

Abstract
CpG oligodeoxynucleotides (CpG-ODNs) fail to elicit antitumor immunity after intravenous administration presumably due to their rapid renal clearance and low tumor accumulation. To address this issue, we tested the hypothesis that endogenous IgG can be used as systemic drug carriers to improve the pharmacokinetics, tumor accumulation, and antitumor activity of intravenously administered CpG-ODNs. To this end, tritium-labeled CpG-ODNs conjugated with one or two dinitrophenyl (DNP) haptens (DNP- and DNP2-[3H]-CpG-ODN) were intravenously dosed into DNP-immunized Balb/c mice bearing subcutaneous CT26 colorectal tumors. Serum and tissue samples for pharmacokinetic and biodistribution profiling were collected at predetermined timepoints and analyzed by liquid scintillation. In antitumor efficacy studies, DNP-immunized, CT26 tumor-bearing mice were intravenously dosed with PBS, CpG-ODN, or DNP-CpG-ODN every five days. Tumor volumes and macroscopic and histological examination of resected solid tumors were used to quantitatively and qualitatively assess tumor growth inhibition. Relative to [3H]-CpG-ODN, dinitrophenylated [3H]-CpG-ODNs displayed substantial increases in systemic exposure (900–1650 fold) and half-life (100–300 fold), marked decreases in systemic clearance (750–1500 fold) and volume of tissue distribution (13–37 fold), as well as substantial and sustained tumor accumulation (~30% vs. <2% injected dose/g). Antitumor efficacy studies demonstrated that DNP-CpG-ODN inhibited tumor growth by up to 60% relative to PBS control whereas CpG-ODN treatment had no apparent effect. Macroscopic and histological examination of harvested tumors at various timepoints revealed the presence of regions of necrotic tissue only in tumors from mice treated with DNP-CpG-ODN. Collectively, these results show the potential of endogenous IgG to mediate the systemic delivery of CpG-ODN to solid tumors and to enhance their antitumor activity following intravenous administration.
Keywords: CpG oligodeoxynucleotides, intravenous pharmacokinetics, monomeric immune complexes, tissue distribution, tumor growth inhibition
1. Introduction
Oligodeoxynucleotides harboring unmethylated cytosine-guanosine motifs (CpG-ODNs) exhibit great potential in the therapeutic treatment of cancer due to their ability to activate innate and adaptive immunity [1, 2]. CpG motifs, which are frequently expressed in the bacterial genome but genomically suppressed in vertebrates, are considered foreign by the immune system and, as a result, stimulate host defense mechanisms [3, 4]. In pre-clinical investigations with tumor-bearing mice, intratumoral or peritumoral CpG-ODN injections often lead to the complete regression of solid tumors [5–6], whereas intravenously administered CpG-ODNs fail to elicit a similarly effective antitumor response [7]. This is likely due to the minimal accumulation of CpG-ODNs in solid tumors following intravenous administration, an effect stemming from the rapid renal and hepatic clearance exhibited by all ODN-based therapeutics [8, 9]. Because intravenous administration may broaden the antitumor therapeutic applicability of CpG-ODNs and facilitate their clinical translation, we intend to use endogenous IgG, elicited through active immunization, as vectors for the systemic delivery of CpG-ODNs to solid tumors.
Endogenous IgG antibodies represent an attractive alternative to other macromolecular drug carriers that are available for systemic drug delivery to solid tumors. Their large molecular weight (MW=150 kDa), long circulation half-life (t1/2> 21 days in humans), high serum concentration (12 mg/ml), and high binding affinity, make these macromolecules ideal for tumor drug delivery via the enhanced permeability and retention (EPR) effect [10–12]. The EPR effect, widely regarded as a passive tumor targeting strategy for macromolecular systemic drug carriers, is based on the enhanced macromolecular vascular permeability and impaired lymphatic drainage displayed by solid tumors [13–15]. Indeed, it has been shown that non-specific IgG substantially accumulates in solid tumors via this process [16–17].
With this in mind, we hypothesized that immune complexes (ICs) between endogenous IgG and CpG-ODNs may also accumulate in solid tumors via the EPR effect. This, however, may ultimately depend on IC binding stoichiometry since monomeric complexes (one IgG per IC) display prolonged systemic half-lives relative to polymeric ICs [18–21]. Since, monomeric IC half-life has been shown to depend on antibody binding affinity and antibody titer [19–20], efforts to optimize CpG-ODN immune complexation were, therefore, focused on maximizing the antibody binding affinity of CpG-ODNs while keeping a constant monomeric IC stoichiometry. Systematic changes in antigen valence, hapten density, and hapten linker flexibility and length in model ODNs lead to the identification of a bivalent ODN ligand with a 66-fold enhancement in binding affinity relative to a monovalent control [22]. The same structural design was subsequently used to conjugate the DNP hapten to CpG-ODNs prior to in vivo testing.
In the present investigations, we evaluate the intravenous pharmacokinetics, tissue distribution, and tumor accumulation of DNP2-CpG-ODN relative to its monovalent control, DNP-CpG-ODN, and its unconjugated control, CpG-ODN, in DNP-immunized, CT26 tumor-bearing mice. Having observed greater systemic exposure, longer systemic half-life, and slower clearance for DNP-CpG-ODN, we subsequently investigated the tumor growth inhibition potential of only DNP-CpG-ODN using the same mouse tumor model. We conclusively demonstrate that, relative to unconjugated CpG-ODN, DNP-CpG-ODN exhibits a prolonged circulation half-life and slowed systemic clearance, significantly accumulates in solid tumors, and effectively inhibits CT26 tumor growth in DNP-immunized mice.
2. Materials and Methods
2.1 Oligodeoxynucleotides, cell lines, and mice
Endotoxin-free, PAGE-purified, phosphorothioate-linked CpG-ODN (ODN 1826: 5′-TCCATGACGTTCCTGACGTT-3′) and its non-CpG control (ODN 1982: 5′-TCCAGGACTTCTCTCAGGTT-3′) were obtained from Coley Pharmaceuticals (Wellesley, MA). Phosphorothioate-linked, 3′ conjugated DNP-CpG-ODN and DNP2-CpG-ODN were supplied at >98% purity by MWG Biotechnology Inc. (High Point, NC); DNP conjugation at the 3′ terminal of CpG-ODN is not expected to affect CpG-ODN pharmacological activity [23]. CpG-ODNs were tritium labeled with [3H]-water (5 Ci/g; Sigma, St Louis, MO) to specific activities of 3.73 × 108 dpm/μmole ([3H]-CpG-ODN), 2.20 × 109 dpm/μmole (DNP-[3H]-CpG-ODN), and 1.88 × 109 dpm/μmole (DNP2-[3H]-CpG-ODN) following the procedure described by Graham et al. [24]. All ODNs were dissolved in sterile phosphate buffered saline (PBS) prior to in vivo use. Balb/c mouse-derived colon adenocarcinoma CT26 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA). Female Balb/c mice, weighing 18–22 grams, were purchased from Jackson Laboratories (Bar Harbor, ME).
2.2 Mouse immunization and tumor development
Primary and secondary vaccines were prepared with DNP-KLH (1 mg/ml; EMD Biosciences, La Jolla, CA) emulsified with Complete or Incomplete Freund’s Adjuvant, respectively, following manufacturer’s instructions (Sigma, St Louis, MO). Mice were immunized intraperitoneally with 200 μL of freshly emulsified primary vaccine and again two weeks later with freshly prepared secondary vaccine. Blood was collected by tail nicking prior to primary vaccination and again 6 weeks later, just prior to tumor cell inoculation. Anti-DNP IgG titers were measured by a direct ELISA assay.
CT26 cells were grown to confluence in DMEM cell media supplemented with 10% FCS and 1% penicillin/streptomycin (Invitrogen Co., Carlsbad, CA). Six weeks after primary vaccination, mice were subcutaneously inoculated in their right hind legs with 2 × 105 CT26 cells suspended in 100 μL HBSS. Tumor size was routinely monitored by visual observation and caliper measurements. Experiments typically started when tumors were 150–200 mm3 in volume and followed the guidelines put forth by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
2.3 Intravenous pharmacokinetics and tissue distribution
As a preliminary experiment, non-immunized, tumor-bearing mice were intravenously dosed with 2.8 nmoles of [3H]-CpG-ODN in 100 uL PBS (17.5 μg, 1.05 × 106 dpm). This dose was chosen to obtain a target signal to noise ratio of 104, since common background levels were found to be in the range of 100–200 dpm. At 1.0, 2.5, 5, 10, 20, 35, 60, and 120 min post-dosing, three mice were either exsanguinated by decapitation (1.0 and 2.5 min timepoints) or anesthetized intraperitoneally with 100 μL ketamine hydrochloride (Vedco, Inc; St. Joseph, MO) and exsanguinated by cardiac puncture. Visceral organs such as the liver, kidneys, lungs, heart, and spleen, as well as solid tumor, contralateral muscle tissue, and injection sites (tail) were subsequently harvested, weighed, flash frozen in liquid nitrogen, and stored at −80°C until analyzed.
Next, DNP-immunized, tumor-bearing mice were intravenously dosed with 2.8 nmoles of [3H]-CpG-ODN, DNP-[3H]-CpG-ODN, or DNP2-[3H]-CpG-ODN in 100 μL PBS. At 0.5, 2, 12, 24, 48, and 72 hours post-dosing, tissues were harvested and processed as described above. In the case of [3H]-CpG-ODN, tissues were collected at 1- and 2-hour timepoints post-dosing solely to corroborate the results obtained during preliminary investigations in non-immunized, tumor-bearing mice. Serum radioactivity was measured by direct liquid scintillation analysis. Organs were homogenized and decolorized prior to radioactivity measurements. All radioactivity values were corrected for quenching and background activity; organ values were further corrected for resident blood volumes [25].
2.4 Tumor growth inhibition studies
DNP-immunized, tumor-bearing mice were intravenously dosed with 100 μL PBS or 15.7 nmole of CpG-ODN or DNP-CpG-ODN in 100 μL PBS on days 0, 5, and 10 (9, 14, and 19 days post-tumor cell inoculation). Tumor growth was monitored every 2–3 days by measuring tumor length and width with an electronic digital caliper. Tumor volumes were calculated using the standard volume formula for an ellipsoid, length × (width)2 × π/6, and reported in cubic millimeters. Mice were euthanized when tumor volumes exceeded 1200 mm3. As a control investigation to assess the effect of immunization on tumor growth, a non-immunized, tumor-bearing mouse was also dosed with 100 μL PBS.
2.5 Macroscopic and histological examination
Livers, spleens, and solid tumors were harvested for gross macroscopic observation and histological analysis. Organs were harvested 4 days after the first treatment (CpG-ODN and DNP-CpG-ODN groups only), 5 days after the second treatment (all groups, including the non immunized mouse), and 8 days after the third treatment (CpG-ODN and DNP-CpG-ODN groups only) following the procedure described on section 2.3. Organs were fixed in phosphate-buffered formalin for 2 days and subsequently photographed with a Canon Powershot A520 4.0 megapixel digital camera, placed in tissue cassettes, and dehydrated in 70% aqueous ethanol. Cassettes were submitted to the UNC Animal Histopathology Core Facility for embedding, sectioning, and hematoxylin/eosin (H&E) staining. Sections were examined with a Nikon Microphot SA upright compound microscope at the UNC Michael Hooker Microscopy Facility and photographed with a Nikon DXM 1200 camera.
2.6 Statistics
An exact Wilcoxon rank sum test was used for comparing median tumor volumes after two doses (on day 9) of DNP-CpG-ODN and PBS against CpG-ODN. Bonferroni correction for multiple comparisons was used for controlling the overall type I error. An exact Kruskal-Wallis test was used to assess any differences in median tumor volume among the three treatment groups before the first dose. Statistical analyses were performed with SAS statistical software, version 9.1, SAS Institute Inc. (Cary, NC) by the Biostatistics Shared Resource of the University of North Carolina Lineberger Comprehensive Cancer Center. In the case of tumor growth inhibition analysis (% tumor growth), a two-tailed homoscedastic student t-Test was used to assess significant differences in % tumor growth in the DNP-CpG-ODN treatment group relative to the PBS control.
3. Results
3.1 Serum pharmacokinetics
The serum pharmacokinetics of [3H]-CpG-ODN and its dinitrophenylated derivatives were evaluated in DNP-immunized, CT26 tumor-bearing Balb/c mice. Initially, baseline pharmacokinetic parameters for [3H]-CpG-ODN were established in non-immunized, tumor-bearing mice. This study was based on the assumption that immunization against the DNP hapten would not affect the pharmacokinetics of unconjugated CpG-ODN; this was later confirmed in a corroborative pharmacokinetic study (data not shown). Subsequent investigations were appropriately conducted in DNP-immunized, tumor-bearing mice.
Concentration-versus-time data, as derived from radioactivity measurements, were fitted to a two-compartment pharmacokinetic model with reversible distribution between central and peripheral compartments and with elimination from the central compartment. As shown in Figure 1, biphasic pharmacokinetic profiles were observed for all three molecules, characterized by rapid tissue distribution and systemic elimination in the case of [3H]-CpG-ODN or, in the case of DNP-[3H]-CpG-ODN and DNP2-[3H]-CpG-ODN, by rapid distribution and slow systemic elimination. Pharmacokinetic parameter estimates, as shown in Table 1, were derived using WinNonLin 5.0.1 (Pharsight Corporation, Cary, NC). A substantial increase in elimination half-life (t1/2,β) was observed when CpG-ODN was administered as a DNP conjugate to DNP-immunized mice. Accordingly, DNP-[3H]-CpG-ODN and DNP2-[3H]-CpG-ODN displayed t1/2,β values of 185 and 75 hours, corresponding to 266- and 108-fold increases in half-life relative to unconjugated CpG-ODN (Table 1). Similarly, systemic clearance (CL) and area under serum concentration-time curve (AUC) also show marked differences relative to unconjugated CpG-ODN. For instance, 1650- and 884-fold increases in AUC values and 1530- and 746-fold decreases in CL values were observed for DNP-[3H]-CpG-ODN and DNP2-[3H]-CpG-ODN, respectively. The serum stability of [3H]-CpG-ODN and DNP-[3H]-CpG-ODN was also tested as part of these investigations by incubating these ODNs in 50% mouse DNP antisera at 37°C for 6 hrs and 72 hours, respectively. SDS-PAGE and liquid scintillation analysis at discrete timepoints revealed only intact [3H]-CpG-ODN and DNP-[3H]-CpG-ODN (results not shown).
Figure 1.
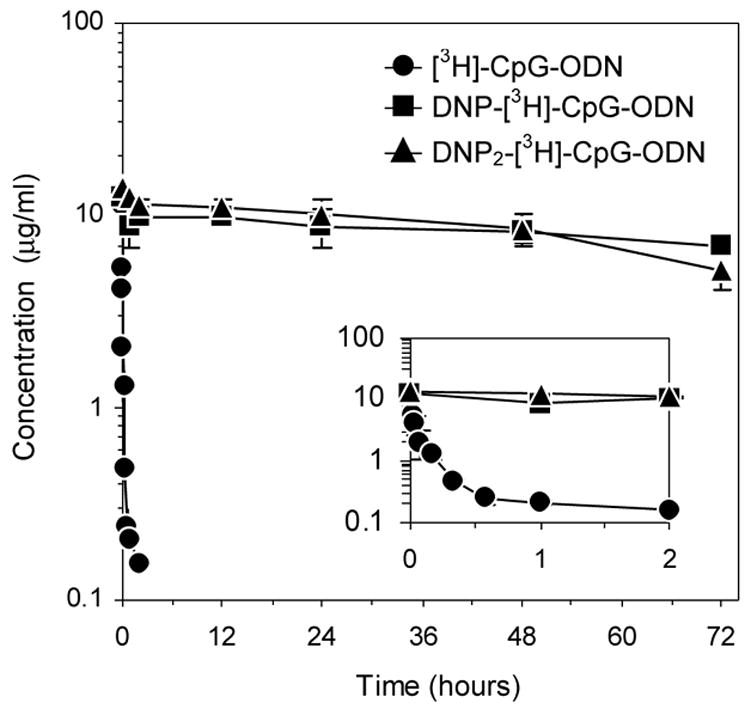
Serum concentration time-course of [3H]-CpG-ODN (●), DNP-[3H]-CpG-ODN (■), and DNP2-[3H]-CpG-ODN (▲) in CT26 tumor-bearing mice. In the case of DNP- and DNP2-[3H]-CpG-ODN, mice were immunized against DNP prior to tumor development. Data at each timepoint (see text) are expressed as mean serum concentration ± SEM, n=3. Inset depicts PK profiles from 0–2 hours.
Table 1.
Serum pharmacokinetic parameters of [3H]-CpG-ODN, DNP-[3H]-CpG-ODN, and DNP2-[3H]-CpG-ODN following a single intravenous bolus injection into DNP-immunized, tumor-bearing mice.
| [3H]-CpG-ODN* | DNP-[3H]-CpG-ODN | DNP2-[3H]-CpG-ODN | |
|---|---|---|---|
| t1/2,β (hr) | 0.694 ± 0.745 | 185 ± 95.8 (266
 )† )†
|
74.6 ± 15.0 (108
) |
| AUC (hr·μg/ml) | 1.46 ± 0.903 | 2410 ± 1120 (1650
) |
1290 ± 214 (884
) |
| V1 (ml) | 1.75 ± 0.131 | 1.54 ± 0.172 | 1.60 ± 0.167 |
| V2 (ml) | 8.46 ± 6.24 | 0.657 ± 0.234 (12.9
 ) ) |
0.228 ± 0.194 (37.0
) |
| CL (ml/hr) | 12.6 ± 7.85 | 0.00825 ± 0.00385 (1530
) |
0.0170 ± 0.00281 (746
) |
| MRT (hr) | 0.807 ± 0.949 | 267 ± 138 (331
) |
108 ± 21.6 (134
) |
In non-immunized, tumor-bearing mice; control ODN is shaded gray
Fold-change relative to control ODN. Arrows indicate direction of change
3.2 Tissue distribution and tumor accumulation
Shown in Figure 2 are the tissue distribution profiles observed for all three CpG-ODNs. [3H]-CpG-ODN was predominantly found in the liver and kidneys, an observation that agrees with the extensive hepatic metabolism and renal clearance typically observed for synthetic ODNs [8]. Other tissues such as the heart, lungs, spleen, and solid tumors displayed minimal radioactivity (<2% ID; Figure 2A). These results, obtained in non-immunized, tumor-bearing mice, were later corroborated in DNP-immunized, tumor-bearing mice (see above section; results not shown). In the case of DNP-[3H]-CpG-ODN and DNP2-[3H]-CpG-ODN, these ODNs showed substantial uptake in solid tumor, spleen, liver, and kidneys (Figure 2B and 2C) and minimal association with the heart and lungs of DNP-immunized, tumor-bearing mice. Tumor accumulation analysis, as shown in Figure 3, revealed their substantial accumulation (up to 30% ID/g) in DNP-immunized, tumor-bearing mice relative to control [3H]-CpG-ODN (<2% ID/g).
Figure 3.
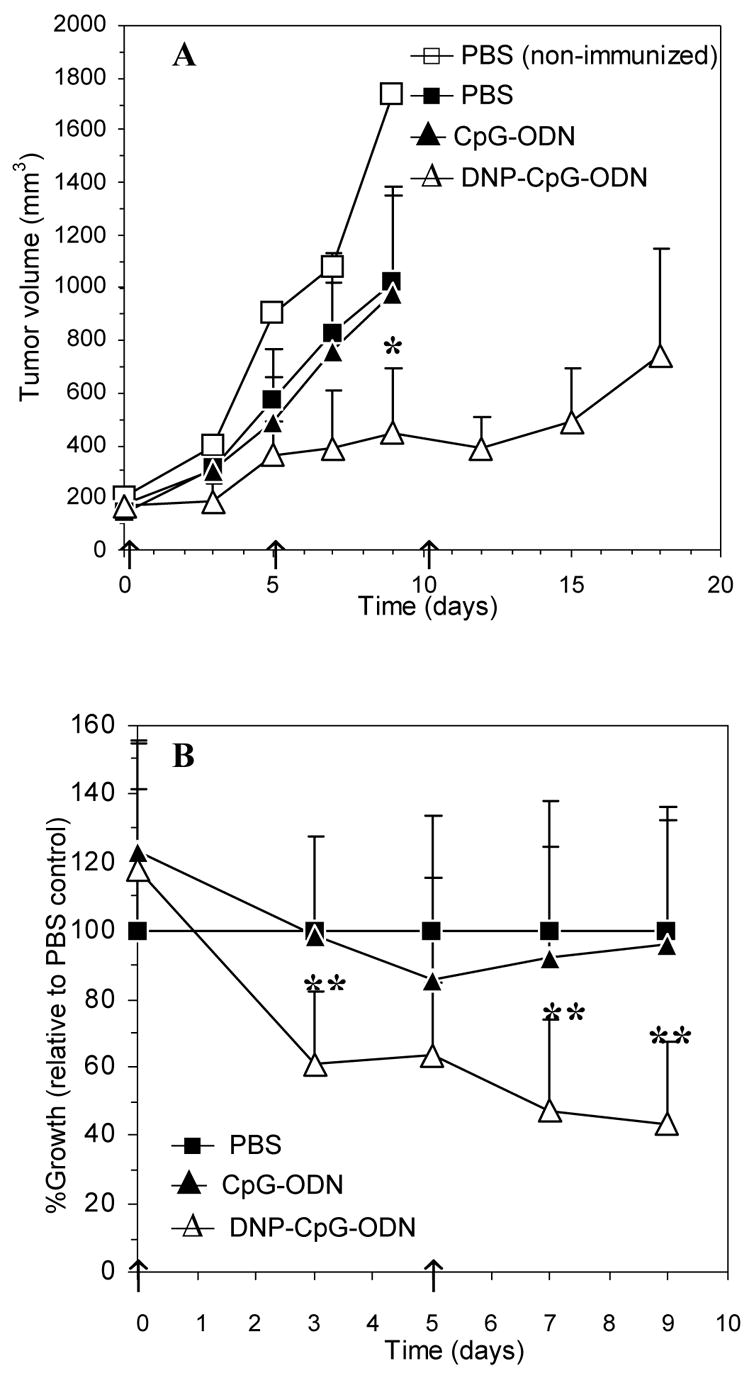
Tumor accumulation of [3H]-CpG-ODN (●), DNP-[3H]-CpG-ODN (■), and DNP2-[3H]-CpG-ODN (▲) in CT26 tumor-bearing mice. In the case of DNP- and DNP2-[3H]-CpG-ODN, mice were immunized against DNP prior to tumor development. Data at each timepoint are shown as the average percent injected dose per gram of tumor (%ID/g) ± SEM, n= 3.
Figure 2.
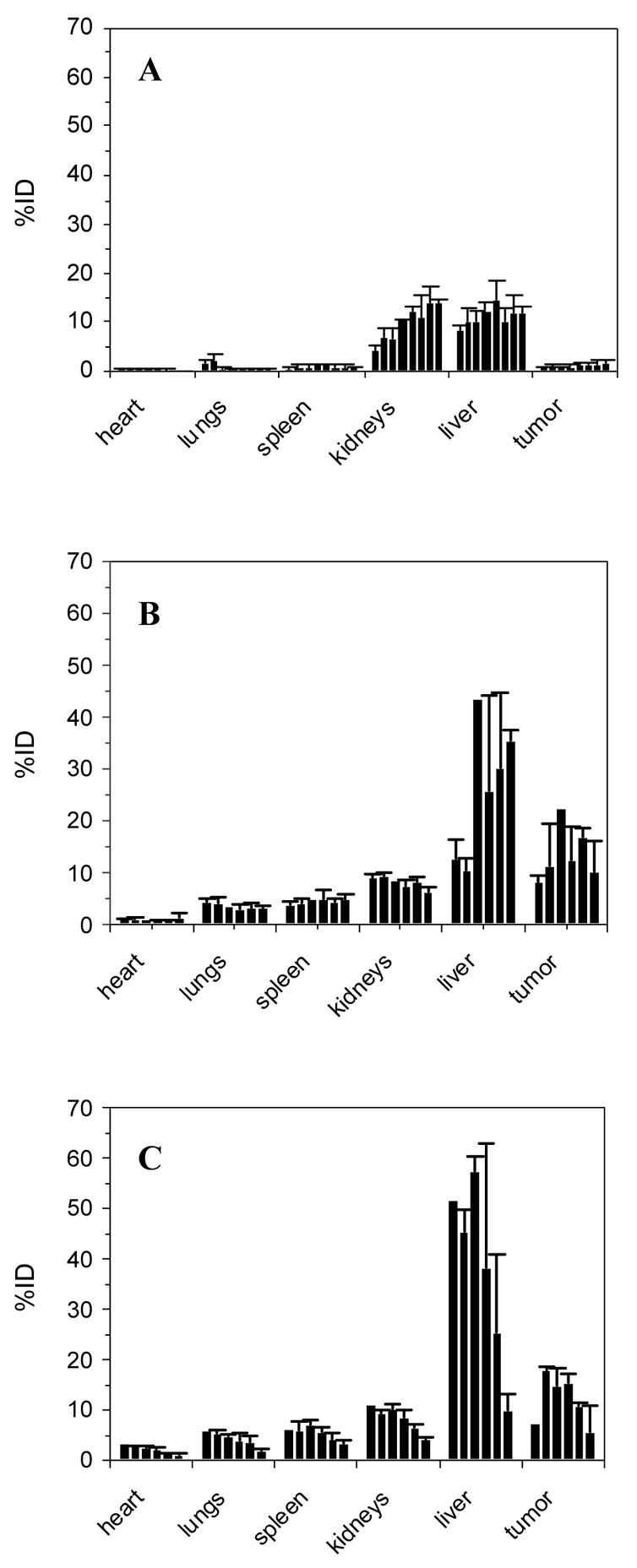
Time-dependent organ uptake of [3H]-CpG-ODN (A), DNP-[3H]-CpG-ODN (B), and DNP2-[3H]-CpG-ODN (C) in CT26 tumor-bearing mice. In the case of DNP- and DNP2-[3H]-CpG-ODN, mice were immunized against DNP prior to tumor development. Data at each timepoint are shown as the average percent injected dose (%ID) ± SEM, n= 3. Timepoints are graphed sequentially from left to right in the following order: 1, 2.5, 5, 10, 20, 35, 60, 120 min (A) and 0.5, 2, 12, 24, 48, 72 hours (B) and (C).
3.3 Tumor growth inhibition
The antitumor activity of intravenous DNP-CpG-ODN was evaluated in DNP-immunized, CT26 tumor-bearing mice dosed every five days with 100 μL PBS (2 doses), 15.7 nmoles of CpG-ODN (2 doses) or DNP-CpG-ODN (3 doses; Figure 4A). Additional dosing was limited by either tail ulcerations resulting from multiple dosing attempts or by institutional tumor size limitations that prompted euthanasia. However, the few mice that received additional doses lacked statistical power (n<3 per dosing group) and their tumor volume data were not included in this report. Accordingly, statistical differences in tumor volume among dosing groups were analyzed at 4 days after the second dose, a junction in the study in which every dosing group had received equivalent total doses. Tumor volume data after the third DNP-CpG-ODN dose are, therefore, only shown for illustrative purposes and are not included in the statistical data analysis (Figure 4A).
Figure 4.
Tumor growth inhibition by intravenously administered DNP-CpG-ODN in DNP-immunized, CT26 tumor-bearing Balb/c mice. Data are represented as tumor volume (A) and % tumor growth (B) relative to time. Tumor volume measurements on dosing days (indicated by arrows) were taken prior to dosing. Data at each timepoint are expressed as mean + SEM. Statistically significant differences relative to median tumor volumes in the CpG-ODN treatment group (A) and % tumor growth in the PBS control group (B) were assessed using an exact Wilcoxon rank sum test and a homoscedastic student t-test, respectively; * p < 0.01, ** p < 0.005.
The selection of 15.7 nmole CpG-ODN as a therapeutic dose was based on tumor growth inhibition studies by Heckelsmiller et al. in which such dose was successfully used for the peritumoral treatment of CT26 tumors in the same animal model [5]. The dosing frequency, on the other hand, was based on the estimated elimination half-life of DNP-CpG-ODN from our pharmacokinetic studies using a 2.8 nmole dose. Although this dose was 5.6-fold lower, dose-dependent changes in pharmacokinetic parameters, i.e. half-life, clearance, were not expected since renal and hepatic clearance mechanisms should remain linear (unsaturated) at both doses.
Prior to the first dose, all mice were found to be similar with respect to their median tumor volumes (p = 0.46, Kruskal-Wallis test). Differences in tumor volume, however, were readily observed between DNP-CpG-ODN and control groups (PBS and CpG-ODN) only three days after the first dose. These differences were significantly magnified by day 9, just prior to the third DNP-CpG-ODN dose, as median tumor volumes in this dosing group were found to be statistically different than tumor volumes from CpG-ODN treated mice (p = 0.006, exact Wilcoxon rank sum test). In contrast, both PBS and CpG-ODN control groups were found to be statistically similar at this timepoint (p = 0.61, exact Wilcoxon rank sum test) and displayed similarly rapid tumor growth profiles throughout the investigation.
Mice treated with DNP-CpG-ODN displayed a much different tumor growth profile than mice treated with controls. Their tumor growth profile was characterized by slowed tumor growth up to 3 days after the first dose, a steady increase in tumor volume from day 3 to day 5, sustained growth inhibition thereafter, and a final tumor volume increase 5 days after the third dose, between days 15 and 18. Since a fourth dose would have been administered on day 15, it is inferred that additional dosing is needed to achieve a sustained therapeutic effect and that a dosing frequency of every five days was sufficient but not optimal to achieve tumor growth inhibition. Nonetheless, as illustrated in Figure 4B, rapid and significant tumor growth inhibition relative to control (p<0.005, homoscedastic Student’s t-test) was observed throughout the whole investigation (up to 60% on day 9). Interestingly, it was also observed that that the non-immunized control mouse dosed with PBS exhibited tumor growth that was seemingly faster than DNP-immunized, tumor-bearing mice dosed with PBS. This single observation, however, cannot establish but only suggest to a therapeutic antitumor effect stemming from DNP immunization alone; this effect will be further investigated. Moreover, in a separate control experiment (results not shown), a DNP conjugate of CpG-ODN 1982 expressing an inactive CpG-motif failed to demonstrate antitumor activity following its intravenous administration into DNP-immunized, CT26 tumor bearing mice.
3.4 Macroscopic and histological examination
Livers, spleens, and solid tumors were harvested at different timepoints during antitumor efficacy studies to qualitatively assess any physiological changes brought about by DNP-CpG-ODN treatment. Livers and spleens were particularly selected because they show preferential DNP-CpG-ODN accumulation during biodistribution studies (Figure 2B) and are critical lymphoid organs susceptible to CpG-ODN pharmacology. Although not shown, hepatic gross physiology appeared unaltered irrespective of treatment. In contrast, CpG-ODN and DNP-CpG-ODN treatments resulted in splenomegaly (results not shown), a common condition associated with CpG-ODN immunotherapy [26–27]. The extent of splenomegaly was, however, larger in DNP-CpG-ODN treated mice, presumably due to the greater accumulation displayed by this DNP conjugate relative to unconjugated CpG-ODN (Figure 2B). Likewise, DNP-CpG-ODN treatment resulted in the appearance of a core of necrotic tissue in solid tumors, an effect that was not observed in control mice and whose extent appears to be time- and dose-dependent (Figure 5).
Figure 5.
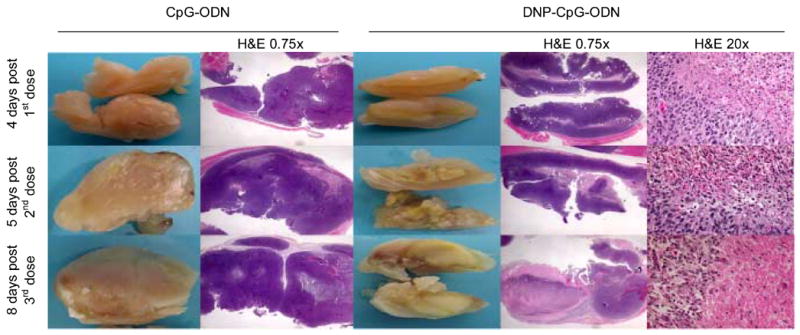
Macroscopic and histological (H&E) examination of solid tumors from DNP-immunized, CT26 tumor-bearing control (CpG-ODN) and test (DNP-CpG-ODN) mice at 4, 5, and 8 days post-first, second, and third doses, respectively. Solid tumors and 0.75x magnified H&E-stained tumor sections are shown adjacently to directly compare macroscopic and histological findings. Twenty-fold magnified sections correspond to the margins between necrotic and adjacent tissue.
Hematoxylin-eosin (H&E) staining of sectioned solid tumors from CpG-ODN and DNP-CpG-ODN treated mice are also shown in Figure 5. Tumor sections are initially shown at 0.75-fold magnification to directly compare histological and macroscopic findings. Viable and necrotic tissues identified during the macroscopic examination of solid tumors visually correspond to the hematoxylin-purple and eosin-pink stained tissue observed during histological analysis, respectively. Accordingly, the predominantly viable tumor tissue from CpG-ODN treated mice exhibited extensive hematoxylin-purple staining whereas necrotic tissue, mainly observed in tumors from DNP-CpG-ODN treated mice, displayed eosin-pink staining.
Tumor sections from DNP-CpG-ODN treated mice were further examined at 20-fold magnification to qualitatively compare the cellular morphology of necrotic (core) and viable (periphery) tumor tissue. Histological differences are evident: viable peripheral cells, as examined after the first dose, exhibited hematoxylin-purple nuclear and eosin-pink cytoplasmic staining while necrotic cells, predominantly seen after the second and third treatment, displayed an eosinophilic cytoplasm, pyknotic nuclei (5 days post-2nd dose), and eventual complete karyolysis (8 days post-3rd dose).
4. Discussion
The primary objective of these investigations is to assess the potential of using endogenous antibodies as systemic drug carriers for CpG-ODN to target solid tumors and elicit an antitumor response. Our approach takes advantage of the prolonged systemic residence and large molecular weight of endogenous antibodies to promote the preferential accumulation of monomerically complexed CpG-ODNs in tumors via the EPR effect. Monomeric immune complexation, which effectively avoids immune clearance mechanisms [18], is made possible by the strategic conjugation of one or two DNP haptens onto the 3′-terminal of the CpG-ODN molecule and by the generation of high systemic concentrations of endogenous anti-DNP IgG via active immunization.
Pharmacokinetically, unconjugated [3H]-CpG-ODN displayed short systemic residence, fast clearance, and minimal systemic exposure in DNP-immunized, tumor bearing mice. DNP-[3H]-CpG-ODN and DNP2-[3H]-CpG-ODN, on the other hand, demonstrated a contrasting systemic disposition, characterized by a long systemic half-life, slow clearance, and prolonged systemic exposure. Estimated half-life values for these molecules, however, extend past the duration of the study (72 hours) and were, therefore, extrapolated from the collected data. Nevertheless, the observed prolongation of systemic half-life is consistent with previous findings in hapten-immunized mice [19–20] and suggests that monomeric ICs may also utilize the neonatal Fc-receptor (FcRn) recycling pathway that grants IgG its long systemic half-life [28–29]. Similarly, differences in clearance and exposure (AUC) estimates may be explained as a direct consequence of the 23-fold increase in apparent MW experienced by the DNP conjugates when complexed to anti-DNP IgG. Such an increase in MW can reduce renal clearance [30] and paracellular extravasation through the vascular endothelium, effectively promoting CpG-ODN residence in the systemic circulation (as monomeric ICs) and reducing their volume of tissue distribution (V2; Table 1).
Based on previous pharmacokinetic investigations [19–20], differences in systemic disposition were also expected between DNP-[3H]-CpG-ODN and DNP2-[3H]-CpG-ODN. Results from such investigations had indicated that a ligand with high antibody binding affinity would persist in circulation for a longer period of time than its lower affinity counterpart. Accordingly, due to its higher in vitro binding affinity, DNP2-[3H]-CpG-ODN was expected to display a longer systemic half-life and slower clearance than DNP-[3H]-CpG-ODN [22]. Since the opposite was observed (Table 1), it is tempting to suggest that DNP2-[3H]-CpG-ODN, which bivalently expresses two copies of the DNP hapten (Figure 6), was in some instances able to bind more than one anti-DNP IgG molecule, triggering the formation of polymeric ICs that can be rapidly cleared from the systemic circulation [31–32]. This, although not observed during in vitro binding affinity studies [22], could be possible in vivo where a high excess of IgG antibodies exists relative to ligand. Nevertheless, due to its longer systemic half-life and slower clearance, DNP-CpG-ODN was used in tumor growth inhibition studies.
Figure 6.
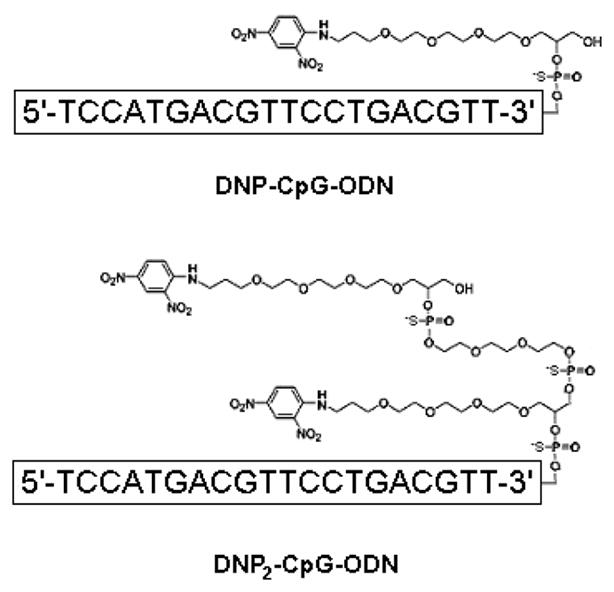
Sequence and chemical structure of dinitrophenylated CpG-ODNs.
The tissue distribution profiles of [3H]-CpG-ODN and its dinitrophenylated conjugates in DNP-immunized, tumor-bearing mice were as contrasting as their systemic pharmacokinetics. In fact, the only similarity among them was their hepatic and renal uptake, a physiological finding that, in the case of CpG-ODN, is attributed to the extensive hepatic metabolism and renal clearance experienced by synthetic ODNs [8]. In the case of DNP-[3H]-CpG-ODN and DNP2-[3H]-CpG-ODN, however, hepatic association is attributed to the combined effect of the high macromolecular vascular permeability, large organ size, and high perfusion rate displayed by the liver, and also to the presence of Kupffer cells which express receptors (FcγR) for the Fc domain of IgG [33–34]. Renal association, on the other hand, is believed to result from the direct binding of monomeric ICs to glomerular epithelial cells via the neonatal Fc receptor, FcRn [35–36]. This event appears more possible than renal excretion since the glomerular renal filter effectively excludes macromolecules with MWs above 60 kDa [30]. Both dinitrophenylated [3H]-CpG-ODNs were also found extensively associated with the spleen and solid tumors. Such tissue distribution is thought to arise from a combination of splenic and tumoral high macromolecular vascular permeability [12, 33], the presence of non-functional lymphatics in tumors [15], and the high content of FcγR-expressing cells residing in both tissues [37–38].
Tumor accumulation also differed greatly between DNP-conjugated and unconjugated CpG-ODNs. While minimal tumor accumulation was observed with [3H]-CpG-ODN, DNP-[3H]-CpG-ODN and DNP2-[3H]-CpG-ODN displayed accumulation values above 30% ID/g (Figure 3). Perhaps more importantly, these high tumor concentrations were achieved within 30 minutes after intravenous administration and did not significantly decrease over time, suggesting that tumor accumulation is a sustained process in dynamic equilibrium with the concentration in the circulation (shown in Figure 1). This is a surprising observation since other macromolecular drug carriers with relatively long circulation half-lives that rely on the EPR effect to passively accumulate in tumors, i.e. PEG-grafted liposomes, polymeric drug carriers, attain maximal levels of 15–20% ID/g tumor approximately 24 hours after dosing and display a steady decrease in concentration after this timepoint [17, 39–40]. Thus, it is unlikely that passive extravasation via the EPR effect completely accounts for the high and sustained level of accumulation observed in the present studies. It is probable, however, that monomeric ICs are actively binding to Fcγ receptors (FcγR) expressed on the surface of tumor-associated APCs and directly delivering dinitrophenylated CpG-ODN to these cells via FcγR-mediated endocytosis. This could account for the observed results, since actively targeted monoclonal antibody-drug conjugates attain tumor levels in excess of 50% ID/g [41]. Fundamental mechanistic studies addressing this hypothesis and the potential of using this approach for targeted drug delivery to tumoral APCs are currently under way.
The apparent pharmacokinetic advantage of DNP-CpG-ODN over its bivalent counterpart, DNP2-CpG-ODN, prompted us to select the former molecule as our lead candidate for tumor growth inhibition studies. Accordingly, pharmacodynamic studies tested the potential of DNP-CpG-ODN to inhibit tumor growth in DNP-immunized, tumor-bearing mice. This was quantitatively measured as a function of tumor volume and qualitatively assessed by macroscopic and histological examination of resected solid tumors. As expected, CpG-ODN, which exhibited a short systemic half-life, fast clearance, and minimal tumor accumulation during pharmacokinetic and biodistribution studies, lacked any antitumor activity. In contrast, DNP-CpG-ODN, which extensively accumulated in solid tumors due to its long circulation half-life and large molecular weight when complexed to anti-DNP IgG, displayed up to 60% tumor growth inhibition relative to PBS control (Figure 4B). Macroscopic and histological examination of solid tumors revealed the extensive tissue necrosis elicited by intravenous DNP-CpG-ODN relative to unconjugated CpG-ODN (Figure 5). Although the observed cellular staining patterns are consistent with previous histological observations on CpG-ODN treated tumors [36], the location of viable and necrotic cells within the tumor is puzzling. Upon DNP-CpG-ODN treatment, tumor necrosis was expected to originate at the tumor periphery and progress inwardly toward the tumor core, perhaps mirroring immune cell infiltration. Since the opposite was observed, it remains to be investigated whether the observed necrosis could have been caused by treatment-induced tumor hypoxia, a condition that is predominantly observed in the tumor core.
The pharmacokinetic and pharmacodynamic investigations presented herein have shown that in DNP-immunized, tumor-bearing mice, endogenous anti-DNP IgG can prolong the systemic residence, restrict the systemic clearance, and promote the accumulation of intravenously administered dinitrophenylated CpG-ODN in solid tumors. Such accumulation creates a high local concentration of CpG-ODN at the target tissue, i.e. the solid tumor, which ultimately leads to tumor necrosis and tumor growth inhibition. The fact that this was achieved by intravenous delivery and not via direct tumoral injections is a novel accomplishment that may facilitate the clinical development of CpG-ODN for the treatment of various oncological malignancies. Intravenous administration not only provides a less invasive and more accessible route of administration but it also, as a result of the sustained tumoral accumulation of this therapeutic ODN in immunized mice, may lessen the dosing frequency and total dose relative to current subcutaneous therapy.
Acknowledgments
The authors acknowledge Dev Chatterji and Jin Lee for their technical expertise during pharmacokinetic investigations. Professors Leaf Huang, Gary Pollack, Dhiren Thakker, Roland Tisch, and Fan Yuan are also gratefully acknowledged for their helpful guidance. Financial support was provided by the NIH (F31 CA099929 and P01 GM059299).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 2.Krieg AM. Antitumor applications of stimulating toll-like receptor 9 with CpG oligodeoxynucleotides. Curr Oncol Rep. 2004;6:88–95. doi: 10.1007/s11912-004-0019-0. [DOI] [PubMed] [Google Scholar]
- 3.Stacey KJ, Young GR, Clark F, Sester DP, Roberts TL, Naik S, et al. The molecular basis for the lack of immunostimulatory activity of vertebrate DNA. J Immunol. 2003;170:3614–3620. doi: 10.4049/jimmunol.170.7.3614. [DOI] [PubMed] [Google Scholar]
- 4.Vollmer J, Weeratna R, Payette P, Jurk M, Schetter C, Laucht M, et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur J Immunol. 2004;34:251–262. doi: 10.1002/eji.200324032. [DOI] [PubMed] [Google Scholar]
- 5.Heckelsmiller K, Rall K, Beck S, Schlamp A, Seiderer J, Jahrsdorfer B, et al. Peritumoral CpG DNA elicits a coordinated response of CD8 T cells and innate effectors to cure established tumors in a murine colon carcinoma model. J Immunol. 2002;169:3892–3899. doi: 10.4049/jimmunol.169.7.3892. [DOI] [PubMed] [Google Scholar]
- 6.Kawarada Y, Ganss R, Garbi N, Sacher T, Arnold B, Hammerling GJ. NK- and CD8+ T cell-mediated eradication of established tumors by peritumoral injection of CpG-containing oligodeoxynucleotides. J Immunol. 2001;167:5247–5253. doi: 10.4049/jimmunol.167.9.5247. [DOI] [PubMed] [Google Scholar]
- 7.Whitmore MM, Li S, Falo L, Jr, Huang L. Systemic administration of LPD prepared with CpG oligonucleotides inhibits the growth of established pulmonary metastases by stimulating innate and acquired antitumor immune responses. Cancer Immunol Immunother. 2001;50:503–514. doi: 10.1007/s002620100227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crooke ST, Graham MJ, Zuckerman JE, Brooks D, Conklin BS, Cummins LL, et al. Pharmacokinetic properties of several novel oligonucleotide analogs in mice. J Pharmacol Exp Ther. 1996;277:923–937. [PubMed] [Google Scholar]
- 9.Mutwiri GK, Nichani AK, Babiuk S, Babiuk LA. Strategies for enhancing the immunostimulatory effects of CpG oligodeoxynucleotides. J Control Rel. 2005;97:1–17. doi: 10.1016/j.jconrel.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 10.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent Smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 11.Noguchi Y, Wu J, Duncan R, Strohalm J, Ulbrich K, Akaike T, et al. Early phase tumor accumulation of macromolecules: a great difference in clearance rate between tumor and normal tissue. Jpn J Cancer Res. 1998;89:307–314. doi: 10.1111/j.1349-7006.1998.tb00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Rel. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 13.Duncan R. Polymer conjugates for tumour targeting and intracytoplasmic delivery. The EPR effect as a common gateway? Pharm Sci Technol Today. 1999;2:441–449. doi: 10.1016/s1461-5347(99)00211-4. [DOI] [PubMed] [Google Scholar]
- 14.McDonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res. 2002;62:5381–5385. [PubMed] [Google Scholar]
- 15.Padera TP, Kadambi A, di Tomaso E, Carreira CM, Brown EB, Boucher Y, et al. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science. 2002;296:1883–1886. doi: 10.1126/science.1071420. [DOI] [PubMed] [Google Scholar]
- 16.Nakahara T, Norberg SM, Shalinsky DR, Hu-Lowe DD, McDonald DM. Effect of inhibition of vascular endothelial growth factor signaling on distribution of extravasated antibodies in tumors. Cancer Res. 2006;66:1434–45. doi: 10.1158/0008-5472.CAN-05-0923. [DOI] [PubMed] [Google Scholar]
- 17.Takakura Y, Fujita T, Hashida M, Sezaki H. Disposition characteristics of macromolecules in tumor-bearing mice. Pharm Res. 1990;7:339–346. doi: 10.1023/a:1015807119753. [DOI] [PubMed] [Google Scholar]
- 18.Skogh T, Stendahl O, Sundqvist T, Edebo L. Physiochemical properties and blood clearance of human serum albumin conjugated to different extents with dinitrophenyl groups. Int Arch Allergy Appl Immun. 1983;70:238–241. doi: 10.1159/000233330. [DOI] [PubMed] [Google Scholar]
- 19.Rehlaender BN, Cho MJ. Anti-drug antibodies as drug carriers I. For small molecules. Pharm Res. 2001;18:745–752. doi: 10.1023/a:1011072009408. [DOI] [PubMed] [Google Scholar]
- 20.Rehlaender BN, Cho MJ. Antibodies as drug carriers. II. For proteins. Pharm Res. 2001;18:753–760. doi: 10.1023/a:1011024126247. [DOI] [PubMed] [Google Scholar]
- 21.O’Hear CE, Foote J. Antibody buffering of a ligand in vivo. Proc Natl Acad Sci U S A. 2005;102:40–44. doi: 10.1073/pnas.0405797102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palma E, Klapper DG, Cho MJ. Antibodies as drug carriers III: design of oligonucleotides with enhanced binding affinity for immunoglobulin G. Pharm Res. 2005;22:122–127. doi: 10.1007/s11095-004-9017-z. [DOI] [PubMed] [Google Scholar]
- 23.Kandimalla ER, Bhagat L, Yu D, Cong Y, Tang J, Agrawal S. Conjugation of ligands at the 5′-end of CpG-DNA affects immunostimulatory activity. Bioconjug Chem. 2002;13:966–974. doi: 10.1021/bc0200374. [DOI] [PubMed] [Google Scholar]
- 24.Graham MJ, Freier SM, Crooke RM, Ecker DJ, Maslova RN, Lesnik EA. Tritium labeling of antisense oligonucleotides by exchange with tritiated water. Nucleic Acids Res. 1993;21:3737–3743. doi: 10.1093/nar/21.16.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fogler WE, Wade R, Brundish DE, Fidler IJ. Distribution and fate of free and liposome-encapsulated [3H]nor-muramyl dipeptide and [3H]muramyl tripeptide phosphatidylethanolamine in mice. J Immunol. 1985;135:1372–1377. [PubMed] [Google Scholar]
- 26.Heikenwalder M, Polymenidou M, Junt T, Sigurdson C, Wagner H, Akira S, et al. Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat Med. 2004;10:187–192. doi: 10.1038/nm987. [DOI] [PubMed] [Google Scholar]
- 27.Sparwasser T, Hultner L, Koch ES, Luz A, Lipford GB, Wagner H. Immunostimulatory CpG-oligodeoxynucleotides cause extramedullary murine hemopoiesis. J Immunol. 1999;162:2368–2374. [PubMed] [Google Scholar]
- 28.Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the b2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci U S A. 1996;93:5512–5516. doi: 10.1073/pnas.93.11.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lencer WI, Blumberg RS. A passionate kiss, then run: exocytosis and recycling of IgG by FcRn. Trends Cell Biol. 2005;15:5–9. doi: 10.1016/j.tcb.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 30.Caliceti P, Veronese FM. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates. Adv Drug Deliv Rev. 2003;55:1261–1277. doi: 10.1016/s0169-409x(03)00108-x. [DOI] [PubMed] [Google Scholar]
- 31.Schumaker VN, Green G, Wilder RL. A theory of bivalent antibody-bivalent hapten interactions. Immunochemistry. 1973;10:521–528. doi: 10.1016/0019-2791(73)90224-3. [DOI] [PubMed] [Google Scholar]
- 32.Wilder RL, Green G, Schumaker VN. Bivalent hapten-antibody interactions – I. A comparison of water soluble and water insoluble bivalent hapten. Immunochemistry. 1975;12:39–47. doi: 10.1016/0019-2791(75)90047-6. [DOI] [PubMed] [Google Scholar]
- 33.Braeckmann R. In: Peptide and Protein Drug Analysis. Reid RE, editor. New York: Marcel Dekker, Inc; 2000. pp. 633–669. [Google Scholar]
- 34.Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54–S62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- 35.Haymann JP, Levraud JP, Bouet S, Kappes V, Hagege J, Nguyen G, et al. Characterization and localization of the neonatal Fc receptor in adult human kidney. J Am Soc Nephrol. 2000;11:632–639. doi: 10.1681/ASN.V114632. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi N, Suzuki Y, Tsuge T, Okumura K, Ra C, Tomino Y. FcRn-mediated transcytosis of immunoglobulin G in human renal proximal tubular epithelial cells. Am J Physiol Renal Physiol. 2002;282:F358–365. doi: 10.1152/ajprenal.0164.2001. [DOI] [PubMed] [Google Scholar]
- 37.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 38.Krall G, Mebius R. New insights into the cell biology of the marginal zone of the spleen. Int Rev Cytol. 2006;250:175–215. doi: 10.1016/S0074-7696(06)50005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seymour LW, Miyamoto Y, Maeda H, Brereton M, Strohalm J, Ulbrich K, et al. Influence of molecular weight on passive tumour accumulation of a soluble macromolecular drug carrier. Eur J Cancer. 1995;31A:766–770. doi: 10.1016/0959-8049(94)00514-6. [DOI] [PubMed] [Google Scholar]
- 40.Ishida O, Maruyama K, Tanahashi H, Iwatsuru M, Sasaki K, Eriguchi M, et al. Liposomes bearing polyethyleneglycol-coupled transferrin with intracellular targeting property to the solid tumors in vivo. Pharm Res. 2001;18:1042–1048. doi: 10.1023/a:1010960900254. [DOI] [PubMed] [Google Scholar]
- 41.Mandler R, Kobayashi H, Hinson ER, Brechbiel MW, Waldmann TA. Herceptin-Geldanamycin immunoconjugates: pharmacokinetics, biodistribution, and enhanced antitumor activity. Cancer Res. 2004;64:1460–1467. doi: 10.1158/0008-5472.can-03-2485. [DOI] [PubMed] [Google Scholar]