer E, Mallon R. Human ras-converting enzyme (hRCE1) endoproteolytic activity on K-ras-derived peptides. Anal Biochem. 2000;286:129–37. doi: 10.1006/abio.2000.4795. [DOI] [PubMed] [Google Scholar]
- [37].Pei J, Grishin NV. Type II CAAX prenyl endopeptidases belong to a novel superfamily of putative membrane-bound metalloproteases. Trends Biochem Sci. 2001;26:275–7. doi: 10.1016/s0968-0004(01)01813-8. [DOI] [PubMed] [Google Scholar]
- [38].Bromme D, Bartels B, Kirschke H, Fittkau S. Peptide methyl ketones as reversible inhibitors of cysteine proteinases. J Enzyme Inhib. 1989;3:13–21. doi: 10.3109/14756368909030360. [DOI] [PubMed] [Google Scholar]
- [39].Krantz A, Copp LJ, Coles PJ, Smith RA, Heard SB. Peptidyl (acyloxy)methyl ketones and the quiescent affinity label concept: the departing group as a variable structural element in the design of inactivators of cysteine proteinases. Biochemistry. 1991;30:4678–87. doi: 10.1021/bi00233a007. [DOI] [PubMed] [Google Scholar]
- [40].Wagner BM, Smith RA, Coles PJ, Copp LJ, Ernest MJ, Krantz A. In vivo inhibition of cathepsin B by peptidyl (acyloxy)methyl ketones. J Med Chem. 1994;37:1833–40. doi: 10.1021/jm00038a012. [DOI] [PubMed] [Google Scholar]
- [41].Kato D, Boatright KM, Berger AB, Nazif T, Blum G, Ryan C, Chehade KA, Salvesen GS, Bogyo M. Activity-based probes that target diverse cysteine protease families. Nat Chem Biol. 2005;1:33–8. doi: 10.1038/nchembio707. [DOI] [PubMed] [Google Scholar]
- [42].Powers S, Michaelis S, Broek D, Santa Anna S, Field J, Herskowitz I, Wigler M. RAM, a gene of yeast required for a functional modification of RAS proteins and for production of mating pheromone a-factor. Cell. 1986;47:413–22. doi: 10.1016/0092-8674(86)90598-2. [DOI] [PubMed] [Google Scholar]
- [43].Elble R. A simple and efficient procedure for transformation of yeasts. BioTechniques. 1992;13:18–20. [PubMed] [Google Scholar]
- [44].Michaelis S, Herskowitz I. The a-factor pheromone of Saccharomyces cerevisiae is essential for mating. Mol. Cell. Biol. 1988;8:1309–18. doi: 10.1128/mcb.8.3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kitz R, Wilson IB. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J Biol Chem. 1962;237:3245–9. [PubMed] [Google Scholar]
- [46].Marcus S, Caldwell GA, Miller D, Xue C-B, Naider F, Becker JM. Significance of C-terminal cysteine modifications to the biological activity of the Saccharomyces cerevisiaea-factor mating pheromone. Mol. Cell. Biol. 1991;11:3603–12. doi: 10.1128/mcb.11.7.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Anderson JL, Frase H, Michaelis S, Hrycyna CA. Purification, functional reconstitution, and characterization of the Saccharomyces cerevisiae isoprenylcysteine carboxylmethyltransferase Ste14p. J Biol Chem. 2005;280:7336–45. doi: 10.1074/jbc.M410292200. [DOI] [PubMed] [Google Scholar]
- [48].Manandhar SP, Schmidt WK. Small molecule inhibitors of the Rce1p CaaX protease. Biochemical Journal. doi: 10.1177/1087057107307226. (submitted).
- [49].Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]