

Abstract
In addition to playing a central role in energy homeostasis, leptin is also an important player in the inflammatory response. Systemic inflammation is accompanied by fever (less severe cases) or hypothermia (more severe cases). In leptin-irresponsive mutants, the hypothermia of systemic inflammation is exaggerated, presumably due to the enhanced production and cryogenic action of tumor necrosis factor (TNF)-α. Mechanisms that exaggerate hypothermia can also attenuate fever, particularly in a cool environment. Another common manifestation of systemic inflammation is behavioral depression. Along with the production of interleukin (IL)-1β, this manifestation is exaggerated in leptin-irresponsive mutants. The enhanced production of TNF-α and IL-1β may be due, at least in part, to insufficient activation of the anti-inflammatory hypothalamo-pituitary-adrenal axis by immune stimuli in the absence of leptin signaling. In experimental animals and humans that are responsive to leptin, suppression of leptin production under conditions of negative energy balance (e.g., fasting) can exaggerate both hypothermia and behavioral depression. Since these manifestations aid energy conservation, exaggeration of these manifestations under conditions of negative energy balance is likely to be beneficial.
Keywords: adipose tissue, obesity, leptin receptor, lipopolysaccharide, LPS, immune-to-brain signaling, fever, hypothermia, sickness behavior, behavioral depression, anorexia
1. Introduction
Inflammation is an important host-defense response [1]. However, when the inflammatory response becomes overwhelming, it disrupts vital homeostatic processes and ultimately results in multiple organ failure and death [2]. It is not surprising, therefore, that severe forms of inflammation are major medical problems. A common complication in hospitalized patients is sepsis (systemic inflammation due to infection). Mortality rate is extremely high in septic patients: it ranges from 30% in those patients that do not develop shock to 70% in those patients that do [3-6]. Sepsis becomes an even more serious threat in view of the fact that its incidence is increasing fast, i.e., at an annual rate of 9% in the United States [7]. The increasing incidence of sepsis seems to be associated with, among other factors, the escalating incidence of metabolic disorders, particularly obesity [8-10]. Clearly, understanding how the mechanisms involved in obesity (and energy homeostasis in general) affect systemic inflammation is important.
The adipocyte-derived hormone, leptin, plays a central role in energy homeostasis, and resistance to its actions is associated with obesity [11-13]. Since it also modulates inflammation [14,15], leptin may integrate energy balance and systemic inflammation. In the present article, we examine this issue. A succinct review of the roles played by leptin in energy homeostasis is followed by an in-depth analysis of the roles of leptin in common physiological manifestations of systemic inflammation. These manifestations include fever (or hypothermia), behavioral depression, and anorexia; they jointly form the so-called sickness syndrome.
2. Leptin and energy homeostasis
2.1. Leptin and its receptors
It has been known for many decades that mice carrying the homozygous obese mutation (ob/ob mice) develop a syndrome that resembles human morbid obesity. However, it was not until the last decade that the obese mutation was identified, and the gene that carries such a mutation was isolated by positional cloning [16]. In wild-type animals, this gene encodes a 167-amino-acid, 16-kDa protein that is produced and secreted primarily by adipocytes [16-19]. Because its absence accounts for the obese phenotype of ob/ob mice, this protein was named leptin (from the Greek, leptós, which means thin); see Ref. 20. Leptin is highly conserved among species [16,17], and its structure resembles that of cytokines, especially interleukin (IL)-6 [21,22].
Not long following the discovery of leptin, the leptin receptor (LR) gene was identified [23-25]. The mRNA encoded by the LR gene is subject to extensive splicing, and each splicing variant is responsible for the translation of a distinct LR isoform. Six LR isoforms have been identified to date (Fig. 1), all of which have identical or nearly identical extracellular domains but differ in their transmembrane and intracellular domains (for reviews, see Refs. 11,12,26). The LRe isoform lacks the transmembrane and intracellular domains altogether and is found dissolved in the extracellular fluid. LRe is thought to sequester circulating leptin, thus limiting its action [27]. The LRa, c, d, and f isoforms have a transmembrane domain, but these membrane-bound isoforms lack a full intracellular domain and display no signal transduction capabilities. These isoforms, especially LRa, appear to be involved in the internalization, transport, and degradation of leptin [28,29]. LRb is the only isoform with complete transmembrane and intracellular domains. It is a class-I cytokine receptor and, as such, signals primarily via the JAK2-STAT3 pathway [30,31]. LRb has been implicated in most biological effects of leptin [32-34]. Importantly, several widely used rodent models of obesity — viz., db/db mice [24,35,36], Zucker fa/fa rats [36-38], and Kotetsky f/f rats [39,40] — carry mutations in the LR gene that result in either absence or dysfunctionality of LRb (Table 1).
Fig 1.

Structures of LR isoforms. All isoforms are products of splicing variants of a single transcript. Each isoform has a conserved region, and may or may not have a variable region. The conserved region forms the extracellular and transmembrane domains as well as a small portion of the intracellular domain; the aminoacid sequence of this region is always constant, although it may be truncated. The variable region forms a large portion of the intracellular domain; the aminoacid sequence in this region is highly sensitive to splicing. The first or last aminoacid of each region and domain are listed at the first (left to right) diagram that contains this region or domain; amino acid numbering is based on the published sequences of mouse LRa-e [23,24] and rat LRf[25]. Box motifs for JAK-STAT phosphorylation are marked. Only when two JAK-STAT box motifs are present (as in LRb) does LR display full signal transduction capability.
Table 1.
LR isoforms in three mutant models of obesity
| Mutant | DNA level | mRNA level | Protein level | |
|---|---|---|---|---|
| LRb | LRa, LRc-f | |||
| db/db mouse | G → T at the region encoding the intracellular domain |
Abnormal splicing | Absent | Functional |
| Zucker fa/fa rat | A → C at the region encoding the extracellular domain |
Glu → Pro codon conversion |
Dysfunctional | Functional |
| Koletsky f/f rat | T → A at the region encoding the extracellular domain |
Early stop codon | Absent | Absent |
LRb is expressed in the brain, especially in areas that are involved in the regulation of appetite and metabolism, such as the arcuate, ventromedial, and dorsomedial hypothalamic nuclei (for a review, see Ref. 41). Circulating leptin crosses the blood-brain barrier and gains access to these areas via a specific, saturable transport mechanism that is believed to involve LRa [42]. Alternatively, leptin can enter the brain via the median eminence. This structure is characterized by a leaky blood-brain barrier, and it is strategically located at the boundaries of the arcuate, ventromedial, and dorsomedial hypothalamic nuclei [43].
2.2. Role of leptin in energy homeostasis
The concentration of leptin in the blood plasma is maintained between 1 and 20 ng/ml in lean animal and human subjects, but it may be as high as 100 ng/ml in obese subjects [44,45]. The circulating level of leptin reflects its production by white adipocytes, and it exhibits both long-term (days) and short-term (hours) changes. Mechanisms that drive changes in fat storage regulate long-term changes in leptin production, as evident from the strong positive correlation between the total size of the body fat stores and the plasma concentration of leptin [44-46]. Not only are more adipocytes expected to produce more leptin, but also adipocytes containing more fat synthesize more leptin [47]. Short-term changes in leptin levels occur independently of body fat mass gain or loss. For example, leptin production is decreased a few hours after initiation of fasting, and refeeding restores its production to prefasting levels [48-52]. The mechanisms of these short-term changes appear to involve insulin, as evidenced by the positive correlation between the plasma levels of leptin and insulin [50,52,53] and by the ability of insulin treatment to increase leptin production [48,54,55].
In lean animal [56-58] and human [59-61] subjects, reduction in plasma leptin (usually to < 4 ng/ml) during fasting or other conditions of negative energy balance (e.g., calorie restriction) is associated with suppression of metabolic rate and stimulation of appetite. These adaptive responses are abolished by supplementation with recombinant leptin at doses that typically elevate the plasma leptin of fasted or calorie restricted subjects to a level that is similar to that found in well-fed subjects, i.e., 4-20 ng/ml [56-58,60,62]. The same doses of leptin, however, produce modest or no effect when given to well-fed subjects [20,56,63-65]. Only when injected at doses known [66] to produce supraphysiological rises in its plasma level (to > 100 ng/ml) does leptin cause marked hypermetabolic and anorexic responses in fed subjects [67,68]. These observations lead to two conclusions: (i) that a fall in circulating leptin during conditions of negative energy balance works as an anti-starvation signal, a signal that suppresses energy expenditure and stimulates appetite; and (ii) that a rise in circulating leptin to a level above that found in lean, fed individuals produces little or no physiological response; see Fig. 2.
Fig. 2.

A simplified schematic of the relationships among plasma leptin, metabolism, and hunger in lean and obese subjects. Please note that quantitative aspects of the relationships shown should be looked upon with great skepticism.
With a few exceptions [69-73], obese patients have plasma levels of leptin that are above those found in lean, fed individuals [44,45]. These patients are even more resistant than lean individuals to the hypermetabolic and anorexic effects of recombinant leptin [74-76]. Likewise, rodents with diet-induced obesity exhibit high plasma leptin and resistance to the hypermetabolic and anorexic effects of leptin [77,78]; see Fig. 2. Resistance to these weight-reducing effects of leptin can contribute to the progression of obesity. Irresponsiveness to leptin appears to involve decreased leptin transport across the blood-brain barrier and impairment of LRb signaling (for a review, see Ref. 13). It may also be due to mutations in the LR gene, but these mutations have been found to be extremely rare in humans [79].
3. Leptin production in inflammation
Parenteral administration of lipopolysaccharide (LPS), a constituent of the outer envelope of Gram-negative bacteria, is commonly used to induce systemic inflammation experimentally. In 1996, Grunfeld and colleagues [80] reported that hamsters respond to intraperitoneal LPS with increases in both the expression of the leptin gene in white fat and the circulating level of leptin. Since then, the effect of LPS on leptin production has been well characterized in mice and rats [81-90]. It is now known that: (i) a rise in plasma leptin may occur as early as 1 h post-LPS; (ii) the rise may last for several hours; (iii) the magnitude of the rise is directly proportional to the LPS dose until a ceiling level of leptin is reached; (iv) as in non-inflammatory states, the ceiling level of leptin is ∼20 ng/ml in lean animals, but it may be higher in obese animals; and (v) the leptin response to LPS occurs in both fasted and fed animals, but the peak leptin level achieved in fasted animals is lower, possibly due to their already reduced basal production of leptin. Inflammatory stimuli other than LPS, specifically turpentine [84] and carrageenan [91], also stimulate leptin production. It is not surprising, therefore, that plasma leptin was found to be high in mice and humans with bacterial sepsis [92,93] as well as in those patients who developed post-surgical inflammation [94].
The fact that many effects of LPS and other microbial products are mediated by pro-inflammatory cytokines [95,96] propelled several studies on the roles of tumor necrosis factor (TNF)-α and IL-1β in the leptin response to inflammatory stimuli. It has been shown that the plasma leptin response of mice to LPS is preceded by an increased production of TNF-α [82]; that systemic administration of TNF-α to mice or hamsters increases the expression of the leptin gene in white fat as well as the circulating level of leptin [80-83]; and that administration of IL-1β to mice, hamsters, or humans increases leptin production [80,81,97]. It has also been shown that the plasma leptin response to LPS or bacterial peritonitis does not occur in mice treated with the TNF-binding protein [92], in mice that lack the IL-1β gene [84], or in rats administered with the IL-1 soluble receptor [98]. These lines of evidence indicate that TNF-α and IL-1β mediate the inflammatory increase in leptin production.
To determine whether LPS and cytokines increase leptin production by acting on adipocytes directly or indirectly, Finck and colleagues [83] treated cultured mouse adipocytes with LPS or TNF-α. They found that only TNF-α is capable of inducing leptin production in their in-vitro model, in spite of the fact that both LPS and TNF-α increase leptin production when administered in vivo. This finding is consistent with the notion that LPS-induced leptin production involves recognition of LPS by cells other than adipocytes (presumably immunocytes), and that TNF-α produced by these cells acts directly on adipocytes to stimulate leptin production. Immune activation of leptin production may also involve neuroendocrine mechanisms [89,99], but these mechanisms remain poorly explored.
Based on the large body of evidence showing that inflammatory insult stimulates leptin production, an involvement of leptin in at least some components of the inflammatory response can be anticipated. Below, we analyze whether and how leptin participates in some of the most common physiological manifestations of systemic inflammation.
4. Leptin and thermoregulatory manifestations of systemic inflammation
4.1. Fever and hypothermia: overview
So strongly is systemic inflammation associated with changes in deep body temperature (Tb) that an altered Tb is included in all definitions of systemic inflammation and sepsis [100-102]. Whereas the majority of septic patients (∼90%) are febrile, a number of them (∼10%) develop hypothermia [3,6]. Importantly, hypothermia occurs in the most severe cases of sepsis, and it is often associated with circulatory shock [3,6]. As humans, experimental animals respond to intravenous administration of LPS with fever or hypothermia; the direction and pattern of the Tb response depends on the LPS dose and ambient temperature (for a review, see Ref. 103). In a thermally neutral environment, fever is the prevailing response; fever is monophasic when the dose of LPS is low (just supra-threshold), but it turns into polyphasic as the dose increases; see Fig. 3 and Refs. 104-107. Three febrile phases have been identified in the rat [107-111] and mouse [106,112,113]. Interestingly, when an LPS dose that normally causes polyphasic fever at a neutral ambient temperature is given to a rat in a cool environment, it causes hypothermia (which may or may not be followed by fever); see Fig. 3 and Refs. 105,110,111,114. More recently, we studied [115,116] the thermoregulatory responses to two doses of LPS in rats that were allowed to select their preferred ambient temperature in a thermally heterogeneous environment, a more natural condition. Whereas a lower dose caused warmth-seeking behavior and polyphasic fever, a higher dose caused cold-seeking behavior and hypothermia. At a later stage of the response to the higher dose, when most of the injected LPS has been cleared from the circulation, warmth-seeking behavior and fever were observed. These data indicate that fever is a natural response to a weaker inflammatory insult, whereas hypothermia is a natural response to a stronger inflammatory insult.
Fig. 3.
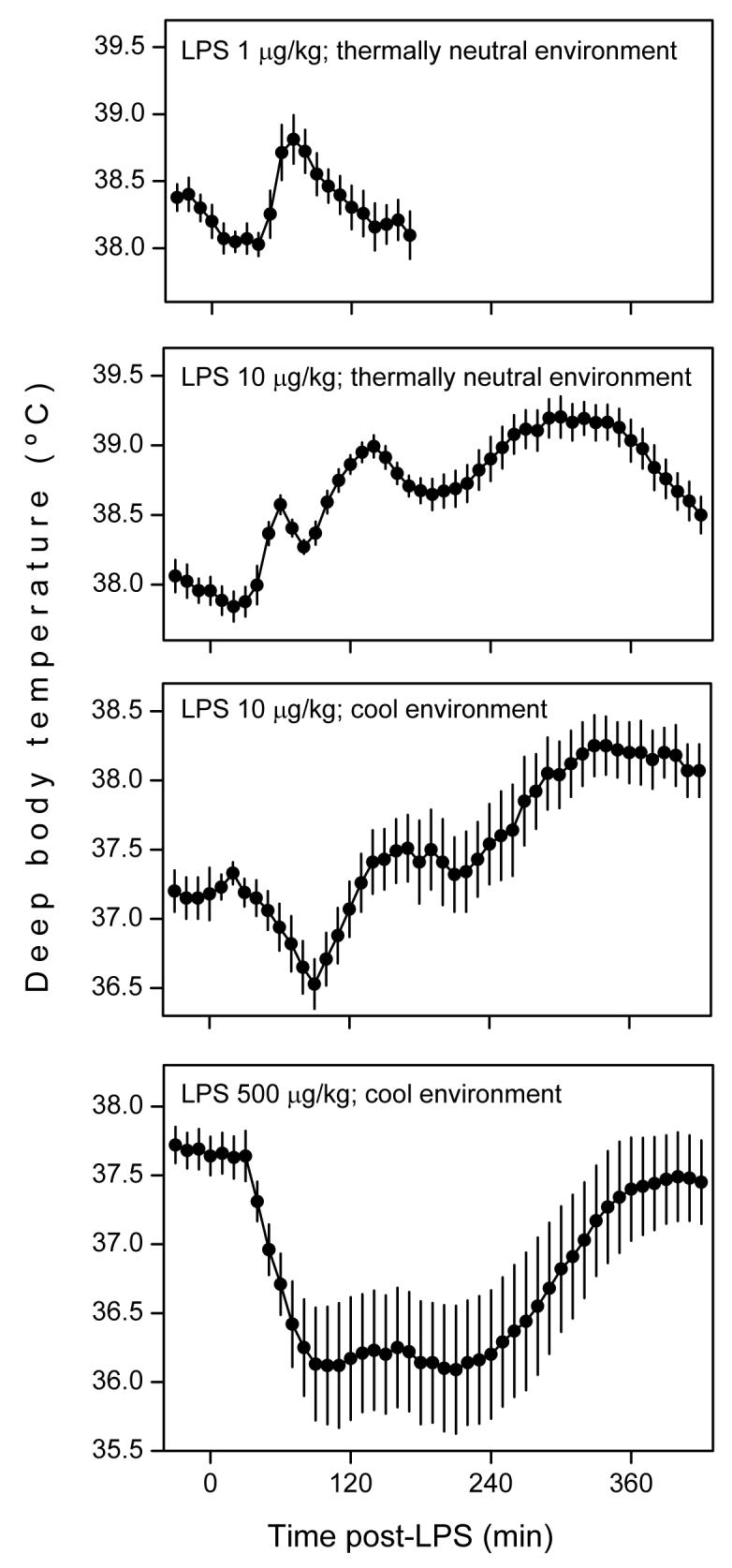
Thermoregulatory responses of the rat to intravenous injection of LPS (doses indicated) in thermally neutral and cool environments. From top to bottom: monophasic fever, polyphasic fever, hypothermia followed by fever, and long-lasting hypothermia. Note that fever prevails when the LPS dose is lower or the environment is warmer, whereas hypothermia prevails when the LPS dose is higher or the environment is cooler. Adapted from Refs. 105,107,108,110 by permissions from the American Physiological Society and the Federation of American Societies for Experimental Biology.
4.2. Leptin and thermoregulation in the absence of inflammation
Before discussing the function of leptin in the thermoregulatory manifestations of systemic inflammation, it is important to consider a few points regarding the role of leptin in thermoregulation under non-inflammatory conditions. Activation of metabolic heat production (nonshivering thermogenesis) is vital for homeothermic organisms (especially small animals) to maintain their Tb at a normothermic level in the cold [117,118]. Therefore, substances that affect metabolic rate invariably have an impact on thermoregulation [119]. In small rodents, leptin raises energy expenditure by increasing the sympathetic outflow to brown adipose tissue [120], which is the primary source of nonshivering thermogenesis in these animals [118]. Elimination of leptin signaling impairs chronic activation of thermogenesis in a cool environment [33,63,64,121,122]. As a result, ob/ob mice [63,64,123], db/db mice [33,65,121,123], Koletsky f/f rats [110], and Zucker fa/fa rats [122,124,125] are hypothermic in a regular laboratory environment, which is often cool for small rodents [126]. The magnitude of the hypothermia varies from a few tenths of a degree Celsius to several degrees; it is inversely proportional to the ambient temperature (as predicted by the Fourier's law for conductive heat transfer). In a thermally neutral environment, Tb is regulated not by activation/inactivation of thermogenesis, but by constriction/dilation of skin vessels [126]. In such an environment, elimination of leptin signaling does not result in hypothermia [110,125].
It is important to note that, despite its critical role in chronic activation of thermogenesis during long-term cold exposure, leptin is apparently uninvolved in acute thermogenic responses. This is evident from the fact that young adult (7-10 week-old) Zucker fa/fa rats are capable of mounting vivid thermogenic responses to short-term cold exposure [127-129], in spite of their irresponsiveness to leptin. Only at an older age (> 10 week-old) do Zucker fa/fa rats display attenuation of acute thermogenic responses, but such an attenuation coincides with involution of the brown adipose tissue [130]. Once this secondary trait is manifested, the thermogenic responses of Zucker fa/fa rats are compromised altogether, regardless of whether or not the responses involve leptin.
4.3. Role of leptin in the fever and hypothermia of systemic inflammation
Activation of thermogenesis is important for fever production, especially in a cool environment [131,132]. It is then natural to ask: can the thermogenic action of leptin contribute to fever production? The answer to this question is probably negative for two reasons. First, whereas fever is brought about by acute activation of thermoeffectors [103], leptin appears to be unimportant for acute activation of thermogenesis [127-129]. Second, the febrile response to LPS is least manifest [133-136] under the condition (negative energy balance) that is most propitious for activation of thermogenesis by a rise in circulating leptin [56-58,60,62]. Most likely, a participation of leptin in fever or hypothermia would be related to the mechanisms of immune-to-brain signaling.
Rosenthal and colleagues [137] were the first to demonstrate that the febrile response to LPS is largely attenuated in Zucker fa/fa rats at room temperature. The attenuation of fever may result from alteration of a leptin-related mechanism of immune-to-brain signaling, but it may also result from factors unrelated to immune-to-brain signaling. As discussed in the previous section, Zucker fa/fa rats of more than 10 weeks of age have an atrophic brown adipose tissue [130]. This trait might result in attenuation of fever at room temperature, because room temperature is often subneutral for rats [126], and because rats develop fever in a subthermoneutral environment by activating thermogenesis [131]. It should be noted, however, that exhaustion of thermogenic reserves is not required for leptin-irresponsive animals to show attenuated febrile responses. This is evident from the fact that young (9 week-old) Koletsky f/f rats do not have exhausted thermogenic reserves, yet their overall febrile response to LPS is attenuated in a cool environment [110]. Excessive fat accumulation and hyperlipidemia are traits that are common to Zucker fa/fa and Koletsky f/f rats, and that might account for their attenuated febrile responses. Each of these traits is independently found in leptin-responsive rat strains: fat accumulation is a characteristic of Otsuka Long-Evans Tokushima fatty rats, and hyperlipidemia is a characteristic of Nagase analbuminemic rats. Since the Tb responses of these rat strains to LPS are not attenuated [138-140], fat accumulation and hyperlipidemia cannot explain the low-grade febrile responses of Zucker fa/fa and Koletsky f/f rats. Therefore, altered immune-to-brain signaling in Zucker fa/fa and Koletsky f/f rats appears to be the most plausible explanation for their attenuated fever. Altered immune-to-brain signaling can also explain the inability of wild-type rats treated with an anti-leptin antibody to mount a full-blown febrile response to LPS [141].
The mechanisms of immune-to-brain signaling can be febrigenic (increase Tb) or cryogenic (decrease Tb). These mechanisms compete with each other, and the balance between the two determines the resulting Tb response [95,142]. Febrigenic mechanisms can inhibit heat loss and activate thermogenesis [143]. Therefore, these mechanisms can drive Tb responses in both cool and warm environments; in the cold, heat loss (which is already at its minimum) cannot be inhibited, but thermogenesis (which is already activated) can be further activated; in the warmth, heat loss (which is already activated) can be inhibited, and thermogenesis (which is at its minimum) can be activated. Cryogenic mechanisms drive strong inhibition of thermogenesis, but have little effect on heat loss [143]. Therefore, they can drive Tb responses in a cool environment (in which thermogenesis is already activated and can be inhibited), but not in a warm environment (in which thermogenesis is at its minimum and cannot be further inhibited). An example of a substance that plays key roles in immune-to-brain signaling is TNF-α. This pro-inflammatory cytokine can be either febrigenic or cryogenic: it mediates the second and later phases of the polyphasic fever to lower LPS doses [144,145]; it also mediates the hypothermic response (and counteracts the febrile response) to higher LPS doses [146-149]. Such a dual role of TNF-α is consistent with the fact that the thermoregulatory response to TNF-α administration shifts from fever to hypothermia as the dose administered increases [150].
To determine whether leptin affects febrigenic or cryogenic mechanisms, we have studied the Tb responses of Zucker fa/fa or Koletsky f/f rats injected with lower or higher doses of LPS in a thermally neutral or cool environment. We have found that Zucker fa/fa rats [125] and Koletsky f/f rats [110] respond to lower doses of LPS with normal polyphasic fever in a thermally neutral environment (in which only those Tb responses that are driven by febrigenic mechanisms are revealed), but with attenuated fever in a cool environment (in which Tb responses driven not only by febrigenic mechanisms, but also by cryogenic mechanisms are revealed). Additionally, we have found that Koletsky f/f rats respond to higher doses of LPS in a cool environment with a dramatically exaggerated hypothermia [110]; see Fig. 4. The exaggerated LPS hypothermia in Koletsky f/f rats is associated with an enhanced rise in the plasma level of TNF-α (Fig. 5). The cryogenic effect of intravenous TNF-α is also markedly prolonged in Koletsky f/f rats. It is concluded from these results that elimination of leptin signaling does not affect febrigenic mechanisms, but it exaggerates cryogenic mechanisms. Among the mechanisms exaggerated in the absence of leptin signaling are the production and the cryogenic action of TNF-α.
Fig. 4.
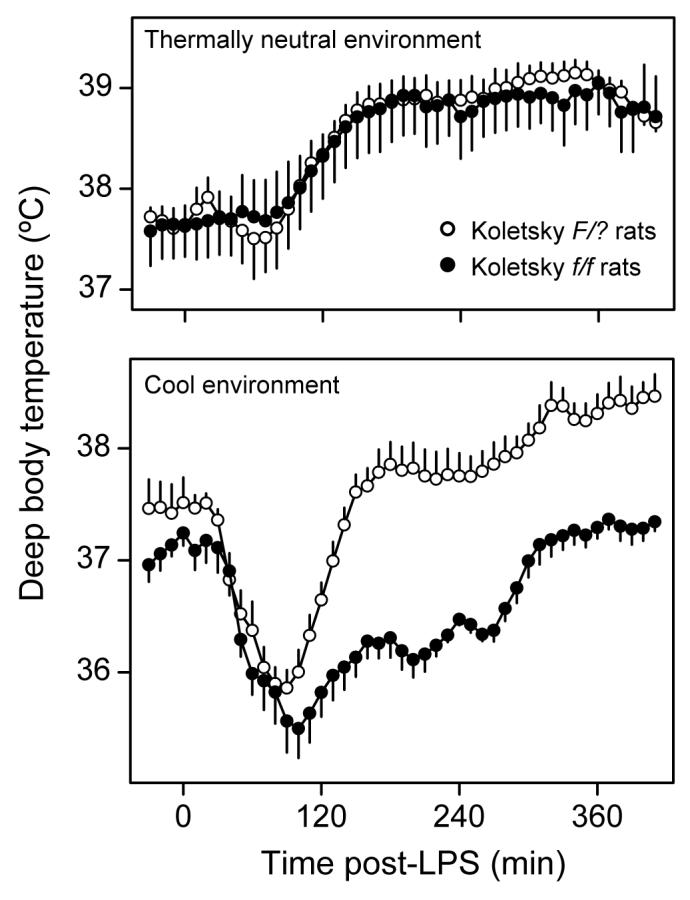
Thermoregulatory responses of Koletsky f/f rats (lack all LR isoforms) and F/? rats (possess all LR isoforms) to intravenous LPS (100 μg/kg) in thermally neutral and cool environments. Reproduced from Ref. 110 by permission from the Federation of American Societies for Experimental Biology.
Fig. 5.
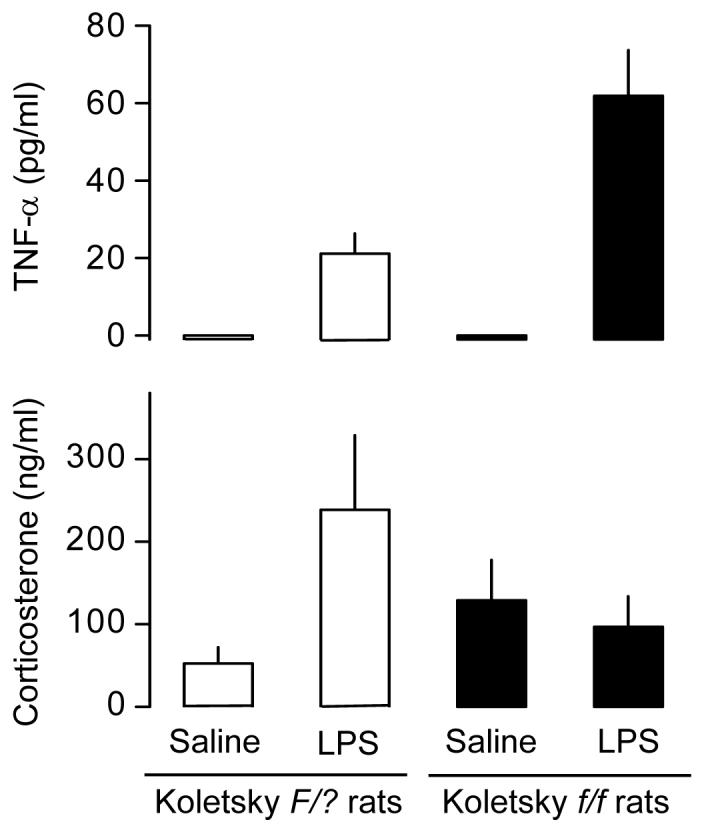
Concentrations of TNF-α and corticosterone in the plasma of Koletsky f/f rats and their wild-type (F/?) counterparts 120 min after intravenous administration of LPS (100 μg/kg) or saline in a cool environment. This time point (120 min) corresponds to the maximal rate of divergence between the hypothermic responses of the f/f and F/? rats shown in Fig. 4. These data suggest that the exaggerated hypothermia of Koletsky f/f rats is associated with an enhanced rise in plasma TNF-α and a lack of activation of the HPA axis. Adapted from Ref. 110 by permission from the Federation of American Societies for Experimental Biology.
Because leptin stimulates TNF-α production by monocytes and macrophages in vitro [151-154], elimination of a direct action of leptin on immune cells is expected to suppress rather than enhance TNF-α production. Other factors must account for the enhanced production of this cytokine in Koletsky f/f rats treated with LPS. A possible link between leptin and the immune response in vivo is the hypothalamo-pituitary-adrenal (HPA) axis. Compared to their leptin-responsive counterparts, undisturbed Koletsky f/f rats [110] and other leptin-irresponsive rodents [85,155] have slightly elevated plasma levels of corticotropin and corticosterone (two major components of the HPA axis). The mechanisms of such an abnormality are not completely understood but may include elimination of leptin signaling in the hypothalamus [156,157] and adrenals [158]. Although the basal activity of their HPA axis is slightly elevated, Koletsky f/f rats are unable to further activate the HPA axis in response to LPS; see Fig. 5 and Ref. 110. Activation of the HPA axis has multiple anti-inflammatory actions [96], and lack of such activation in the Koletsky f/f rats is likely to contribute to their exaggerated production of TNF-α in response to LPS and to their hypersensitivity to the cryogenic action of this cytokine. The revealed irresponsiveness of the HPA axis to LPS in Koletsky f/f rats agrees with the fact that LPS does not activate the HPA axis in ob/ob mice [85], and that corticosterone responses of wild-type mice and rats to LPS and IL-1β are enhanced by leptin administration [87,159,160]. Because leptin can trigger the release of corticotropin-releasing factor by hypothalamic neurons [161,162], and because several substances only stimulate the release of corticotropin-releasing factor in the presence of a functional leptin-LR system [163,164], it is possible that the participation of leptin in the immune activation of the HPA axis is related to regulation of the production and release of corticotropin-releasing factor.
5. Leptin and non-thermoregulatory manifestations of systemic inflammation
5.1. Leptin and behavioral depression
The thermoregulatory manifestations of systemic inflammation are not isolated events. They occur within the sickness syndrome, along with its behavioral manifestations [165,166]. As proposed by Romanovsky and colleagues [104,167,168], the sickness syndrome is a dynamic entity, i.e., its symptoms change drastically as the syndrome progresses. Like the thermoregulatory manifestations, the behavioral manifestations of systemic inflammation are dynamic. At the onset of a systemic inflammatory response (e.g., associated with a viral infection), humans feel uncomfortable and restless. As the disease progresses, this hyperactive state turns into a longer-lasting hypoactive state characterized by general weakness, sleepiness, and inability (or unwillingness) to perform normal daily routines. A similar behavioral pattern exists in experimental animals. Locomotor excitation has been occasionally reported in LPS-treated rats [104] and guinea pigs [169]. This excitation is short-lasting; it occurs during the course of the inflammatory response to low (just supra-threshold) doses of LPS or during the early phase of the response to moderate and high doses; and it coincides with monophasic fever or the first phase of polyphasic fever [104]. Behavioral excitation belongs to the so-called early sickness syndrome, which is aimed at actively fighting infection [104,167,168]. Most frequently, however, experimental animals challenged with microbial products, including LPS, are reported to have reduced locomotor activity and to spend more time sleeping [170-178]. This behavioral depression is long-lasting; it occurs during the later stages of the inflammatory response to moderate and high LPS doses; and it coincides with the second and subsequent phases of polyphasic fever or with hypothermia [104]. Behavioral depression belongs to the so-called late sickness syndrome, which is aimed at energy conservation [104,167,168].
Leptin is involved in the regulation of locomotor behavior. The spontaneous locomotor activity of ob/ob and db/db mice is depressed in several experimental paradigms [33,63,179-182]. As expected, leptin supplementation reverses the locomotor depression of ob/ob mice, but not that of db/db mice [63,179]. O'Connor and colleagues [183] determined how intraperitoneal administration of LPS affects exploratory behavior to introduction of a new cage mate in db/db mice or wild-type mice. In the wild-type mice, LPS caused a dose-dependent depression of exploratory behavior. In the db/db mice, LPS also induced behavioral depression, but this response was augmented in both magnitude and duration when compared to the response of the wild-type mice (Fig. 6). In a separate experiment, O'Connor and colleagues [183] showed that the augmented behavioral depression of db/db mice is associated with enhanced production of IL-1β (Fig. 6), a known mediator of LPS-induced depression [184,185].
Fig. 6.

Exploratory behavior and level of IL-1β in the peritoneal lavage of db/db mice (lack LRb) and db/+ mice (possess LRb) injected intraperitoneally with LPS (5 μg/mouse). Reproduced from Ref. 183; copyright 2005 by the American Association of Immunologists, Inc.
Because exaggeration of the hypothermic and TNF-α responses to LPS in Koletsky f/f rats is associated with inability of these animals to respond to inflammatory stimuli with activation of the HPA axis [110], it is reasonable to conjecture that exaggeration of the behavioral and IL-1β responses to LPS in db/db mice is also related to the HPA axis. Compared to wild-type mice, the circulating level of corticosterone is higher in db/db mice before inflammatory challenge, but it tends to be lower after challenge with a lethal dose of LPS [85], thus suggesting that LPS-induced HPA axis activation in db/db mice is less pronounced. Perhaps, such an activation is even less pronounced when non-lethal doses of LPS are used, like the doses used in the study by O'Connor and colleagues [183]. It should be considered, however, that HPA axis-unrelated factors might also contribute to the exaggeration of the IL-1β response, as suggested by the in-vitro observation that db/db mice-derived macrophages produce more IL-1β than wild-type macrophages in response to LPS [183].
5.2. Leptin and anorexia
Another prominent manifestation of systemic inflammation is anorexia. Loss of appetite and consequent decrease in food intake is commonly observed in humans with infections of different etiologies. Likewise, a reduction in food intake has been reported in rats, mice, sheep, and pigs challenged with microbial products, including LPS (for reviews, see Refs. 165,186,187). In rats, suppression of food intake usually starts 2-4 h after LPS administration [188-191], and it is associated with symptoms of the late sickness syndrome, viz., the second and subsequent phases of polyphasic fever and behavioral depression [188,191]. Importantly, though, LPS-induced suppression of feeding is not a result of fever or behavioral depression, since it can occur even when the febrile or behavioral response is experimentally abolished [188,191,192]. In fact, the inflammatory suppression of feeding is related to a specific aspect of appetite regulation, as evident from the fact that peripheral LPS reduces meal frequency, but does not affect meal size or duration [193]. Like other manifestations of the sickness syndrome, anorexia is a well-defined strategy to cope with infection [165,194,195]. A decreased motivation for food reduces the possibility of predation while an animal is less vigilant due to disease [165]. Another benefit of anorexia may lie in the fact that the plasma levels of trace metals that are essential for bacterial growth drop abruptly upon suppression of food intake [196]. Additional benefits are likely to exist, considering that anorexia is a phylogenetically conserved host-defense response.
Leptin may participate in the anorexia of systemic inflammation in at least two distinct ways. First, leptin may mediate inflammatory anorexia; it can do so by acting directly on the hypothalamic neurons involved in appetite regulation [41]. Second, leptin may counteract inflammatory anorexia; it can do so by promoting activation of the HPA axis [85,110] and, consequently, limiting the production and action of cytokines (e.g., TNF-α, and IL-1β) that mediate inflammatory anorexia. Whether these mechanisms are involved in inflammatory anorexia is unknown. It is unclear why the anorexic effect of LPS was suppressed in db/db mice, whereas it was enhanced in ob/ob mice under the same experimental conditions [197]. It is also unclear why LPS-induced anorexia was unaffected in Zucker fa/fa rats [198], but was attenuated in wild-type rats treated with an anti-leptin antibody [141]. Future studies are clearly necessary to determine what role, if any, leptin plays in the anorexia of systemic inflammation.
6. An integrated view: leptin, energy balance, and systemic inflammation
6.1. When leptin and its receptors function normally
A fall in the circulating level of leptin during conditions of negative energy balance (e.g., calorie restriction and fasting) works as an anti-starvation signal, and this is believed to be the main physiological function of leptin (see section 2.2). Conditions of negative energy balance also reduce the peak level of leptin achieved during systemic inflammation [87]. It is, therefore, tempting to speculate that the roles of leptin in systemic inflammation may be generally related to regulation of energy balance.
At doses that are highly febrigenic in fed animals, LPS produces low-grade fever or hypothermia in fasted animals [133-136]. A shift from fever to hypothermia in fasted animals may be related to their lowered leptin production, as suggested by the fact that fever is often attenuated and hypothermia is exaggerated in leptin-irresponsive mutants (see section 4.3). As in leptin-irresponsive mutants, LPS-induced TNF-α production is enhanced in fasted animals whose plasma leptin is allowed to drop [87]. It remains to the determined, however, whether this enhanced TNF-α production accounts for prevalence of hypothermia over fever in fasted animals. Regardless of the mechanisms involved, a shift from fever to hypothermia is likely to be adaptive under conditions of negative energy balance. Although fever has antimicrobial and immunostimulating effects [199,200], its benefits may be offset by the high energetic cost associated with maintaining a high Tb [167,201,202]. On the other hand, lowering Tb is an energy conservation strategy that is widely used by vertebrates and invertebrates under conditions of imminent or enduring energy deficit [167,203,204].
Like hypothermia, behavioral depression is a manifestation of systemic inflammation that is augmented in the absence of leptin signaling (see section 5.1) and aimed at energy conservation [104,165,167]. It is, therefore, possible that suppression of leptin production under conditions of negative energy balance augments behavioral depression as well. This possibility has not yet been tested in a direct experiment, but it finds support in the observation that malnourished rats respond to LPS with an exaggerated production of IL-1β [205], a key mediator of behavioral depression [184,185]. The key role of leptin in regulating both energy balance and systemic inflammation is illustrated in Fig. 7.
Fig. 7.

Mechanisms of energy balance and systemic inflammation are interconnected via leptin. The fat white arrows show changes or manifestations of the processes indicated. The thin black arrows show stimulating (+) or inhibitory (−) effects.
6.2. Leptin irresponsiveness
Humans with idiopathic obesity and experimental animals with diet-induced obesity are typically irresponsive to leptin, and this irresponsiveness seems to be a cause of their low energy expenditure and relentless hunger (see section 2.2). Whereas inhibition of energy expenditure and stimulation of appetite are adaptive under conditions of negative energy balance, they are maladaptive in obesity, a persistent state of positive energy balance. Likewise, exaggeration of inflammation-associated hypothermia and behavioral depression looses its purpose in obese subjects. However, obese subjects may suffer from the detrimental effects of an exaggerated production of some inflammatory mediators. Indeed, LPS is more lethal in ob/ob mice than in their lean counterparts [85]. The ob/ob mice respond to LPS with an excessive production of TNF-α [85]. They are also more sensitive to the detrimental effects of TNF-α, as are db/db mice and Zucker fa/fa rats [206,207]. These findings are consistent with clinical studies [8-10,208,209] showing that obesity is often associated with a poor outcome in inflammatory disorders such as sepsis, lupus erythematosus, and severe pancreatitis.
7. Summary
Mutant rats that are irresponsive to leptin due to an absent or a dysfunctional LRb show attenuated fever or exaggerated hypothermia in a cool environment, but not in a warm environment. This shift from fever to hypothermia is likely to be due to amplification of cryogenic signaling. Indeed, both the production and cryogenic action of TNF-α are enhanced in leptin-irresponsive mutants.
In mutant mice that are irresponsive to leptin due to an absent LRb, the behavioral depression of systemic inflammation is exaggerated. This exaggeration is associated with enhancement of IL-1β production.
Leptin-irresponsive mutants are unable to fully activate the HPA axis in response to inflammatory stimuli. The HPA axis has multiple anti-inflammatory actions, and its incomplete activation may account for the exaggerated production of TNF-α and IL-1β in leptin-irresponsive rats and mice.
In lean subjects (experimental animals and humans), the leptin-LR system is functional. When energy balance in these subjects is negative (e.g., during fasting or calorie restriction), leptin production is suppressed, which can exaggerate both the hypothermia and behavioral depression of systemic inflammation. The biological value of this exaggeration is likely to be energy conservation (Fig. 7).
Experimental animals with diet-induced obesity and humans with idiopathic obesity are irresponsive to leptin. This dysfunctionality can result in augmentation of the hypothermia and behavioral depression of systemic inflammation. These manifestations, however, aid no benefit in obesity, a persistent state of positive energy balance.
An involvement of leptin in inflammatory anorexia is also possible, but decisive evidence for such an involvement is still missing.
Acknowledgements
The authors' research reviewed in this article has been supported by grants from the National Institute of Neurological Disorders and Stroke (NS41233), Arizona Biomedical Research Commission (8016), and St. Joseph's Foundation. The editorial assistance of Faye Farmer is greatly appreciated.
Abbreviations
- HPA
hypothalamo-pituitary-adrenal
- IL
interleukin
- LPS
lipopolysaccharide
- LR
leptin receptor
- TNF
tumor necrosis factor
- Tb
deep body temperature
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
8. References
- 1.Larsen GL, Henson PM. Mediators of inflammation. Annu Rev Immunol. 1983;1:335–59. doi: 10.1146/annurev.iy.01.040183.002003. [DOI] [PubMed] [Google Scholar]
- 2.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–91. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 3.Clemmer TP, Fisher CJ, Jr., Bone RC, Slotman GJ, Metz CA, Thomas FO. Hypothermia in the sepsis syndrome and clinical outcome. The Methylprednisolone Severe Sepsis Study Group. Crit Care Med. 1992;20:1395–401. doi: 10.1097/00003246-199210000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Brun-Buisson C, Doyon F, Carlet J, et al. Incidence, risk factors, and outcome of severe sepsis and septic shock in adults. A multicenter prospective study in intensive care units. French ICU Group for Severe Sepsis. JAMA. 1995;274:968–74. [PubMed] [Google Scholar]
- 5.Dhainaut JF, Tenaillon A, Hemmer M, et al. Confirmatory platelet-activating factor receptor antagonist trial in patients with severe gram-negative bacterial sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. BN 52021 Sepsis Investigator Group. Crit Care Med. 1998;26:1963–71. doi: 10.1097/00003246-199812000-00021. [DOI] [PubMed] [Google Scholar]
- 6.Arons MM, Wheeler AP, Bernard GR, et al. Effects of ibuprofen on the physiology and survival of hypothermic sepsis. Ibuprofen in Sepsis Study Group. Crit Care Med. 1999;27:699–707. doi: 10.1097/00003246-199904000-00020. [DOI] [PubMed] [Google Scholar]
- 7.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–54. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 8.Gottschlich MM, Mayes T, Khoury JC, Warden GD. Significance of obesity on nutritional, immunologic, hormonal, and clinical outcome parameters in burns. J Am Diet Assoc. 1993;93:1261–8. doi: 10.1016/0002-8223(93)91952-m. [DOI] [PubMed] [Google Scholar]
- 9.El-Solh A, Sikka P, Bozkanat E, Jaafar W, Davies J. Morbid obesity in the medical ICU. Chest. 2001;120:1989–97. doi: 10.1378/chest.120.6.1989. [DOI] [PubMed] [Google Scholar]
- 10.Yaegashi M, Jean R, Zuriqat M, Noack S, Homel P. Outcome of morbid obesity in the intensive care unit. J Intensive Care Med. 2005;20:147–54. doi: 10.1177/0885066605275314. [DOI] [PubMed] [Google Scholar]
- 11.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–70. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 12.Ahima RS, Flier JS. Leptin. Annu Rev Physiol. 2000;62:413–37. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- 13.Munzberg H, Myers MG., Jr Molecular and anatomical determinants of central leptin resistance. Nat Neurosci. 2005;8:566–70. doi: 10.1038/nn1454. [DOI] [PubMed] [Google Scholar]
- 14.Faggioni R, Feingold KR, Grunfeld C. Leptin regulation of the immune response and the immunodeficiency of malnutrition. FASEB J. 2001;15:2565–71. doi: 10.1096/fj.01-0431rev. [DOI] [PubMed] [Google Scholar]
- 15.Matarese G, La Cava A. Immune responses in obesity models. Drug Discov Today Dis Models. 2005;2:177–81. [Google Scholar]
- 16.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–32. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 17.Green ED, Maffei M, Braden VV, et al. The human obese (OB) gene: RNA expression pattern and mapping on the physical, cytogenetic, and genetic maps of chromosome 7. Genome Res. 1995;5:5–12. doi: 10.1101/gr.5.1.5. [DOI] [PubMed] [Google Scholar]
- 18.Maffei M, Fei H, Lee GH, et al. Increased expression in adipocytes of ob RNA in mice with lesions of the hypothalamus and with mutations at the db locus. Proc Natl Acad Sci U S A. 1995;92:6957–60. doi: 10.1073/pnas.92.15.6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masuzaki H, Ogawa Y, Isse N, et al. Human obese gene expression. Adipocyte-specific expression and regional differences in the adipose tissue. Diabetes. 1995;44:855–8. doi: 10.2337/diab.44.7.855. [DOI] [PubMed] [Google Scholar]
- 20.Halaas JL, Gajiwala KS, Maffei M, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–6. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 21.Madej T, Boguski MS, Bryant SH. Threading analysis suggests that the obese gene product may be a helical cytokine. FEBS Lett. 1995;373:13–8. doi: 10.1016/0014-5793(95)00977-h. [DOI] [PubMed] [Google Scholar]
- 22.Zhang F, Basinski MB, Beals JM, et al. Crystal structure of the obese protein leptin-E100. Nature. 1997;387:206–9. doi: 10.1038/387206a0. [DOI] [PubMed] [Google Scholar]
- 23.Tartaglia LA, Dembski M, Weng X, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–71. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 24.Lee GH, Proenca R, Montez JM, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–5. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 25.Wang MY, Zhou YT, Newgard CB, Unger RH. A novel leptin receptor isoform in rat. FEBS Lett. 1996;392:87–90. doi: 10.1016/0014-5793(96)00790-9. [DOI] [PubMed] [Google Scholar]
- 26.Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093–6. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- 27.Yang G, Ge H, Boucher A, Yu X, Li C. Modulation of direct leptin signaling by soluble leptin receptor. Mol Endocrinol. 2004;18:1354–62. doi: 10.1210/me.2004-0027. [DOI] [PubMed] [Google Scholar]
- 28.Golden PL, Maccagnan TJ, Pardridge WM. Human blood-brain barrier leptin receptor. Binding and endocytosis in isolated human brain microvessels. J Clin Invest. 1997;99:14–8. doi: 10.1172/JCI119125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uotani S, Bjorbaek C, Tornoe J, Flier JS. Functional properties of leptin receptor isoforms: internalization and degradation of leptin and ligand-induced receptor downregulation. Diabetes. 1999;48:279–86. doi: 10.2337/diabetes.48.2.279. [DOI] [PubMed] [Google Scholar]
- 30.White DW, Kuropatwinski KK, Devos R, Baumann H, Tartaglia LA. Leptin receptor (OB-R) signaling. Cytoplasmic domain mutational analysis and evidence for receptor homo-oligomerization. J Biol Chem. 1997;272:4065–71. doi: 10.1074/jbc.272.7.4065. [DOI] [PubMed] [Google Scholar]
- 31.Banks AS, Davis SM, Bates SH, Myers MG., Jr Activation of downstream signals by the long form of the leptin receptor. J Biol Chem. 2000;275:14563–72. doi: 10.1074/jbc.275.19.14563. [DOI] [PubMed] [Google Scholar]
- 32.Bates SH, Stearns WH, Dundon TA, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–9. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- 33.Bates SH, Dundon TA, Seifert M, Carlson M, Maratos-Flier E, Myers MG., Jr LRb-STAT3 signaling is required for the neuroendocrine regulation of energy expenditure by leptin. Diabetes. 2004;53:3067–73. doi: 10.2337/diabetes.53.12.3067. [DOI] [PubMed] [Google Scholar]
- 34.de Luca C, Kowalski TJ, Zhang Y, et al. Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J Clin Invest. 2005;115:3484–93. doi: 10.1172/JCI24059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen H, Charlat O, Tartaglia LA, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84:491–5. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 36.Chua SC, Jr., Chung WK, Wu-Peng XS, et al. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science. 1996;271:994–6. doi: 10.1126/science.271.5251.994. [DOI] [PubMed] [Google Scholar]
- 37.Chua SC, Jr., White DW, Wu-Peng XS, et al. Phenotype of fatty due to Gln269Pro mutation in the leptin receptor (Lepr) Diabetes. 1996;45:1141–3. doi: 10.2337/diab.45.8.1141. [DOI] [PubMed] [Google Scholar]
- 38.Phillips MS, Liu Q, Hammond HA, et al. Leptin receptor missense mutation in the fatty Zucker rat. Nat Genet. 1996;13:18–9. doi: 10.1038/ng0596-18. [DOI] [PubMed] [Google Scholar]
- 39.Takaya K, Ogawa Y, Hiraoka J, et al. Nonsense mutation of leptin receptor in the obese spontaneously hypertensive Koletsky rat. Nat Genet. 1996;14:130–1. doi: 10.1038/ng1096-130. [DOI] [PubMed] [Google Scholar]
- 40.Wu-Peng XS, Chua SC, Jr., Okada N, Liu SM, Nicolson M, Leibel RL. Phenotype of the obese Koletsky (f) rat due to Tyr763Stop mutation in the extracellular domain of the leptin receptor (Lepr): evidence for deficient plasma-to-CSF transport of leptin in both the Zucker and Koletsky obese rat. Diabetes. 1997;46:513–8. doi: 10.2337/diab.46.3.513. [DOI] [PubMed] [Google Scholar]
- 41.Elmquist JK, Maratos-Flier E, Saper CB, Flier JS. Unraveling the central nervous system pathways underlying responses to leptin. Nat Neurosci. 1998;1:445–50. doi: 10.1038/2164. [DOI] [PubMed] [Google Scholar]
- 42.Banks WA, Kastin AJ, Huang W, Jaspan JB, Maness LM. Leptin enters the brain by a saturable system independent of insulin. Peptides. 1996;17:305–11. doi: 10.1016/0196-9781(96)00025-3. [DOI] [PubMed] [Google Scholar]
- 43.Elias CF, Kelly JF, Lee CE, et al. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol. 2000;423:261–81. [PubMed] [Google Scholar]
- 44.Maffei M, Halaas J, Ravussin E, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–61. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 45.Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–5. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 46.Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–4. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- 47.Hamilton BS, Paglia D, Kwan AY, Deitel M. Increased obese mRNA expression in omental fat cells from massively obese humans. Nat Med. 1995;1:953–6. doi: 10.1038/nm0995-953. [DOI] [PubMed] [Google Scholar]
- 48.Saladin R, De Vos P, Guerre-Millo M, et al. Transient increase in obese gene expression after food intake or insulin administration. Nature. 1995;377:527–9. doi: 10.1038/377527a0. [DOI] [PubMed] [Google Scholar]
- 49.Kolaczynski JW, Considine RV, Ohannesian J, et al. Responses of leptin to short-term fasting and refeeding in humans: a link with ketogenesis but not ketones themselves. Diabetes. 1996;45:1511–5. doi: 10.2337/diab.45.11.1511. [DOI] [PubMed] [Google Scholar]
- 50.Boden G, Chen X, Mozzoli M, Ryan I. Effect of fasting on serum leptin in normal human subjects. J Clin Endocrinol Metab. 1996;81:3419–23. doi: 10.1210/jcem.81.9.8784108. [DOI] [PubMed] [Google Scholar]
- 51.Korbonits M, Trainer PJ, Little JA, et al. Leptin levels do not change acutely with food administration in normal or obese subjects, but are negatively correlated with pituitary-adrenal activity. Clin Endocrinol. 1997;46:751–7. doi: 10.1046/j.1365-2265.1997.1820979.x. [DOI] [PubMed] [Google Scholar]
- 52.Ahren B, Mansson S, Gingerich RL, Havel PJ. Regulation of plasma leptin in mice: influence of age, high-fat diet, and fasting. Am J Physiol. 1997;273:R113–20. doi: 10.1152/ajpregu.1997.273.1.R113. [DOI] [PubMed] [Google Scholar]
- 53.Havel PJ, Townsend R, Chaump L, Teff K. High-fat meals reduce 24-h circulating leptin concentrations in women. Diabetes. 1999;48:334–41. doi: 10.2337/diabetes.48.2.334. [DOI] [PubMed] [Google Scholar]
- 54.Mueller WM, Gregoire FM, Stanhope KL, et al. Evidence that glucose metabolism regulates leptin secretion from cultured rat adipocytes. Endocrinology. 1998;139:551–8. doi: 10.1210/endo.139.2.5716. [DOI] [PubMed] [Google Scholar]
- 55.Wellhoener P, Fruehwald-Schultes B, Kern W, et al. Glucose metabolism rather than insulin is a main determinant of leptin secretion in humans. J Clin Endocrinol Metab. 2000;85:1267–71. doi: 10.1210/jcem.85.3.6483. [DOI] [PubMed] [Google Scholar]
- 56.Doring H, Schwarzer K, Nuesslein-Hildesheim B, Schmidt I. Leptin selectively increases energy expenditure of food-restricted lean mice. Int J Obes Relat Metab Disord. 1998;22:83–8. doi: 10.1038/sj.ijo.0800547. [DOI] [PubMed] [Google Scholar]
- 57.Montez JM, Soukas A, Asilmaz E, Fayzikhodjaeva G, Fantuzzi G, Friedman JM. Acute leptin deficiency, leptin resistance, and the physiologic response to leptin withdrawal. Proc Natl Acad Sci U S A. 2005;102:2537–42. doi: 10.1073/pnas.0409530102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freeman DA, Lewis DA, Kauffman AS, Blum RM, Dark J. Reduced leptin concentrations are permissive for display of torpor in Siberian hamsters. Am J Physiol Regul Integr Comp Physiol. 2004;287:R97–R103. doi: 10.1152/ajpregu.00716.2003. [DOI] [PubMed] [Google Scholar]
- 59.Keim NL, Stern JS, Havel PJ. Relation between circulating leptin concentrations and appetite during a prolonged, moderate energy deficit in women. Am J Clin Nutr. 1998;68:794–801. doi: 10.1093/ajcn/68.4.794. [DOI] [PubMed] [Google Scholar]
- 60.Rosenbaum M, Murphy EM, Heymsfield SB, Matthews DE, Leibel RL. Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulating concentrations of thyroid hormones. J Clin Endocrinol Metab. 2002;87:2391–4. doi: 10.1210/jcem.87.5.8628. [DOI] [PubMed] [Google Scholar]
- 61.Mars M, de Graaf C, de Groot CP, van Rossum CT, Kok FJ. Fasting leptin and appetite responses induced by a 4-day 65%-energy-restricted diet. Int J Obes. 2006;30:122–8. doi: 10.1038/sj.ijo.0803070. [DOI] [PubMed] [Google Scholar]
- 62.Gavrilova O, Leon LR, Marcus-Samuels B, et al. Torpor in mice is induced by both leptin-dependent and -independent mechanisms. Proc Natl Acad Sci U S A. 1999;96:14623–8. doi: 10.1073/pnas.96.25.14623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pelleymounter MA, Cullen MJ, Baker MB, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–3. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 64.Harris RB, Zhou J, Redmann SM, Jr., et al. A leptin dose-response study in obese (ob/ob) and lean (+/?) mice. Endocrinology. 1998;139:8–19. doi: 10.1210/endo.139.1.5675. [DOI] [PubMed] [Google Scholar]
- 65.Harris RB, Mitchell TD, Yan X, Simpson JS, Redmann SM., Jr. Metabolic responses to leptin in obese db/db mice are strain dependent. Am J Physiol Regul Integr Comp Physiol. 2001;281:R115–32. doi: 10.1152/ajpregu.2001.281.1.R115. [DOI] [PubMed] [Google Scholar]
- 66.Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI. Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest. 1997;100:270–8. doi: 10.1172/JCI119532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luheshi GN, Gardner JD, Rushforth DA, Loudon AS, Rothwell NJ. Leptin actions on food intake and body temperature are mediated by IL-1. Proc Natl Acad Sci U S A. 1999;96:7047–52. doi: 10.1073/pnas.96.12.7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morrison SF. Activation of 5-HT1A receptors in raphe pallidus inhibits leptin-evoked increases in brown adipose tissue thermogenesis. Am J Physiol Regul Integr Comp Physiol. 2004;286:R832–7. doi: 10.1152/ajpregu.00678.2003. [DOI] [PubMed] [Google Scholar]
- 69.Farooqi IS, Keogh JM, Kamath S, et al. Partial leptin deficiency and human adiposity. Nature. 2001;414:34–5. doi: 10.1038/35102112. [DOI] [PubMed] [Google Scholar]
- 70.Montague CT, Farooqi IS, Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–8. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 71.Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat Genet. 1998;18:213–5. doi: 10.1038/ng0398-213. [DOI] [PubMed] [Google Scholar]
- 72.Farooqi IS, Matarese G, Lord GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Licinio J, Caglayan S, Ozata M, et al. Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behavior in leptin-deficient adults. Proc Natl Acad Sci U S A. 2004;101:4531–6. doi: 10.1073/pnas.0308767101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heymsfield SB, Greenberg AS, Fujioka K, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282:1568–75. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- 75.Hukshorn CJ, van Dielen FM, Buurman WA, Westerterp-Plantenga MS, Campfield LA, Saris WH. The effect of pegylated recombinant human leptin (PEG-OB) on weight loss and inflammatory status in obese subjects. Int J Obes Relat Metab Disord. 2002;26:504–9. doi: 10.1038/sj.ijo.0801952. [DOI] [PubMed] [Google Scholar]
- 76.Zelissen PM, Stenlof K, Lean ME, et al. Effect of three treatment schedules of recombinant methionyl human leptin on body weight in obese adults: a randomized, placebo-controlled trial. Diabetes Obes Metab. 2005;7:755–61. doi: 10.1111/j.1463-1326.2005.00468.x. [DOI] [PubMed] [Google Scholar]
- 77.Van Heek M, Compton DS, France CF, et al. Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J Clin Invest. 1997;99:385–90. doi: 10.1172/JCI119171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin S, Thomas TC, Storlien LH, Huang XF. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int J Obes Relat Metab Disord. 2000;24:639–46. doi: 10.1038/sj.ijo.0801209. [DOI] [PubMed] [Google Scholar]
- 79.Clement K, Vaisse C, Lahlou N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 80.Grunfeld C, Zhao C, Fuller J, et al. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J Clin Invest. 1996;97:2152–7. doi: 10.1172/JCI118653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sarraf P, Frederich RC, Turner EM, et al. Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. J Exp Med. 1997;185:171–5. doi: 10.1084/jem.185.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Berkowitz DE, Brown D, Lee KM, et al. Endotoxin-induced alteration in the expression of leptin and β3-adrenergic receptor in adipose tissue. Am J Physiol. 1998;274:E992–7. doi: 10.1152/ajpendo.1998.274.6.E992. [DOI] [PubMed] [Google Scholar]
- 83.Finck BN, Kelley KW, Dantzer R, Johnson RW. In vivo and in vitro evidence for the involvement of tumor necrosis factor-α in the induction of leptin by lipopolysaccharide. Endocrinology. 1998;139:2278–83. doi: 10.1210/endo.139.5.6012. [DOI] [PubMed] [Google Scholar]
- 84.Faggioni R, Fantuzzi G, Fuller J, Dinarello CA, Feingold KR, Grunfeld C. IL-1β mediates leptin induction during inflammation. Am J Physiol. 1998;274:R204–8. doi: 10.1152/ajpregu.1998.274.1.R204. [DOI] [PubMed] [Google Scholar]
- 85.Faggioni R, Fantuzzi G, Gabay C, et al. Leptin deficiency enhances sensitivity to endotoxin-induced lethality. Am J Physiol. 1999;276:R136–42. doi: 10.1152/ajpregu.1999.276.1.R136. [DOI] [PubMed] [Google Scholar]
- 86.Chautard T, Spinedi E, Voirol M, Pralong FP, Gaillard RC. Role of glucocorticoids in the response of the hypothalamo-corticotrope, immune and adipose systems to repeated endotoxin administration. Neuroendocrinology. 1999;69:360–9. doi: 10.1159/000054438. [DOI] [PubMed] [Google Scholar]
- 87.Faggioni R, Moser A, Feingold KR, Grunfeld C. Reduced leptin levels in starvation increase susceptibility to endotoxic shock. Am J Pathol. 2000;156:1781–7. doi: 10.1016/S0002-9440(10)65049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mastronardi CA, Yu WH, Rettori V, McCann S. Lipopolysaccharide-induced leptin release is not mediated by nitric oxide, but is blocked by dexamethasone. Neuroimmunomodulation. 2000;8:91–7. doi: 10.1159/000026458. [DOI] [PubMed] [Google Scholar]
- 89.Mastronardi CA, Yu WH, Srivastava VK, Dees WL, McCann SM. Lipopolysaccharide-induced leptin release is neurally controlled. Proc Natl Acad Sci U S A. 2001;98:14720–5. doi: 10.1073/pnas.251543598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang W, Poole B, Mitra A, et al. Role of leptin deficiency in early acute renal failure during endotoxemia in ob/ob mice. J Am Soc Nephrol. 2004;15:645–9. doi: 10.1097/01.asn.0000113551.14276.0b. [DOI] [PubMed] [Google Scholar]
- 91.Gualillo O, Eiras S, Lago F, Dieguez C, Casanueva FF. Elevated serum leptin concentrations induced by experimental acute inflammation. Life Sci. 2000;67:2433–41. doi: 10.1016/s0024-3205(00)00827-4. [DOI] [PubMed] [Google Scholar]
- 92.Moshyedi AK, Josephs MD, Abdalla EK, et al. Increased leptin expression in mice with bacterial peritonitis is partially regulated by tumor necrosis factor alpha. Infect Immun. 1998;66:1800–2. doi: 10.1128/iai.66.4.1800-1802.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arnalich F, Lopez J, Codoceo R, Jim nez M, Madero R, Montiel C. Relationship of plasma leptin to plasma cytokines and human survivalin sepsis and septic shock. J Infect Dis. 1999;180:908–11. doi: 10.1086/314963. [DOI] [PubMed] [Google Scholar]
- 94.Moses AG, Dowidar N, Holloway B, Waddell I, Fearon KC, Ross JA. Leptin and its relation to weight loss, ob gene expression and the acute-phase response in surgical patients. Br J Surg. 2001;88:588–93. doi: 10.1046/j.1365-2168.2001.01743.x. [DOI] [PubMed] [Google Scholar]
- 95.Kluger MJ. Fever: role of pyrogens and cryogens. Physiol Rev. 1991;71:93–127. doi: 10.1152/physrev.1991.71.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Turnbull AV, Rivier CL. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action. Physiol Rev. 1999;79:1–71. doi: 10.1152/physrev.1999.79.1.1. [DOI] [PubMed] [Google Scholar]
- 97.Janik JE, Curti BD, Considine RV, et al. Interleukin 1α increases serum leptin concentrations in humans. J Clin Endocrinol Metab. 1997;82:3084–6. doi: 10.1210/jcem.82.9.4214. [DOI] [PubMed] [Google Scholar]
- 98.Francis J, MohanKumar PS, MohanKumar SM, Quadri SK. Systemic administration of lipopolysaccharide increases plasma leptin levels: blockade by soluble interleukin-1 receptor. Endocrine. 1999;10:291–5. doi: 10.1007/BF02738628. [DOI] [PubMed] [Google Scholar]
- 99.Finck BN, Johnson RW. Intracerebroventricular injection of lipopolysaccharide increases plasma leptin levels. Neuroreport. 1999;10:153–6. doi: 10.1097/00001756-199901180-00029. [DOI] [PubMed] [Google Scholar]
- 100.American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864–74. [PubMed] [Google Scholar]
- 101.Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–6. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- 102.Goldstein B, Giroir B, Randolph A. International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med. 2005;6:2–8. doi: 10.1097/01.PCC.0000149131.72248.E6. [DOI] [PubMed] [Google Scholar]
- 103.Romanovsky AA, Almeida MC, Aronoff DM, et al. Fever and hypothermia in systemic inflammation: recent discoveries and revisions. Front Biosci. 2005;10:2193–216. doi: 10.2741/1690. [DOI] [PubMed] [Google Scholar]
- 104.Romanovsky AA, Kulchitsky VA, Akulich NV, et al. First and second phases of biphasic fever: two sequential stages of the sickness syndrome? Am J Physiol. 1996;271:R244–53. doi: 10.1152/ajpregu.1996.271.1.R244. [DOI] [PubMed] [Google Scholar]
- 105.Romanovsky AA, Simons CT, Szekely M, Kulchitsky VA. The vagus nerve in the thermoregulatory response to systemic inflammation. Am J Physiol. 1997;273:R407–13. doi: 10.1152/ajpregu.1997.273.1.R407. [DOI] [PubMed] [Google Scholar]
- 106.Rudaya AY, Steiner AA, Robbins JR, Dragic AS, Romanovsky AA. Thermoregulatory responses to lipopolysaccharide in the mouse: dependence on the dose and ambient temperature. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1244–52. doi: 10.1152/ajpregu.00370.2005. [DOI] [PubMed] [Google Scholar]
- 107.Steiner AA, Chakravarty S, Robbins JR, et al. Thermoregulatory responses of rats to conventional preparations of lipopolysaccharide are caused by lipopolysaccharide per se - not by lipoprotein contaminants. Am J Physiol Regul Integr Comp Physiol. 2005;289:R348–R52. doi: 10.1152/ajpregu.00223.2005. [DOI] [PubMed] [Google Scholar]
- 108.Romanovsky AA, Simons CT, Kulchitsky VA. “Biphasic” fevers often consist of more than two phases. Am J Physiol. 1998;275:R323–31. doi: 10.1152/ajpregu.1998.275.1.R323. [DOI] [PubMed] [Google Scholar]
- 109.Romanovsky AA, Kulchitsky VA, Simons CT, Sugimoto N. Methodology of fever research: why are polyphasic fevers often thought to be biphasic? Am J Physiol. 1998;275:R332–8. doi: 10.1152/ajpregu.1998.275.1.R332. [DOI] [PubMed] [Google Scholar]
- 110.Steiner AA, Dogan MD, Ivanov AI, et al. A new function of the leptin receptor: mediation of the recovery from lipopolysaccharide-induced hypothermia. FASEB J. 2004;18:1949–51. doi: 10.1096/fj.04-2295fje. [DOI] [PubMed] [Google Scholar]
- 111.Steiner AA, Rudaya AY, Ivanov AI, Romanovsky AA. Febrigenic signaling to the brain does not involve nitric oxide. Br J Pharmacol. 2004;141:1204–13. doi: 10.1038/sj.bjp.0705713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Oka T, Oka K, Kobayashi T, et al. Characteristics of thermoregulatory and febrile responses in mice deficient in prostaglandin EP1 and EP3 receptors. J Physiol. 2003;551:945–54. doi: 10.1113/jphysiol.2003.048140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Steiner AA, Chakravarty S, Rudaya AY, Herkenham M, Romanovsky AA. Bacterial lipopolysaccharide fever is initiated via Toll-like receptor 4 on hematopoietic cells. Blood. 2006;107:4000–2. doi: 10.1182/blood-2005-11-4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Romanovsky AA, Shido O, Sakurada S, Sugimoto N, Nagasaka T. Endotoxin shock: thermoregulatory mechanisms. Am J Physiol. 1996;270:R693–703. doi: 10.1152/ajpregu.1996.270.4.R693. [DOI] [PubMed] [Google Scholar]
- 115.Almeida MC, Steiner AA, Branco LG, Romanovsky AA. Cold-seeking behavior as a thermoregulatory strategy in systemic inflammation. Eur J Neurosci. 2006;23:3359–67. doi: 10.1111/j.1460-9568.2006.04854.x. [DOI] [PubMed] [Google Scholar]
- 116.Almeida MC, Steiner AA, Branco LG, Romanovsky AA. Neural substrate of cold-seeking behavior in endotoxin shock. PLoS ONE. 2006;1:e1. doi: 10.1371/journal.pone.0000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gordon CJ. Thermal biology of the laboratory rat. Physiol Behav. 1990;47:963–91. doi: 10.1016/0031-9384(90)90025-y. [DOI] [PubMed] [Google Scholar]
- 118.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 119.Szekely M, Szelenyi Z. Regulation of energy balance by peptides: a review. Curr Protein Pept Sci. 2005;6:327–53. doi: 10.2174/1389203054546343. [DOI] [PubMed] [Google Scholar]
- 120.Collins S, Kuhn CM, Petro AE, Swick AG, Chrunyk BA, Surwit RS. Role of leptin in fat regulation. Nature. 1996;380:677. doi: 10.1038/380677a0. [DOI] [PubMed] [Google Scholar]
- 121.Gao Q, Wolfgang MJ, Neschen S, et al. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A. 2004;101:4661–6. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maskrey M, Megirian D, Farkas GA. Effect of changing body temperature on the ventilatory and metabolic responses of lean and obese Zucker rats. Am J Physiol. 1998;275:R531–40. doi: 10.1152/ajpregu.1998.275.2.R531. [DOI] [PubMed] [Google Scholar]
- 123.Harris RB. Parabiosis between db/db and ob/ob or db/+ mice. Endocrinology. 1999;140:138–45. doi: 10.1210/endo.140.1.6449. [DOI] [PubMed] [Google Scholar]
- 124.Murakami DM, Horwitz BA, Fuller CA. Circadian rhythms of temperature and activity in obese and lean Zucker rats. Am J Physiol. 1995;269:R1038–43. doi: 10.1152/ajpregu.1995.269.5.R1038. [DOI] [PubMed] [Google Scholar]
- 125.Ivanov AI, Romanovsky AA. Fever responses of Zucker rats with and without fatty mutation of the leptin receptor. Am J Physiol Regul Integr Comp Physiol. 2002;282:R311–6. doi: 10.1152/ajpregu.00376.2001. [DOI] [PubMed] [Google Scholar]
- 126.Romanovsky AA, Ivanov AI, Shimansky YP. Selected contribution: ambient temperature for experiments in rats: a new method for determining the zone of thermal neutrality. J Appl Physiol. 2002;92:2667–79. doi: 10.1152/japplphysiol.01173.2001. [DOI] [PubMed] [Google Scholar]
- 127.Holt S, York DA, Fitzsimons JT. The effects of corticosterone, cold exposure and overfeeding with sucrose on brown adipose tissue of obese Zucker rats (fa/fa) Biochem J. 1983;214:215–23. doi: 10.1042/bj2140215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Triandafillou J, Himms-Hagen J. Brown adipose tissue in genetically obese (fa/fa) rats: response to cold and diet. Am J Physiol. 1983;244:E145–50. doi: 10.1152/ajpendo.1983.244.2.E145. [DOI] [PubMed] [Google Scholar]
- 129.Girardier L, Clark MG, Seydoux J. Thermogenesis associated with spontaneous activity: an important component of thermoregulatory needs in rats. J Physiol. 1995;488:779–87. doi: 10.1113/jphysiol.1995.sp021009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Seydoux J, Benzi RH, Shibata M, Girardier L. Underlying mechanisms of atrophic state of brown adipose tissue in obese Zucker rats. Am J Physiol. 1990;259:R61–9. doi: 10.1152/ajpregu.1990.259.1.R61. [DOI] [PubMed] [Google Scholar]
- 131.Szekely M, Szelenyi Z. Endotoxin fever in the rat. Acta Physiol Acad Sci Hung. 1979;53:265–77. [PubMed] [Google Scholar]
- 132.Morimoto A, Nakamori T, Watanabe T, Ono T, Murakami N. Pattern differences in experimental fevers induced by endotoxin, endogenous pyrogen, and prostaglandins. Am J Physiol. 1988;254:R633–40. doi: 10.1152/ajpregu.1988.254.4.R633. [DOI] [PubMed] [Google Scholar]
- 133.Szekely M. Nutritional state and endotoxin fever of newborn rabbits. Acta Physiol Acad Sci Hung. 1979;53:279–83. [PubMed] [Google Scholar]
- 134.Molnar D, Milner RD. Metabolic and hormonal response to endotoxin fever in fed and starved one-week rabbits. Biol Neonate. 1983;44:309–14. doi: 10.1159/000241732. [DOI] [PubMed] [Google Scholar]
- 135.Shojoony MJ. Effects of fasting on heat balance and nonshivering thermogenesis in febrile adult guinea pigs. J Therm Biol. 1985;10:239–43. [Google Scholar]
- 136.Shido O, Nagasaka T, Watanabe T. Blunted febrile response to intravenous endotoxin in starved rats. J Appl Physiol. 1989;67:963–9. doi: 10.1152/jappl.1989.67.3.963. [DOI] [PubMed] [Google Scholar]
- 137.Rosenthal M, Roth J, Storr B, Zeisberger E. Fever response in lean (Fa/-) and obese (fa/fa) Zucker rats and its lack to repeated injections of LPS. Physiol Behav. 1996;59:787–93. doi: 10.1016/0031-9384(95)02158-2. [DOI] [PubMed] [Google Scholar]
- 138.Ivanov AI, Kulchitsky VA, Romanovsky AA. Does obesity affect febrile responsiveness? Int J Obes Relat Metab Disord. 2001;25:586–9. doi: 10.1038/sj.ijo.0801523. [DOI] [PubMed] [Google Scholar]
- 139.Ivanov AI, Kulchitsky VA, Romanovsky AA. Role for the cholecystokinin-A receptor in fever: a study of a mutant rat strain and a pharmacological analysis. J Physiol. 2003;547:941–9. doi: 10.1113/jphysiol.2002.033183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ivanov AI, Steiner AA, Patel S, Rudaya AY, Romanovsky AA. Albumin is not an irreplaceable carrier for amphipathic mediators of thermoregulatory responses to LPS: compensatory role of α1-acid glycoprotein. Am J Physiol Regul Integr Comp Physiol. 2005;288:R872–8. doi: 10.1152/ajpregu.00514.2004. [DOI] [PubMed] [Google Scholar]
- 141.Sachot C, Poole S, Luheshi GN. Circulating leptin mediates lipopolysaccharide-induced anorexia and fever in rats. J Physiol. 2004;561:263–72. doi: 10.1113/jphysiol.2004.074351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Szekely M, Romanovsky AA. Pyretic and antipyretic signals within and without fever: a possible interplay. Med Hypotheses. 1998;50:213–8. doi: 10.1016/s0306-9877(98)90021-4. [DOI] [PubMed] [Google Scholar]
- 143.Romanovsky AA. Do fever and anapyrexia exist? Analysis of set point-based definitions. Am J Physiol Regul Integr Comp Physiol. 2004;287:R992–5. doi: 10.1152/ajpregu.00068.2004. [DOI] [PubMed] [Google Scholar]
- 144.Nagai M, Saigusa T, Shimada Y, Inagawa H, Oshima H, Iriki M. Antibody to tumor necrosis factor (TNF) reduces endotoxin fever. Experientia. 1988;44:606–7. doi: 10.1007/BF01953311. [DOI] [PubMed] [Google Scholar]
- 145.Roth J, Martin D, Storr B, Zeisberger E. Neutralization of pyrogen-induced tumour necrosis factor by its type 1 soluble receptor in guinea-pigs: effects on fever and interleukin-6 release. J Physiol. 1998;509:267–75. doi: 10.1111/j.1469-7793.1998.267bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Derijk RH, Berkenbosch F. Hypothermia to endotoxin involves the cytokine tumor necrosis factor and the neuropeptide vasopressin in rats. Am J Physiol. 1994;266:R9–14. doi: 10.1152/ajpregu.1994.266.1.R9. [DOI] [PubMed] [Google Scholar]
- 147.Kozak W, Conn CA, Klir JJ, Wong GH, Kluger MJ. TNF soluble receptor and antiserum against TNF enhance lipopolysaccharide fever in mice. Am J Physiol. 1995;269:R23–9. doi: 10.1152/ajpregu.1995.269.1.R23. [DOI] [PubMed] [Google Scholar]
- 148.Leon LR, Kozak W, Peschon J, Kluger MJ. Exacerbated febrile responses to LPS, but not turpentine, in TNF double receptor-knockout mice. Am J Physiol. 1997;272:R563–9. doi: 10.1152/ajpregu.1997.272.2.R563. [DOI] [PubMed] [Google Scholar]
- 149.Tollner B, Roth J, Storr B, Martin D, Voigt K, Zeisberger E. The role of tumor necrosis factor (TNF) in the febrile and metabolic responses of rats to intraperitoneal injection of a high dose of lipopolysaccharide. Pflugers Arch. 2000;440:925–32. doi: 10.1007/s004240000386. [DOI] [PubMed] [Google Scholar]
- 150.Bibby DC, Grimble RF. Temperature and metabolic changes in rats after various doses of tumour necrosis factor α. J Physiol. 1989;410:367–80. doi: 10.1113/jphysiol.1989.sp017538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Loffreda S, Yang SQ, Lin HZ, et al. Leptin regulates proinflammatory immune responses. FASEB J. 1998;12:57–65. [PubMed] [Google Scholar]
- 152.Zarkesh-Esfahani H, Pockley G, Metcalfe RA, et al. High-dose leptin activates human leukocytes via receptor expression on monocytes. J Immunol. 2001;167:4593–9. doi: 10.4049/jimmunol.167.8.4593. [DOI] [PubMed] [Google Scholar]
- 153.Zarkesh-Esfahani H, Pockley AG, Wu Z, Hellewell PG, Weetman AP, Ross RJ. Leptin indirectly activates human neutrophils via induction of TNF-α. J Immunol. 2004;172:1809–14. doi: 10.4049/jimmunol.172.3.1809. [DOI] [PubMed] [Google Scholar]
- 154.Shen J, Sakaida I, Uchida K, Terai S, Okita K. Leptin enhances TNF-α production via p38 and JNK MAPK in LPS-stimulated Kupffer cells. Life Sci. 2005;77:1502–15. doi: 10.1016/j.lfs.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 155.Livingstone DE, Jones GC, Smith K, et al. Understanding the role of glucocorticoids in obesity: tissue-specific alterations of corticosterone metabolism in obese Zucker rats. Endocrinology. 2000;141:560–3. doi: 10.1210/endo.141.2.7297. [DOI] [PubMed] [Google Scholar]
- 156.Liu Y, Nakagawa Y, Wang Y, et al. Leptin activation of corticosterone production in hepatocytes may contribute to the reversal of obesity and hyperglycemia in leptin-deficient ob/ob mice. Diabetes. 2003;52:1409–16. doi: 10.2337/diabetes.52.6.1409. [DOI] [PubMed] [Google Scholar]
- 157.Huang Q, Rivest R, Richard D. Effects of leptin on corticotropin-releasing factor (CRF) synthesis and CRF neuron activation in the paraventricular hypothalamic nucleus of obese (ob/ob) mice. Endocrinology. 1998;139:1524–32. doi: 10.1210/endo.139.4.5889. [DOI] [PubMed] [Google Scholar]
- 158.Gaillard RC, Spinedi E, Chautard T, Pralong FP. Cytokines, leptin, and the hypothalamo-pituitary-adrenal axis. Ann N Y Acad Sci. 2000;917:647–57. doi: 10.1111/j.1749-6632.2000.tb05430.x. [DOI] [PubMed] [Google Scholar]
- 159.Bik W, Wolinska-Witort E, Chmielowska M, Rusiecka-Kuczalek E, Baranowska B. Does leptin modulate immune and endocrine response in the time of LPS-induced acute inflammation? Neuro Endocrinol Lett. 2001;22:208–14. [PubMed] [Google Scholar]
- 160.Agnello D, Meazza C, Rowan CG, Villa P, Ghezzi P, Senaldi G. Leptin causes body weight loss in the absence of in vivo activities typical of cytokines of the IL-6 family. Am J Physiol. 1998;275:R913–9. doi: 10.1152/ajpregu.1998.275.3.R913. [DOI] [PubMed] [Google Scholar]
- 161.Costa A, Nappi RE, Smeraldi A, Bergamaschi M, Navarra P, Grossman A. Novel regulators of the in vitro release of hypothalamic corticotrophin-releasing hormone two decades after its discovery: a review. Funct Neurol. 2001;16:205–16. [PubMed] [Google Scholar]
- 162.Jethwa PH, Smith KL, Small CJ, et al. Neuromedin U partially mediates leptin-induced hypothalamo-pituitary adrenal (HPA) stimulation and has a physiological role in the regulation of the HPA axis in the rat. Endocrinology. 2006;147:2886–92. doi: 10.1210/en.2005-0983. [DOI] [PubMed] [Google Scholar]
- 163.Plotsky PM, Thrivikraman KV, Watts AG, Hauger RL. Hypothalamic-pituitary-adrenal axis function in the Zucker obese rat. Endocrinology. 1992;130:1931–41. doi: 10.1210/endo.130.4.1312431. [DOI] [PubMed] [Google Scholar]
- 164.Cabot C, del Mar Grasa M, Estruch J, Fernandez-Lopez JA, Remesar X, Alemany M. Zucker obese rats are insensitive to the CRH-increasing effect of oleoyl-estrone. Brain Res Bull. 1998;46:529–34. doi: 10.1016/s0361-9230(98)00050-1. [DOI] [PubMed] [Google Scholar]
- 165.Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12:123–37. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- 166.Kent S, Bluthe RM, Kelley KW, Dantzer R. Sickness behavior as a new target for drug development. Trends Pharmacol Sci. 1992;13:24–8. doi: 10.1016/0165-6147(92)90012-u. [DOI] [PubMed] [Google Scholar]
- 167.Romanovsky AA, Szekely M. Fever and hypothermia: two adaptive thermoregulatory responses to systemic inflammation. Med Hypotheses. 1998;50:219–26. doi: 10.1016/s0306-9877(98)90022-6. [DOI] [PubMed] [Google Scholar]
- 168.Romanovsky AA. Signaling the brain in the early sickness syndrome: are sensory nerves involved? Front Biosci. 2004;9:494–504. doi: 10.2741/1247. [DOI] [PubMed] [Google Scholar]
- 169.Laughter JS, Jr., Blatteis CM. A system for the study of behavioral thermoregulation of small animals. Physiol Behav. 1985;35:993–7. doi: 10.1016/0031-9384(85)90271-9. [DOI] [PubMed] [Google Scholar]
- 170.Krueger JM, Kubillus S, Shoham S, Davenne D. Enhancement of slow-wave sleep by endotoxin and lipid A. Am J Physiol. 1986;251:R591–7. doi: 10.1152/ajpregu.1986.251.3.R591. [DOI] [PubMed] [Google Scholar]
- 171.Shoham S, Davenne D, Krueger JM. Muramyl dipeptide, amphetamine, and physostigmine: effects on sleep of rabbits. Physiol Behav. 1987;41:179–85. doi: 10.1016/0031-9384(87)90149-1. [DOI] [PubMed] [Google Scholar]
- 172.Shoham S, Krueger JM. Muramyl dipeptide-induced sleep and fever: effects of ambient temperature and time of injections. Am J Physiol. 1988;255:R157–65. doi: 10.1152/ajpregu.1988.255.1.R157. [DOI] [PubMed] [Google Scholar]
- 173.Kapas L, Hansen MK, Chang HY, Krueger JM. Vagotomy attenuates but does not prevent the somnogenic and febrile effects of lipopolysaccharide in rats. Am J Physiol. 1998;274:R406–11. doi: 10.1152/ajpregu.1998.274.2.R406. [DOI] [PubMed] [Google Scholar]
- 174.Opp MR, Toth LA. Somnogenic and pyrogenic effects of interleukin-1β and lipopolysaccharide in intact and vagotomized rats. Life Sci. 1998;62:923–36. doi: 10.1016/s0024-3205(98)00010-1. [DOI] [PubMed] [Google Scholar]
- 175.Mathias S, Schiffelholz T, Linthorst AC, Pollmacher T, Lancel M. Diurnal variations in lipopolysaccharide-induced sleep, sickness behavior and changes in corticosterone levels in the rat. Neuroendocrinology. 2000;71:375–85. doi: 10.1159/000054558. [DOI] [PubMed] [Google Scholar]
- 176.du Plessis I, Mitchell D, Laburn HP, Cartmell T. Fever and lethargy induced by subcutaneous pyrogen infusion in unrestrained rats. Can J Physiol Pharmacol. 2005;83:1007–14. doi: 10.1139/y05-065. [DOI] [PubMed] [Google Scholar]
- 177.Kozak W, Conn CA, Kluger MJ. Lipopolysaccharide induces fever and depresses locomotor activity in unrestrained mice. Am J Physiol. 1994;266:R125–35. doi: 10.1152/ajpregu.1994.266.1.R125. [DOI] [PubMed] [Google Scholar]
- 178.Johnson RW, Gheusi G, Segreti S, Dantzer R, Kelley KW. C3H/HeJ mice are refractory to lipopolysaccharide in the brain. Brain Res. 1997;752:219–26. doi: 10.1016/s0006-8993(96)01454-0. [DOI] [PubMed] [Google Scholar]
- 179.Ahima RS, Bjorbaek C, Osei S, Flier JS. Regulation of neuronal and glial proteins by leptin: implications for brain development. Endocrinology. 1999;140:2755–62. doi: 10.1210/endo.140.6.6774. [DOI] [PubMed] [Google Scholar]
- 180.Segal-Lieberman G, Bradley RL, Kokkotou E, et al. Melanin-concentrating hormone is a critical mediator of the leptin-deficient phenotype. Proc Natl Acad Sci U S A. 2003;100:10085–90. doi: 10.1073/pnas.1633636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kudo T, Akiyama M, Kuriyama K, Sudo M, Moriya T, Shibata S. Night-time restricted feeding normalises clock genes and Pai-1 gene expression in the db/db mouse liver. Diabetologia. 2004;47:1425–36. doi: 10.1007/s00125-004-1461-0. [DOI] [PubMed] [Google Scholar]
- 182.Jurgens HS, Schurmann A, Kluge R, et al. Hyperphagia, lower body temperature, and reduced running wheel activity precede development of morbid obesity in New Zealand obese mice. Physiol Genomics. 2006;25:234–41. doi: 10.1152/physiolgenomics.00252.2005. [DOI] [PubMed] [Google Scholar]
- 183.O'Connor JC, Satpathy A, Hartman ME, et al. IL-1β-mediated innate immunity is amplified in the db/db mouse model of type 2 diabetes. J Immunol. 2005;174:4991–7. doi: 10.4049/jimmunol.174.8.4991. [DOI] [PubMed] [Google Scholar]
- 184.Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. 2002;25:154–9. doi: 10.1016/s0166-2236(00)02088-9. [DOI] [PubMed] [Google Scholar]
- 185.Dantzer R. Cytokine, sickness behavior, and depression. Neurol Clin. 2006;24:441–60. doi: 10.1016/j.ncl.2006.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Langhans W. Bacterial products and the control of ingestive behavior: clinical implications. Nutrition. 1996;12:303–15. doi: 10.1016/s0899-9007(96)80052-9. [DOI] [PubMed] [Google Scholar]
- 187.Konsman JP, Dantzer R. How the immune and nervous systems interact during disease-associated anorexia. Nutrition. 2001;17:664–8. doi: 10.1016/s0899-9007(01)00602-5. [DOI] [PubMed] [Google Scholar]
- 188.McCarthy DO, Kluger MJ, Vander AJ. The role of fever in appetite suppression after endotoxin administration. Am J Clin Nutr. 1984;40:310–6. doi: 10.1093/ajcn/40.2.310. [DOI] [PubMed] [Google Scholar]
- 189.Langhans W, Balkowski G, Savoldelli D. Differential feeding responses to bacterial lipopolysaccharide and muramyl dipeptide. Am J Physiol. 1991;261:R659–64. doi: 10.1152/ajpregu.1991.261.3.R659. [DOI] [PubMed] [Google Scholar]
- 190.Porter MH, Arnold M, Langhans W. Lipopolysaccharide-induced anorexia following hepatic portal vein and vena cava administration. Physiol Behav. 1998;64:581–4. doi: 10.1016/s0031-9384(98)00082-1. [DOI] [PubMed] [Google Scholar]
- 191.Huang QH, Hruby VJ, Tatro JB. Role of central melanocortins in endotoxin-induced anorexia. Am J Physiol. 1999;276:R864–71. doi: 10.1152/ajpregu.1999.276.3.R864. [DOI] [PubMed] [Google Scholar]
- 192.O'Reilly B, Vander AJ, Kluger MJ. Effects of chronic infusion of lipopolysaccharide on food intake and body temperature of the rat. Physiol Behav. 1988;42:287–91. doi: 10.1016/0031-9384(88)90084-4. [DOI] [PubMed] [Google Scholar]
- 193.Langhans W, Harlacher R, Scharrer E. Verapamil and indomethacin attenuate endotoxin-induced anorexia. Physiol Behav. 1989;46:535–9. doi: 10.1016/0031-9384(89)90032-2. [DOI] [PubMed] [Google Scholar]
- 194.Murray MJ, Murray AB. Anorexia of infection as a mechanism of host defense. Am J Clin Nutr. 1979;32:593–6. doi: 10.1093/ajcn/32.3.593. [DOI] [PubMed] [Google Scholar]
- 195.Wing EJ, Young JB. Acute starvation protects mice against Listeria monocytogenes. Infect Immun. 1980;28:771–6. doi: 10.1128/iai.28.3.771-776.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Exton MS. Infection-induced anorexia: active host defence strategy. Appetite. 1997;29:369–83. doi: 10.1006/appe.1997.0116. [DOI] [PubMed] [Google Scholar]
- 197.Faggioni R, Fuller J, Moser A, Feingold KR, Grunfeld C. LPS-induced anorexia in leptin-deficient (ob/ob) and leptin receptor-deficient (db/db) mice. Am J Physiol. 1997;273:R181–6. doi: 10.1152/ajpregu.1997.273.1.R181. [DOI] [PubMed] [Google Scholar]
- 198.Lugarini F, Hrupka BJ, Schwartz GJ, Plata-Salaman CR, Langhans W. Acute and chronic administration of immunomodulators induces anorexia in Zucker rats. Physiol Behav. 2005;84:165–73. doi: 10.1016/j.physbeh.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 199.Kluger MJ, Ringler DH, Anver MR. Fever and survival. Science. 1975;188:166–8. [PubMed] [Google Scholar]
- 200.Hanson DF. Fever and the immune response. The effects of physiological temperatures on primary murine splenic T-cell responses in vitro. J Immunol. 1993;151:436–48. [PubMed] [Google Scholar]
- 201.Baracos VE, Whitmore WT, Gale R. The metabolic cost of fever. Can J Physiol Pharmacol. 1987;65:1248–54. doi: 10.1139/y87-199. [DOI] [PubMed] [Google Scholar]
- 202.Aronoff DM, Neilson EG. Antipyretics: mechanisms of action and clinical use in fever suppression. Am J Med. 2001;111:304–15. doi: 10.1016/s0002-9343(01)00834-8. [DOI] [PubMed] [Google Scholar]
- 203.Romanovsky AA, Shido O, Sakurada S, Sugimoto N, Nagasaka T. Endotoxin shock-associated hypothermia. How and why does it occur? Ann N Y Acad Sci. 1997;813:733–7. doi: 10.1111/j.1749-6632.1997.tb51775.x. [DOI] [PubMed] [Google Scholar]
- 204.Steiner AA, Branco LG. Hypoxia-induced anapyrexia: implications and putative mediators. Annu Rev Physiol. 2002;64:263–88. doi: 10.1146/annurev.physiol.64.081501.155856. [DOI] [PubMed] [Google Scholar]
- 205.Li J, Quan N, Bray TM. Supplementation of N-acetylcysteine normalizes lipopolysaccharide-induced nuclear factor κB activation and proinflammatory cytokine production during early rehabilitation of protein malnourished mice. J Nutr. 2002;132:3286–92. doi: 10.1093/jn/132.11.3286. [DOI] [PubMed] [Google Scholar]
- 206.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci U S A. 1997;94:2557–62. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Takahashi N, Waelput W, Guisez Y. Leptin is an endogenous protective protein against the toxicity exerted by tumor necrosis factor. J Exp Med. 1999;189:207–12. [PMC free article] [PubMed] [Google Scholar]
- 208.Oeser A, Chung CP, Asanuma Y, Avalos I. Stein CM. Obesity is an independent contributor to functional capacity and inflammation in systemic lupus erythematosus. Arthritis Rheum. 2005;52:3651–9. doi: 10.1002/art.21400. [DOI] [PubMed] [Google Scholar]
- 209.Martinez J, Sanchez-Paya J, Palazon JM, Aparicio JR, Pico A, Perez-Mateo M. Obesity: a prognostic factor of severity in acute pancreatitis. Pancreas. 1999;19:15–20. [PubMed] [Google Scholar]