

Abstract
The host innate response to viral infection includes the production of interferons, which is dependent on the coordinated activity of multiple transcription factors. Herpes simplex virus 1 (HSV-1) has been shown to block efficient interferon expression by multiple mechanisms. We and others have demonstrated that HSV-1 can inhibit the transcription of genes promoted by Interferon Regulatory Factor-3 (IRF-3), including interferon beta (IFN-β), and that the immediate-early ICP0 protein is sufficient for this function. However, the exact mechanism by which ICP0 blocks IRF-3 activity has yet to be determined. Unlike some other viral proteins that inhibit IRF-3 activity, ICP0 does not appear to affect phosphorylation and dimerization of IRF-3. Here, we show that a portion of activated IRF-3 co-localizes with nuclear foci containing ICP0 at early times after virus infection. Co-localization to ICP0-containing foci is also seen with the IRF-3-binding partners and transcriptional co-activators, CBP and p300. In addition, using immunoprecipitation of infected cell lysates, we can immunoprecipitate a complex containing ICP0, IRF-3, and CBP. Thus we hypothesize that ICP0 recruits activated IRF-3 and CBP/p300 to nuclear structures, away from the host chromatin. This leads to the inactivation and accelerated degradation of IRF-3, resulting in reduced transcription of IFN-β and an inhibition of the host response. Therefore, ICP0 provides an example of how viruses can block IFN-β induction by sequestration of important transcription factors essential for the host response.
Introduction
Innate immune responses to viral infection are multi-step processes requiring the recognition of virus infection, activation of multiple signal transduction cascades, and the transcription of antiviral genes. One of the most important proteins produced following infection, and a critical component of the innate immune response, is interferon beta (IFN-β). IFN-β is a cytokine that acts in an autocrine and paracrine manner to upregulate the expression of a number of cellular proteins with anti-viral activity (Biron and Sen, 2001). Expression of IFN-β is an essential component of the innate immune response to viral infection, as demonstrated by studies in animals and humans that have defects in the ability to make or respond to interferons (Dupuis et al., 2003; Hwang et al., 1995; Muller et al., 1994). The importance of interferon production to host defense is also apparent from the number of ways in which viruses have evolved to inhibit this response.
Because there are multiple proteins involved in the signaling pathways that lead to IFN-β production, viruses have many opportunities to target cellular proteins for inhibition. Some viruses can block recognition of viral infection, by inhibiting the signaling from Toll-like receptors (TLRs) or other pattern recognition receptors. For example, vaccinia virus and hepatitis C virus can interfere with the signaling of TLR-interacting proteins (Li et al., 2005; Stack et al., 2005; Unterstab et al., 2005). Another efficient way that viruses block the initial transcription of IFN-β is to inhibit the activity of a crucial transcription factor, interferon regulatory factor-3 (IRF-3). Functional IRF-3 is absolutely required for the immediate transcription of interferon resulting from virus infection (Sato et al., 2000; Yeow et al., 2001). Activation of IRF-3 requires phosphorylation of the C-terminus by one of the IκB kinase (IKK)-related kinases, IKKε or TANK-binding kinase 1 (Fitzgerald et al., 2003; Sharma et al., 2003). Following phosphorylation, IRF-3 undergoes a series of post-translational modifications in the process of localizing to the nucleus and associating with the CBP/p300 acetyltransferase (Wathelet et al., 1998; Yang et al., 2002; Yang et al., 2004; Yoneyama et al., 1998). IRF-3 signaling is inhibited by a wide variety of viruses at one or more steps in the activation pathway. Viruses have been shown to inhibit phosphorylation (Basler et al., 2003; Brzozka, Finke, and Conzelmann, 2005; Foy et al., 2003), dimerization (Jennings et al., 2005), nuclear translocation (Donelan et al., 2004), interaction with CBP/p300 (Burysek et al., 1999; Chakravarti et al., 1999; Jennings et al., 2005), or to decrease the cellular levels of IRF-3 (Barro and Patton, 2005; La Rocca et al., 2005). Understanding the mechanisms by which viruses inhibit IRF-3 provides insight into how viruses block the interferon response, and ultimately, of how viruses replicate successfully.
Herpes simplex virus 1 (HSV-1) is a common human pathogen that can lytically infect epithelial cells and establish latency in neurons. Soon after infection, the VP16 (α-TIF) protein stimulates the transcription of the five immediate-early (IE) genes; ICP0, -4, -22, -27, and -47 (Batterson and Roizman, 1983; Campbell, Palfreyman, and Preston, 1984; Spear and Roizman, 1972). Like other viruses, HSV-1 activates the innate immune response through interactions with TLRs on the host cell (Krug et al., 2004; Kurt-Jones et al., 2004; Lund et al., 2003). HSV-1 activates IRF-3 in a cell-type dependent manner (Preston, Harman, and Nicholl, 2001). However, activation of the immune response is far more robust in the absence of viral IE protein synthesis (Mossman et al., 2001; Nicholl, Robinson, and Preston, 2000; Preston, Harman, and Nicholl, 2001). Pretreatment of cells with interferons has been shown to decrease the expression of IE genes, and thus blocks viral replication (Mittnacht et al., 1988; Oberman and Panet, 1988; Oberman and Panet, 1989).
Multiple studies have demonstrated that the immediate-early protein ICP0 can inhibit transcription regulated by IRF-3, thereby decreasing host interferon production and attenuating the innate immune response (Eidson et al., 2002; Lin et al., 2004; Melroe, DeLuca, and Knipe, 2004). We investigated the mechanism by which HSV-1 could inhibit the activity of IRF-3 induced following activation by another virus. We previously used Sendai virus (SeV) to activate the IRF-3 signaling pathway and generate IFN-β (Melroe, DeLuca, and Knipe, 2004). We determined that HSV-1 infection can inhibit the production of interferon induced by SeV infection in HEC-1-B cells. This inhibition is enhanced by the presence of ICP0, but ICP0 is not required for this effect. We observed that the presence of ICP0 causes increased degradation of activated IRF-3 following SeV infection. In addition, activated IRF-3 does not efficiently accumulate in the nucleus of infected cells at late times post-infection in the presence of ICP0, perhaps as a consequence of the increased degradation. Studies by Mossman’s group demonstrated that HSV-1 infection can block the transcription from interferon-stimulated gene (ISG)-specific promoters (Lin et al., 2004). In their system, HSV-1 infection of A549 cells, IRF-3 fails to become hyperphosphorylated and activated following HSV-1 infection. Thus, the ICP0-enhanced destabilization of IRF-3 may involve activated and/or nuclear IRF-3.
In these studies, we further investigated the effects of ICP0 production on IRF-3 activity after phosphorylation, dimerization, and nuclear accumulation. We focused our investigation on ICP0 by using the d106 HSV-1 mutant virus, which expresses ICP0 and no other IE protein (Samaniego, Neiderhiser, and DeLuca, 1998). In addition, we used a panel of ICP0 nonsense mutants to determine the regions required for IRF-3 inhibition. We observed a change in the normal localization patterns of IRF-3 and the IRF-3-binding partner, CBP/p300, following d106 infection and ICP0 production. These cellular proteins, which are required for efficient interferon transcription, localize to punctate nuclear domains that contain ICP0. These results suggest that ICP0 functions, in part, to sequester certain cellular proteins required for efficient IFN-β gene transcription. We propose that this sequestration affects the ability of IRF-3 and CBP/p300 to promote the transcription of cellular genes, resulting in a sub-optimal interferon response.
Results
ICP0 expressed by d106 can enhance the degradation of IRF-3
We observed previously that activation of IRF-3 in the presence of ICP0 leads to increased IRF-3 degradation (Melroe, DeLuca, and Knipe, 2004). To determine if ICP0 was sufficient for this observed loss of IRF-3, we used the HSV-1 d106 virus (Samaniego, Neiderhiser, and DeLuca, 1998), which expresses ICP0 in the absence of other viral IE proteins and can block interferon induction (Eidson et al., 2002; Melroe, DeLuca, and Knipe, 2004).
We first examined IRF-3 levels in cells infected with SeV, d106, or both. At 2 and 4 hours post SeV infection, IRF-3 showed decreased mobility but constant levels (Fig. 1, lanes 2 and 3). However, at 6 and 8 hours post-infection, the amount of IRF-3 began to decrease (Fig. 1, lanes 6 and 8). With cells infected with both SeV and d106, we saw a greater decrease in levels of IRF-3 at 6 and 8 hours post-infection (Fig. 1, lanes 5 and 7). The quantification shown is representative of results obtained from multiple experiments. This decrease in IRF-3 levels was similar to what we observed previously when we infected cells with SeV and wild-type HSV-1 (Melroe, DeLuca, and Knipe, 2004). We also observed decreased levels of DNA-PKcs (Fig. 1, lanes 5 to 7), a protein whose degradation is promoted by ICP0 (Lees-Miller et al., 1996; Parkinson, Lees-Miller, and Everett, 1999). The levels of NF-κB p65 subunit remained constant (Fig. 1).
FIG. 1.


IRF-3 levels are reduced in d106-infected cells. (A) Western blot showing total levels of IRF-3 at different time points post-infection following exposure to SeV in the presence of absence of d106. Proteins in cell lysates were separated by SDS-PAGE and detected with antibodies towards DNA-PKcs, NF-kB (p65 subunit), and IRF-3. Individual bands corresponding to the proteins were scanned, and the intensity was quantified by computer analysis. The protein amounts were normalized to the amount present in mock-infected cells. The data presented are representative of multiple experiments. (B) Indirect immunofluorescence was used to determine the localization and relative amount of IRF-3 at different time points post-infection. After fixation, the cells were incubated with the SL12.1 antibody to IRF-3, followed by an Alexa Fluor 488 goat anti-mouse secondary antibody. Image exposure time was set by using the SeV-infected cells at 4 hpi and was kept constant in the other samples.
We next examined the levels and distribution of IRF-3 by using an antibody that recognizes nuclear IRF-3 (Melroe, DeLuca, and Knipe, 2004). In SeV-infected cells, IRF-3 accumulated in the nucleus by 2 hpi, but decreased over time (Fig. 1B, a to e). In cells co-infected with SeV and d106, nuclear IRF-3 was apparent by 2 hpi but then decreased by 4 hpi (Fig. 1B, g to j), consistent with our Western blot data. At 2 hpi, activated IRF-3 appeared to have two distinct localization patterns depending on the infection condition. In cells infected with SeV alone, IRF-3 was largely diffuse (Fig. 1B, b). In contrast, in cells co-infected with SeV and d106, much of the IRF-3 was localized to a number of punctate structures at 2 hpi (Fig. 1B, g).
These results indicated that ICP0, in the absence of the other IE proteins, can accelerate the degradation of activated IRF-3. In addition, we observed that infection with d106 caused a change in the distribution of IRF-3 at early times after activation.
Co-localization of ICP0, activated IRF-3, and CBP/p300 at early times post-infection
ICP0 promotes the degradation of a variety of proteins in the host cell (Hagglund and Roizman, 2004). Many cellular proteins affected by ICP0 have been found to localize to similar nuclear domains following infection, while not necessarily binding directly to ICP0 (Burch and Weller, 2004; Lomonte et al., 2004). We wished to investigate possible mechanisms by which ICP0 could affect the degradation and nuclear accumulation of IRF-3 we observed. To determine if IRF-3 co-localized with ICP0 at early times post-infection, we examined the distribution of the two proteins by immunofluorescence microscopy. SeV infection caused IRF-3 to accumulate in the nucleus (Fig. 2A, b). Cells infected with d106 alone showed no nuclear IRF-3, and therefore no co-localization with ICP0 (not shown). Following infection with both d106 and SeV, we observed IRF-3 in punctate nuclear structures (Fig. 2A, c). These areas of enhanced IRF-3 accumulation overlapped with the staining for nuclear ICP0 (Fig. 2A, f), but not all nuclear ICP0 co-localized with the nuclear IRF-3 (Fig. 2A, i). In addition, there was still a noticeable amount of diffuse IRF-3 not found in the ICP0-containing foci at these early times. These data showed that, following activation, a fraction of nuclear IRF-3 co-localized with a subset of ICP0-containing nuclear bodies.
FIG. 2.

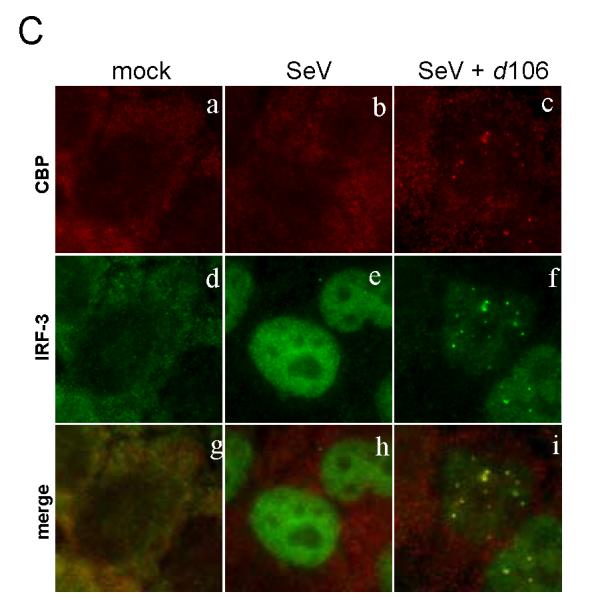
Change of IRF-3 localization in the presence of ICP0. HEC-1-B cells were infected with SeV in the presence or absence of d106. (A) The cells were fixed at 2.5 h post infection and stained with mouse anti-ICP0 and rabbit anti-IRF-3 antibodies with appropriate secondary antibodies. (B) The cells were stained with mouse anti-p300 and rabbit anti-IRF-3 antibodies with appropriate secondary antibodies. (C) The cells were stained with mouse anti-CBP and rabbit anti-IRF-3 antibodies with appropriate secondary antibodies.
Upon entering the nucleus, the activated IRF-3 dimer associates with transcriptional co-activators, either CBP or p300 (Sato et al., 1998; Wathelet et al., 1998; Yoneyama et al., 1998). Because this association follows IRF-3 activation, we determined the localization of CBP and p300 to see if ICP0 could influence IRF-3 co-localization with CBP/p300. To examine the localization of CBP and p300 following infection, we performed immunofluorescence microscopy using monoclonal antibodies specific for p300 (RW128) or CBP (AC238). The p300 monoclonal antibody was very effective at staining the nuclei of cells (Fig. 2B, a). Infection of cells with SeV alone did not change the nuclear distribution of p300 (Fig. 2B, b). Following d106 co-infection with SeV, we observed that IRF-3 localized to punctate nuclear structures (Fig. 2B, f). P300 localized to punctate nuclear structures that co-localized with the nuclear IRF-3 (Fig. 2B, i). Similar co-localization results were obtained when the CBP antibody was used (Fig. 2C). In the presence of d106 co-infection, CBP localized to punctate dots that co-localized with IRF-3 (Fig. 2C, i). This particular CBP antibody did not specifically stain the nucleus of the cells (Fig. 2C, a, b), but it was able to react with CBP by Western blot and immunoprecipitate CBP (not shown). In total, each IRF-3-containing nuclear structure also contained p300 and CBP. Therefore, it appeared that ICP0 co-localized with a fraction of activated IRF-3, and that IRF-3, in turn, co-localized with CBP and p300. Thus, ICP0 did not appear to disrupt IRF-3 and CBP/p300 association.
Treatment with MG132 prevents the d106-induced loss of IRF-3
ICP0 is known to influence the degradation of numerous cellular proteins. In addition, it has been demonstrated that the RING domain of ICP0 is required for the inhibition of IRF-3 activity (Lin et al., 2004). To determine if the loss of nuclear IRF-3 was dependent on proteasomal activity, we performed infections in the presence or absence of the proteasomal inhibitor MG132. In the absence of MG132, SeV infection caused an increase in the nuclear accumulation of IRF-3 (Fig. 3b), while co-infection with d106 inhibited efficient IRF-3 nuclear accumulation (Fig. 3c). However, in the presence of MG132, the level of nuclear IRF-3 found in cells infected with SeV and d106 was approximately equal to that of cells infected with SeV alone (Fig. 3d). MG132 treatment also prevented the localization of ICP0 to punctate structures, causing it to be diffuse (Fig. 3h). These data indicated that proteasomal activity was required for ICP0-mediated inhibition of IRF-3 nuclear accumulation.
FIG. 3.
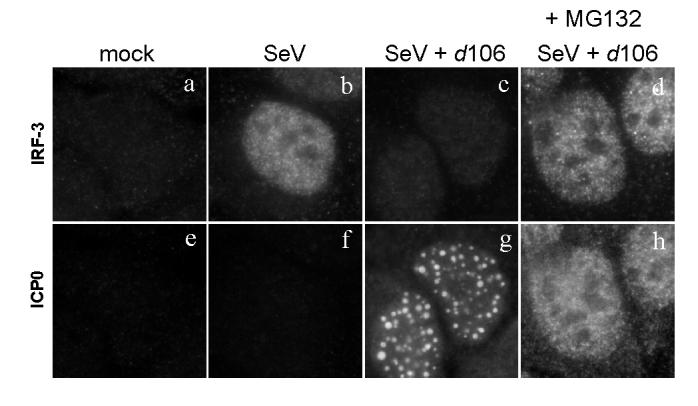
Effect of MG132 on nuclear accumulation of IRF-3. Infection of HEC-1-B cells was carried out as before, only this time the proteasomal inhibitor MG132 was added to select samples. MG132 (10μM) was added at 1 hour post infection and maintained in the medium until fixation at 6 hpi. The cells were stained with mouse anti-IRF-3 and rabbit anti-ICP0 antibodies with appropriate secondary antibodies.
Association of ICP0 with activated IRF-3 and CBP/p300 at early times post-infection
To investigate if ICP0 associated with CBP and IRF-3 in infected cells, we performed immunoprecipitation experiments with lysates from HEC-1-B cells infected by SeV with or without d106 co-infection. We observed that IRF-3 co-precipitated with the CBP immune complex when cells were infected with SeV alone (Fig. 4, lane 8), but not in mock-infected cells (lane 7). However, when cells were infected with both SeV and d106, we observed both ICP0 and IRF-3 in the immunoprecipitate (Fig. 4, lane 9). When we immunoprecipitated IRF-3 from infected cell extracts (Fig. 4, lanes 10-12), we observed that the IRF-3 antibody was able to immunoprecipitate a complex that contained CBP from SeV-infected cell lysate (lane 11), but not from mock-infected lysates (lane 10). In addition, both ICP0 and CBP associated with IRF-3 following IRF-3 immunoprecipitation from the lysates of cells co-infected with SeV and d106 (lane 12). The co-immunoprecipitation between these proteins was not disrupted by the addition of ethidium bromide or DNase treatment of the immunoprecipitation reaction (not shown), indicating that the association was not via DNA. These data argued that associations between ICP0 and activated IRF-3 and CBP/p300 could explain the recruitment of these proteins to ICP0-containing foci.
FIG. 4.

Association of ICP0 with CBP and IRF-3 in infected cells. HEC-1-B cells were infected with the indicated viruses (SeV and/or d106) and harvested at 4 hpi. Shown is the Western blot analysis of proteins immunoprecipitated using polyclonal antibodies against CBP or IRF-3. Lanes 1-6 show a sample of the whole cell lysate (5% total). The arrow indicates a cellular protein band that cross-reacts with the CBP antibody. Lanes 7-9 are lanes with proteins immunoprecipitated using an anti-CBP antibody. Lanes 10-12 are lanes with proteins immunoprecipitated using an anti-IRF-3 antibody. The blot was probed with antibodies specific for CBP (top panel), ICP0 (middle panel) or IRF-3 (lower panel).
IRF-3 co-localizes with ICP0 expressed from a transfected plasmid
To determine if ICP0, in the absence of any other SeV or HSV-1 proteins, could recruit activated IRF-3, we transfected cells with a plasmid expressing ICP0. To ensure that proteins produced by SeV were not affecting ICP0 activity, we activated IRF-3 using polyinosinic-polycytidylic acid (poly I:C), an artificial mimic of double-stranded RNA treatment (Wathelet et al., 1988). At eighteen hours after HEC-1-B cells were transfected with an ICP0-expressing plasmid, the cells were treated with poly I:C. Treatment with poly I:C led to the activation of IRF-3 in about 30% of the cells (Fig. 5b). Transfection with the ICP0-expressing plasmid alone did not lead to the activation of IRF-3 so there was no co-localization between the two proteins (Fig. 5k). Much of the IRF-3 activated by poly I:C in the presence of ICP0 localized to distinct nuclear foci (Fig. 5d). These foci localized with or near the nuclear ICP0 foci (Fig. 5f).
FIG. 5.
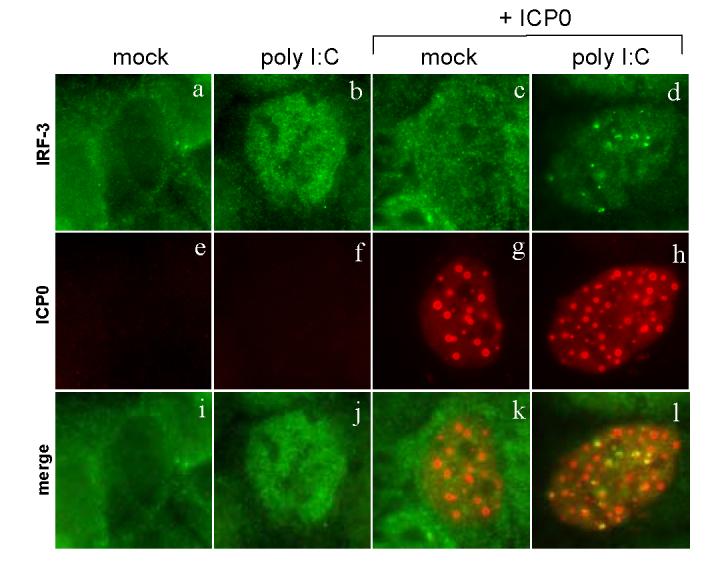
Change of IRF-3 localization in the presence of ICP0 only. HEC-1-B cells were transfected with an ICP0-expressing plasmid at 18 hours prior to treatment. The cells were treated with poly I:C (100 μg/ml) in the presence or absence of transfected ICP0. The cells were fixed at 4 h post treatment and stained with mouse anti-ICP0 and rabbit anti-IRF-3 antibodies with appropriate secondary antibodies.
These results indicated that no other herpes viral proteins were required for the co-localization of ICP0 and IRF-3. In addition, these data argued that the association between ICP0 and IRF-3 was not an artifact of SeV co-infection.
Activation of IRF-3 is required for co-localization
Non-activated IRF-3 is in a conformation thought to be folded upon itself, hiding the domains required for IRF-3 dimerization and DNA binding. This non-active form of IRF-3 shuttles in and out of the nucleus (Lin et al., 1998; Lin, Mamane, and Hiscott, 1999; Yoneyama et al., 1998)). To determine if IRF-3 needs to be activated to co-localize with ICP0, we treated infected cells with an inhibitor of IRF-3 nuclear export, leptomycin B (LepB), which causes nuclear accumulation of non-activated IRF-3 (Karpova et al., 2002).
Cells were transfected with plasmids expressing IRF-3 or both ICP0 and IRF-3, treated with LepB (Fig. 6, right panels) or left untreated (left panels), and infected with SeV as indicated. In cells transfected with IRF-3 alone, IRF-3 localized mostly to the cytoplasm (Fig. 6i). In cells treated with LepB, IRF-3 localized strongly to the nucleus, but there was still a considerable amount in the cytosol (Fig. 6m). SeV infection increased the amount of IRF-3 found in the nucleus of LepB treated cells (Fig. 6n). The nuclear IRF-3 induced by LepB treatment did not co-localize with ICP0 (Fig. 6o). This nuclear IRF-3 was diffuse and did not localize to distinct nuclear structures. In contrast, ICP0 and IRF-3 co-localized when IRF-3 was activated by SeV infection (Fig. 6, t and x). These data supported the hypothesis that IRF-3 must be activated to co-localize with ICP0.
FIG. 6.

ICP0 co-localizes only with activated IRF-3. HEC-1-B cells were infected with SeV (indicated at the top of the columns) or mock infected. Cells transfected with IRF-3 (pcDNA-IRF-3) and ICP0 (pICP0) expression plasmids were infected at 18 hours following transfection. Cells were treated with leptomycin B (LepB) for 2 h before infection (right panels). LepB levels were maintained during and after infection. Cells infected with SeV were fixed at 3 h after infection and stained with mouse anti-ICP0 and rabbit anti-IRF-3 anitbodies with appropriate secondary antibodies.
Activated IRF-3 co-localizes with ICP0 in HSV-1 infected cells
To ensure that the association between ICP0 and IRF-3 occurs during wild-type HSV-1 infection, we used a cell type, human BJ fibroblasts, in which HSV-1 can activate the interferon response (Preston, Harman, and Nicholl, 2001), and we used immunofluorescence microscopy to view the localization of IRF-3 and ICP0 in BJ fibroblasts infected under a variety of conditions. Mock-infected cells showed a cytoplasmic localization of IRF-3 (Fig. 7a). In BJ fibroblasts infected with wt-HSV-1, we observed no nuclear accumulation of IRF-3 (Fig. 7b). We did observe ICP0 production and localization to the cytoplasm (Fig. 7g). The addition of CHX at the time of HSV-1 infection blocked viral protein expression (Fig. 7h), which allowed activation of IRF-3 (Fig. 7c).
FIG. 7.

Localization of IRF-3 to early sites of viral replication. HEp-2 cells were infected with wild-type HSV-1 (KOS) in the presence or absence of cycloheximide treatment (50 μg/ml). The CHX treatment was either maintained throughout infection (KOS +) or removed and replaced with normal media at 3.5 hpi (mock and KOS +/-). At 6 hours post infection or mock treatment, the cells were stained with mouse anti-ICP0 and rabbit anti-IRF-3 antibodies with appropriate secondary antibodies.
To determine if activated IRF-3 co-localized with ICP0, we varied the infection conditions so that we would see IRF-3 activation as well as ICP0 production. To do this, we infected cells with HSV-1 in the presence of CHX for 3.5 hours, then removed the CHX to allow translation to take place for 2.5 hours. Mock-infected cells treated with CHX did not show IRF-3 activation after CHX removal (Fig. 7d). In the HSV-1-infected cells, we observed IRF-3 activation and ICP0 production (Fig. 7, e and j). A large portion of the activated IRF-3 co-localized with the ICP0 foci (Fig. 7o). Thus, activated IRF-3 co-localized with ICP0 in HSV-1 infected cells. This recruitment required IRF-3 to be activated, as no recruitment was seen in cells infected with HSV-1 in the absence of CHX.
Residues 680-720 of ICP0 are required for inhibition of IRF-3 nuclear accumulation
To define the domains of ICP0 needed for IRF-3 inhibition, we tested several nonsense mutant forms of ICP0 (Cai et al., 1993; Cai and Schaffer, 1992). Immunofluorescence microscopy was performed at 6 hpi to determine the levels and localization of IRF-3 in cells infected with the various viruses. Co-staining with ICP8 antibody was performed to verify virus infection. Of the nonsense mutants tested, only n720 and n770 blocked IRF-3 nuclear accumulation like wild-type HSV-1 (Fig. 8, f and g). All of the other mutants, while they were able to infect cells and produce ICP8 (Fig. 8, i to l), were not able to inhibit the nuclear accumulation of IRF-3 (Fig. 8, b to e). In most of these cases, the nuclear IRF-3 co-localized with ICP8 (Fig. 8, b to d), while co-infection with n680 led to a diffuse nulcear IRF-3 localization (Fig. 8e). Thus, residues 680-720 of ICP0 are required for the inhibition of IRF-3 nuclear accumulation. This portion of ICP0 contains a region required for localization of ICP0 to ND10 foci (Ciufo, Mullen, and Hayward, 1994).
FIG. 8.

Effect of ICP0-mutant virus infection on IRF-3 nuclear accumulation. HEC-1-B cells were infected with SeV in the presence of a panel of ICP0 nonsense mutant viruses (n212, n428, n525, n680, n720, and n770). The cells were fixed at 6 hours post co-infection and stained with mouse anti-IRF-3 and rabbit anti-ICP8 antibodies with appropriate secondary antibodies. Shown also is the merged image.
To determine if n680 ICP0 also lacked the ability to localize to nuclear structure, we characterized the ICP0 nonsense mutants for their ability to co-localize with IRF-3 at an early time post infection. Following HSV-1 infection of HEp-2 cells, a very small percentage of the cells showed IRF-3 activation. Thus, in HEp-2 cells infected with wt HSV-1, we observed IRF-3 activation in only approximately 1 - 3% of the cells. In the cases where IRF-3 was activated, it accumulated in the nucleus and localized to punctate structures (Fig. 9c). These structures co-localized with the nuclear ICP0 (Fig. 9o). We observed that the n680 ICP0 did not co-localize with IRF-3 at early times post infection (Fig. 9p). In this case, the ICP0 did not localize to punctate structures in the nucleus (Fig. 9j). Thus, localization of ICP0 to nuclear foci correlates with its inhibition of IRF-3 accumulation.
FIG. 9.
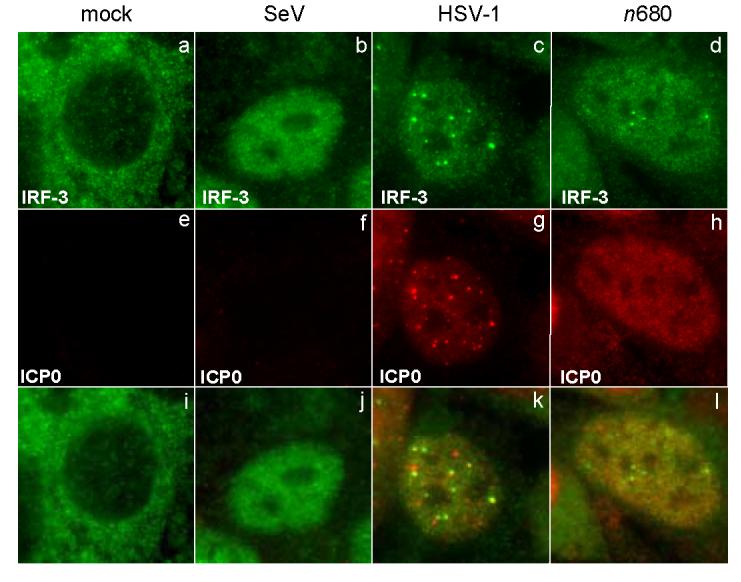
Effect of ICP0-mutant virus infection on localization of activated IRF-3. HEp-2 cells were infected with either SeV, HSV-1 (KOS strain), or n680. The cells were fixed at 2.5 hours post infection and stained with mouse anti-ICP0 and rabbit anti-IRF-3 antibodies with appropriate secondary antibodies. Shown also is the merged image.
Absence of recruitment of other enhanceosome proteins
A variety of proteins are recruited to ICP0-containing bodies in the infected cell nucleas (Burch and Weller, 2004; Everett, 2000; Hagglund and Roizman, 2004). To examine the specificity of the recruitment of CBP/p300 to activated IRF-3 in the nuclear foci, we looked at the localization of other members of the IFN-β transcriptional complex. Both NF-κB (p65 subunit) and a member of the AP-1 complex, ATF2, are activated following SeV infection and should form a complex with activated IRF-3 and CBP/p300 to cooperatively enhance the transcription of the IFN-β gene (Merika and Thanos, 2001).
In cells co-infected with SeV and HSV-1, we observed that IRF-3 localized to the nucleus and co-localed with ICP0 (Fig. 10g). However neither NF-κB (Fig. 10e) nor ATF2 (Fig. 10f) co-localized with IRF-3; instead, they were localized diffusely throughout the nucleus. If NF-kB or ATF2 were being recruited to ICP0 foci, we would have expected to see a co-localization with IRF-3.
FIG. 10.
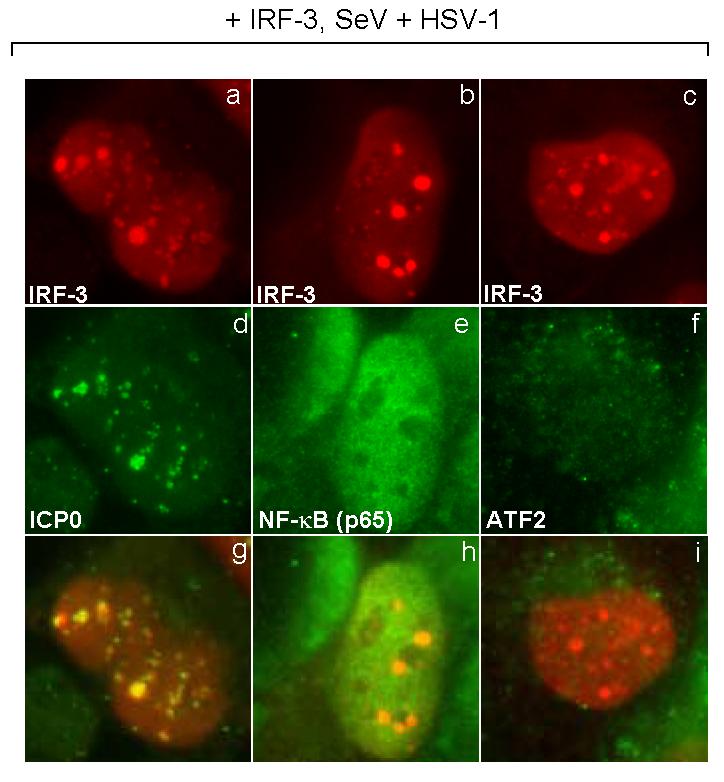
Localization of members of the enhanceosome complex. HEC-1-B cells were transfected with a plasmid expressing IRF-3. At eighteen hours after transfection, the cells were infected with HSV-1 (KOS strain) and SeV. The cells were fixed at 3 hours post-infection and stained with mouse anti-IRF-3 (a to c) and rabbit anti-ICP0 (d), -NF-kB (p65 subunit) (e), or -ATF2 (f) anitbodies with appropriate secondary antibodies. Shown also is the merged image.
In the absence of ICP0, activated IRF-3 is segregated from the host cell chromatin during HSV-1 infection
We have shown that ICP0-mutant viruses are still able to inhibit IRF-3 activity induced by SeV infection (Melroe, DeLuca, and Knipe, 2004). There are multiple ways by which this inhibition may occur. We therefore used immunofluorescence microscopy to determine the localization of IRF-3 at later times after infection. Histone H1 antibody staining was used as a marker for the location of host cell chromatin. In mock-infected cells, histone staining was primarily nuclear (Fig. 11e), while IRF-3 staining was cytoplasmic (Fig. 11a). Following SeV infection, IRF-3 entered the nucleus and co-localized with histones (Fig. 11j). In cells infected with both SeV and HSV-1, we observed a loss of nuclear IRF-3 (Fig. 11c) and marginalization of the histones to the periphery of the nucleus (Fig. 11g). Co-infection with the ICP0-mutant virus (7134) did not lead to a loss of nuclear IRF-3 (Fig. 11d). Instead, IRF-3 localized to viral replication compartments, segregated from the host cell histones (Fig. 11).
FIG. 11.
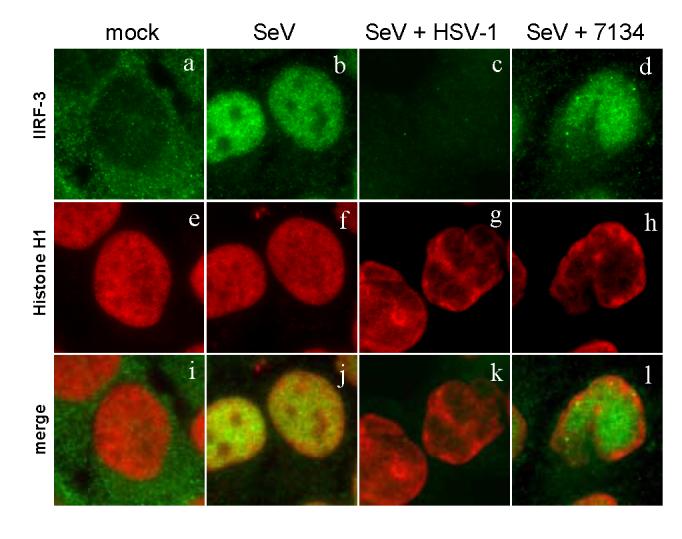
Sequestration of IRF-3 in cells infected with an ICP0 mutant. HEC-1-B cells were infected with various virus combinations as indicated at the top of each column. At 6 hours post infection or mock treatment, the cells were fixed and stained with mouse anti-Histone H1 and rabbit anti-IRF-3 antibodies with appropriate secondary antibodies.
Discussion
HSV ICP0, in addition to functioning as a promiscuous transcriptional transactivator (Everett, 1984; Gelman and Silverstein, 1985; Nabel et al., 1988; O′Hare and Hayward, 1985; Quinlan and Knipe, 1985), has been shown to play a central role in the resistance of HSV-1 to interferon-mediated anti-viral activity (Harle et al., 2002; Mossman, Saffran, and Smiley, 2000). What is not known, however, is the mechanism by which ICP0 performs this function. We had previously shown that ICP0 can block nuclear accumulation of IRF-3 and IFN-β induction (Melroe, DeLuca, and Knipe, 2004). We demonstrate here that ICP0 associates with both CBP/p300 and IRF-3 following the activation of IRF-3. We observed that activated IRF-3 enters the nucleus, co-localizes with ICP0, and then becomes destroyed in a proteasomal-dependent fashion. This series of events is seen under a variety of different conditions of ICP0 expression and IRF-3 activation. These data therefore argue that one possible mechanism by which ICP0 inhibits the efficient transcription of interferon is by recruiting activated IRF-3 and CBP away from host cellular promoters at early times after infection and then promoting their inactivation. This inhibition could be due to the degradation of IRF-3 and/or an undefined ICP0-dependent post-translational modification of IRF-3 or CBP/p300.
Recruitment of activated IRF-3 to nuclear ICP0 sites
The co-localization and association of activated IRF-3 and ICPO that we have observed suggests that it is possible that IRF-3 is associating directly with ICP0. Another possibility is that IRF-3 is interacting with another protein recruited to ICP0-containing foci. We speculate that the efficient recruitment ability of ICP0 depends on at least two domains: the RING domain and the C-terminal multimerization domain. The RING family of zinc finger domains have been reported to play a role in the formation of large multi-protein complexes (Matthews and sunce, 2002).
Mossman’s group reported that the RING finger domain is required for the efficient inhibition of IRF-3 by ICP0, although they see, at most, a five-fold inhibition of IRF-3 activity (Lin et al., 2004). Disruption of the RING finger domain ablates nearly all functions of ICP0 measured to date; therefore, the finding that it is required for the inhibition of IRF-3 could be expected (Everett, 2000).
ICP0 localizes to PML-containing ND10 domains at early times of infection (Everett and Maul, 1994; Maul and Everett, 1994; Maul, Guldner, and Spivack, 1993). However, IRF-3 has never been shown to accumulate at these domains following activation. Due to the large quantities of CBP at ND10 domains (LaMorte et al., 1998), it is possible that IRF-3 normally localizes transiently to these foci after activation. Therefore, the possibility exists that PML, the scaffold of ND10 domains, is required for the efficient association between ICP0 and IRF-3. We believe this requirement for PML is unlikely for two reasons: (i) we observed that the association between ICP0 and IRF-3 occurs after the destruction of PML and (ii) others have reported that ICP0 can form non-ND10-associated foci (Everett et al., 1999; Morency et al., 2005).
In our immunofluorescence studies we do not see a total sequestration of activated IRF-3 with ICP0 at early times after infection, arguing for a case of gradual recruitment. One possible reason for the diffuse localization is that the amount of nuclear IRF-3 could be higher than the amount of nuclear ICP0 at very early times after infection. It may take time for ICP0 to clear the larger quantity of nuclear IRF-3. That initial period where IRF-3 is diffuse throughout the nucleus could explain the observation that ICP0 only inhibits the activity of IRF-3 by, at most, five-fold (Lin et al., 2004). Alternatively, it is still possible that the diffuse ICP0 is interacting with the diffuse IRF-3. In the context of cellular infection, these early interactions may be enough to give HSV-1 the head start it needs to replicate and spread. Thus, the spatial and temporal regulation of ICP0 and IRF-3 are important factors to consider.
We observed that CBP/p300 can be found in the complex that contains both ICP0 and IRF-3. Recently it has been reported that the bovine herpesvirus 1 bICP0 protein interacts with p300 (Zhang et al., 2006). The histone deacetylase and scaffolding functions of CBP and p300 are very important for the efficient transcription of numerous cellular proteins (Chan and La Thangue, 2001; Kalkhoven, 2004). Reducing the amount of CBP available for cellular transcription may help to create an environment more suitable for HSV-1 replication. As stated previously, CBP/p300 is thought to be used by HSV-1 during infection (Herrera and Triezenberg, 2004). Our findings support the possibility that there is competition for these transcription factors following IRF-3 activation.
Physical separation of IRF-3 from the host chromatin is also observed when cells are infected with an ICP0 mutant virus. This altered localization may play a role in the inhibition of interferon transcription seen previously (Melroe, DeLuca, and Knipe, 2004). These results compliment previous work in that they provide an explanation as to how HSV-1 can inhibit IRF-3 activity in the absence of ICP0.
The ability to recruit IRF-3 to nuclear sites away from cellular chromatin is a new mechanism by which ICP0 can inhibit or alter the function of select cellular proteins. ICP0 has been shown to recruit other cellular proteins without greatly affecting their stability (Burch and Weller, 2004). Hence, it is likely that the RING finger domain of ICP0 functions as a domain promoting the formation of larger complexes. The evidence that neither NF-kB (p65) nor ATF2 co-localize with the activated IRF-3 in ICP0-foci argue against the non-specific recruitment of proteins to this area. The question remains as to how ICP0 is able to specifically recruit certain cellular proteins while excluding others. Identification of more proteins that localize to these structures may give a better picture of the function of these domains and this specificity.
Degradation of activated IRF-3 is promoted by ICP0
Following the dsRNA-induced activation of IRF-3, its degradation occurs in a proteasomal-dependent fashion as a means to attenuate the immune response (Lin et al., 1998; Saitoh et al., 2006). During HSV-1 infection, following the recruitment of activated IRF-3, we believe ICP0 is promoting the negative regulation of IRF-3. ICP0 is likely using its E3 ubiquitin-ligase activity to positively influence the ubiquitination and degradation of IRF-3 using the host proteasomal machinery. The reported presence of the proteasomal proteins in ICP0-containing foci may explain the higher rate of degradation of select cellular proteins (Burch and Weller, 2004).
Alternate explanations for the inhibition of nuclear accumulation is that the association of IRF-3 with ICP0 may result in a post-translational modification that would send IRF-3 back to the cytoplasm, thus reducing the nuclear levels or ICP0 may shuttle IRF-3 to the ctyoplasm. A similar situation is seen when IRF-3 is activated in the absence of DNA-PK in some cells (Karpova et al., 2002).
The localization of nuclear IRF-3 during infection with an ICP0-mutant virus helps to explain how HSV-1 inhibits the activity of IRF-3 in the absence of degradation. The incorporation of IRF-3 into replication compartments during DNA replication would make it unavailable to promote the transcription of interferon at later times. IRF-3 may be able to promote transcription at earlier times, before the formation of replication compartments. It is likely that the vhs protein would destroy these transcripts (Elgadi and Smiley, 1999; Lin et al., 2004; Strelow and Leib, 1995). In addition, it is possible that this replication compartment-associated IRF-3 negatively affects HSV-1 replication.
The failure of the n680 mutant to inhibit IRF-3 nuclear accumulation suggests that association with ND10 nuclear structures is a necessary prerequisite for IRF-3 inhibition. Another study tested a variety of ICP0 mutants, including C-terminal truncation mutants containing only the first 593 residues of ICP0, and observed that the only region of ICP0 required for the inhibition of IRF-3 activity is the N-terminal RING finger domain (Lin et al., 2004). If ICP0-IRF-3 association is required for efficient inhibition of interferon production, why are C-terminal mutants still able to inhibit IRF-3 activity? The answer may lie in the ways the experiments were conducted. In transfection systems, there is a lot more ICP0 present than what is found in infected cells. This is especially true for cells infected with a virus at a low multiplicity of infection when the defect found in ICP0 mutants is most apparent. Furthermore, in transfected cells, the ICP0 has been present for a much longer time, amplifying its possible deleterious effects on the cell. Therefore, we believe that in infected cells, the multimerization/ND10 localization domain of ICP0 is required for optimal ICP0 inhibition of IRF-3 activity. However, this requirement may not be apparent in a transfection situation.
Our proposed mechanism by which ICP0 inhibits IRF-3 activity may be a common scheme used by other proteins as well. It has been recently shown that the host Pin1 protein will specifically interact with activated IRF-3 and, following an induced conformational change, make IRF-3 more susceptible to destruction (Saitoh et al., 2006). The rotavirus NSP1 protein has been shown to associate with IRF-3 by yeast two-hybrid (Graff et al., 2002) and this association leads to the increased degradation of activated IRF-3 (Barro and Patton, 2005). Overall, the presence of the NSP1 protein is responsible for the inhibition of interferon production.
These results provide insight into the sequestration ability of ICP0 and how it contributes to the inhibition of the innate immune response. Recruitment of activated IRF-3 and CBP/p300 and accelerated degradation of IRF-3 joins the growing list of reported functions of ICP0 and allows us to hypothesize a mechanism by which ICP0 inhibits IRF-3 function and creates a favorable environment for viral transcription and replication.
Materials and Methods
Cells and viruses
Human endometrial adenocarcinoma (HEC-1-B), BJ fibroblast, and HEp-2 cells were obtained from the American Type Culture Collection (Manassas, VA) (ATCC numbers HTB-113, CRL-2522, and CCL-23, respectively). The cells were grown as monolayer cultures in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 5% heat-inactivated bovine calf serum (HyClone), penicillin (100 U/ml), and streptomycin (100 μg/ml) in a humidified 5% CO2 atmosphere at 37°C.
The HSV-1 wild type (wt) KOS virus strain was propagated and titrated as described previously (Knipe and Spang, 1982). The 7134 (ICP0 null) virus (Cai and Schaffer, 1989) and the ICP0 nonsense mutants (n212, n428, n525, n680, n720, and n770) were described in the indicated references. The HSV-1 d106 virus has been described elsewhere (Samaniego, Neiderhiser, and DeLuca, 1998). Sendai virus (SeV) Cantel strain was obtained from Charles River SPAFAS (Wilmington, MA).
Plasmids and Antibodies
The ICP0 expression vector (pICP0) was constructed by inserting the KpnI fragment from pSG1-ES1 (Quinlan and Knipe, 1985) containing the ICP0 gene into pUC19 (New England Biolabs, Beverly, MA). The IRF-3 expression vector was provided by Peter Howley (Harvard Medial School, Boston, MA). Transfections were carried out using Lipofectamine 2000 following the manufacturer’s protocol (Invitrogen, San Diego, CA).
Mouse monoclonal antibodies were used as follows: Anti-ICP0 (5H7 - East Coast Biologics, Inc., North Berwick, ME) used at 1:200 dilution for immunofluorescence, and 1:1000 dilution for Western blots. Anti-IRF-3 (SL12.1 -BD PharMingen, San Diego, CA) used at 1:100 for immunofluorescence, and 1:1000 for Western blots. Anti-CBP (C-1 -Santa Cruz Biotechnology, Inc., Santa Cruz, CA) used at 1:50 for immunofluorescence. Anti-CBP (AC238, provided by James DeCaprio, Dana Farber Cancer Institute, Boston, MA) used at 1:10 for immunofluorescence. Anti-p300 (RW128, provided by James DeCaprio, Dana Farber Cancer Institute, Boston, MA) used at 1:10 for immunofluorescence.
Rabbit polyclonal antibodies were used as follows: Anti-ICP0 (provided by Bernard Roizman, The University of Chicago, Chicago, IL) used at 1:1000 for immunofluorescence. Anti-IRF-3 (FL425 - Santa Cruz Biotechnology, Inc., Santa Cruz, CA) used at 1:50 for immunofluorescence. Anti-CBP (A-22 - Santa Cruz Biotechnology, Inc., Santa Cruz, CA) used at 1:50 for immunofluorescence, and 1:500 dilution for Western blots. Anti-ICP8 (3-83; (Knipe et al., 1987) used at 1:200 for immunofluorescence. Anti-p300 (N-15 - Santa Cruz Biotechnology, Inc., Santa Cruz, CA) used at 1:50 for immunofluorescence. Anti-NF-kB p65 (C-20 - Santa Cruz Biotechnology, Inc., Santa Cruz, CA) used at 1:50 for immunofluorescence. Anti-ATF2 (C-19 - Santa Cruz Biotechnology, Inc., Santa Cruz, CA) used at 1:50 for immunofluorescence.
Viral infections
HEC-1-B cells were plated into 24 well culture dishes on 12 mm circle cover glass (for immunofluorescence), 100 mm culture dishes (for immunoprecipitation), or 6 well culture dishes (for Western blot) 24 h prior to infection to obtain 90% confluence at the time of infection. Cells were infected with either wild-type or mutant HSV-1 virus strains at a multiplicity of infection of 20 plaque-forming units (PFU, as titrated on Vero cells (KOS) or V529 cells (d106)) per cell in cold phosphate-buffered saline (PBS) containing 0.1% glucose and 1% heat-inactivated fetal bovine serum. As indicated, cells were infected with 100 hemagglutination units (HAU)/ml of SeV (as determined by Charles River Labs). After 1 h of adsorption at 37°C, cells were overlaid with Dulbecco’s modified Eagle’s medium containing 1% heat-inactivated fetal bovine serum and maintained at 37°C.
Immunofluorescence
For indirect immunofluorescence, HEC-1-B monolayers were fixed for 15 min in PBS with 3.7% formaldehyde and permeabilized for 2 min with ice-cold methanol. Alexa Fluor 488- or Alexa Fluor 594-conjugated goat anti-rabbit or goat anti-mouse secondary antibodies (Molecular Probes, Eugene, OR) were used at a 1:1000 dilution. Coverslips were mounted on glass slides in ProLong antifade agent (Molecular Probes, Eugene, OR). Images were obtained with a Zeiss Axioplan 2 microscope using a Hamamatsu digital camera (C4742-95) and OpenLab software (version 3.1.7; Improvision, Lexington, MA).
Immunoprecipitation and western blot analyses
Cells were harvested by scraping into media. After two washes in cold PBS, cells from each dish were incubated on ice for 30 min in 1 ml of immunoprecipitation (IP) lysis buffer (20 mM Tris-HCl [pH 7.5], 50 mM NaCl, 0.1% NP-40, 10 mM β-glycerophosphate, 5 mM NaF, 1 mM PMSF, 2.5% glycerol, and 1 Complete mini protease inhibitor cocktail tablet [Roche Applied Science, Indianapolis, Ind.] per 10 ml). Cell lysates were clarified by centrifugation at 10K X g at 4°C for 5 min, and precleared overnight by incubating with protein A-agarose beads at 4°C. Forty microliters of precleared lysate was set aside, and immunoprecipitation was carried out with either the polyclonal IRF-3 or CBP antibody (1:50 dilution) and protein A-agarose beads at 4°C for 2-4 h. After four washes in washing buffer (20 mM Tris-HCl [pH 7.5], 50 mM NaCl, 0.1% NP-40, and 1 mM PMSF), the immunoprecipitates were dissolved in gel sample buffer (Knipe and Spang, 1982) for separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in a 9% bis-crosslinked polyacrylamide gel.
After electrophoresis, the proteins were transferred onto a nitrocellulose membrane (Protran; Schleicher & Schuell Bioscience, Keene, NH) by electroblotting at 40V overnight. The membranes were blocked in 5% milk in Tris-buffered saline (TBS), probed for the appropriate antibody in TBS containing 0.1% Tween 20, and stained with enhanced chemiluminescence (ECL) Western blotting detection reagents (PerkinElmer, Boston, MA) in accordance with the manufacturer’s protocol.
Western blot for IRF-3 levels
Cells were harvested at various time points post infection by scraping into media. After two washes in cold PBS, cells from each well were incubated on ice for 30 min in immunoprecipitation (IP) lysis buffer. Cell lysates were clarified by centrifugation at 10K X g at 4°C for 5 min, and the resulting extract was dissolved in gel sample buffer (Knipe and Spang, 1982) for separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in a 9% bis-crosslinked polyacrylamide gel. Protein transfer and blotting was performed as described above.
Quantification of relative cellular levels of IRF-3 and DNA-PKcs by measurement of intensity of Western blot staining has been described previously (Melroe, DeLuca, and Knipe, 2004). Briefly, the developed film was converted into a high-resolution digital image (UMAX scanner UTA-MII). The intensity of each band was determined by using image-scanning densitometer software (ImageQuant TL; GE Healthcare, Piscataway, NJ).
Acknowledgments
This research was supported by the National Institutes of Health grant AI20530 to DMK.
We thank Bernard Roizman for providing the polyclonal ICP0 antibody, James DeCaprio for providing the monoclonal CBP and p300 antibodies, Nicholas Nagykery for technical assistance, and Lisa Holik for help in preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Barro M, Patton JT. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc Nat Acad Sci USA. 2005;102:4114–9. doi: 10.1073/pnas.0408376102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, Klenk HD, Palese P, Garcia-Sastre A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol. 2003;77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batterson W, Roizman B. Characterization of the herpes simplex virionassociated factor responsible for the induction of alpha genes. J Virol. 1983;46:371–377. doi: 10.1128/jvi.46.2.371-377.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron CA, Sen GC. Interferons and other cytokines. In: Knipe DM, Howley PM, editors. “Fields Virology”. 4th ed. Vol. 1. Lippincott Williams & Wilkins; Philadelphia, PA.: 2001. pp. 321–351. [Google Scholar]
- Brzozka K, Finke S, Conzelmann KK. Identification of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J Virol. 2005;79:7673–81. doi: 10.1128/JVI.79.12.7673-7681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch AD, Weller SK. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J Virol. 2004;78:7175–85. doi: 10.1128/JVI.78.13.7175-7185.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burysek L, Yeow WS, Lubyova B, Kellum M, Schafer SL, Huang YQ, Pitha PM. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J Virol. 1999;73:7334–42. doi: 10.1128/jvi.73.9.7334-7342.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Astor TL, Liptak LM, Cho C, Coen DM, Schaffer PA. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J Virol. 1993;67(12):7501–7512. doi: 10.1128/jvi.67.12.7501-7512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Schaffer PA. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J Virol. 1992;66:2904–2915. doi: 10.1128/jvi.66.5.2904-2915.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai WZ, Schaffer PA. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J Virol. 1989;63:4579–4589. doi: 10.1128/jvi.63.11.4579-4589.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MEM, Palfreyman JW, Preston CM. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. Journal of Molecular Biology. 1984;180:1–19. doi: 10.1016/0022-2836(84)90427-3. [DOI] [PubMed] [Google Scholar]
- Chakravarti D, Ogryzko V, Kao HY, Nash A, Chen H, Nakatani Y, Evans RM. A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell. 1999;96:393–403. doi: 10.1016/s0092-8674(00)80552-8. [DOI] [PubMed] [Google Scholar]
- Chan HM, La Thangue, N. B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363–73. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- Ciufo DM, Mullen MA, Hayward GS. Identification of a dimerization domain in the C-terminal segment of the IE110 transactivator protein from herpes simplex virus. J Virol. 1994;68(5):3267–3282. doi: 10.1128/jvi.68.5.3267-3282.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donelan NR, Dauber B, Wang X, Basler CF, Wolff T, Garcia-Sastre A. The N- and C-terminal domains of the NS1 protein of influenza B virus can independently inhibit IRF-3 and beta interferon promoter activation. J Virol. 2004;78:11574–82. doi: 10.1128/JVI.78.21.11574-11582.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, Al Ghonaium A, Tufenkeji H, Frayha H, Al-Gazlan S, Al-Rayes H, Schreiber RD, Gresser I, Casanova JL. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33:388–91. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- Eidson KM, Hobbs WE, Manning BJ, Carlson P, DeLuca NA. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral Infection. J Virol. 2002;76:2180–2191. doi: 10.1128/jvi.76.5.2180-2191.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgadi MM, Smiley JR. Picornavirus internal ribosome entry site elements target RNA cleavage events induced by the herpes simplex virus virion host shutoff protein. J Virol. 1999;73:9222–9231. doi: 10.1128/jvi.73.11.9222-9231.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD. Transactivation of transcription by herpes virus products: requirement for two HSV-1 immediate-early polypeptides for maximum activity. EMBO Journal. 1984;3:3135–3141. doi: 10.1002/j.1460-2075.1984.tb02270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays. 2000;22:761–70. doi: 10.1002/1521-1878(200008)22:8<761::AID-BIES10>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Everett RD, Earnshaw WC, Findlay J, Lomonte P. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO Journal. 1999;18:1526–1538. doi: 10.1093/emboj/18.6.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Maul GG. HSV-1 IE protein VMW 110 causes redistribution of PML. EMBO Journal. 1994;13:5062–5069. doi: 10.1002/j.1460-2075.1994.tb06835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nature Immunology. 2003;4:491–6. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Foy E, Li K, Wang C, Sumpter RJ, Ikeda M, Lemon SM, Gale MJ. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- Gelman IH, Silverstein S. Identification of immediate early genes from herpes simplex virus that transactivate the virus thymidine kinase gene. Proc Natl Acad Sci U S A. 1985;82:5265–5269. doi: 10.1073/pnas.82.16.5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff JW, Mitzel DN, Weisend CM, Flenniken ML, Hardy ME. Interferon regulatory factor 3 is a cellular partner of rotavirus NSP1. J Virol. 2002;76:9545–50. doi: 10.1128/JVI.76.18.9545-9550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagglund R, Roizman B. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J Virol. 2004;78:2169–2178. doi: 10.1128/JVI.78.5.2169-2178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harle P, Sainz B, Jr., Carr DJ, Halford WP. The immediate-early protein, ICP0, is essential for the resistance of herpes simplex virus to interferon-alpha/beta. Virology. 2002;293:295–304. doi: 10.1006/viro.2001.1280. [DOI] [PubMed] [Google Scholar]
- Herrera FJ, Triezenberg SJ. VP16-dependent association of chromatinmodifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J Virol. 2004;78:9689–96. doi: 10.1128/JVI.78.18.9689-9696.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SY, Hertzog PJ, Holland KA, Sumarsono SH, Tymms MJ, Hamilton JA, Whitty G, Bertoncello I, Kola I. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci U S A. 1995;92:11284–8. doi: 10.1073/pnas.92.24.11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings S, Martinez-Sobrido L, Garcia-Sastre A, Weber F, Kochs G. Thogoto virus ML protein suppresses IRF3 function. Virology. 2005;331:63–72. doi: 10.1016/j.virol.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Kalkhoven E. CBP and p300: HATs for different occasions. Biochem Pharmacol. 2004;68:1145–55. doi: 10.1016/j.bcp.2004.03.045. [DOI] [PubMed] [Google Scholar]
- Karpova AY, Trost M, Murray JM, Cantley LC, Howley PM. Interferon regulatory factor-3 is an in vivo target of DNA-PK. Proc Natl Acad Sci USA. 2002;99:2818–2823. doi: 10.1073/pnas.052713899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe DM, Senechek D, Rice SA, Smith JL. Stages in the nuclear association of the herpes simplex virus transcriptional activator protein ICP4. J Virol. 1987;61:276–284. doi: 10.1128/jvi.61.2.276-284.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe DM, Spang AE. Definition of a series of stages in the association of two herpesviral proteins with the cell nucleus. J Virol. 1982;43:314–324. doi: 10.1128/jvi.43.1.314-324.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood. 2004;103:1433–7. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci USA. 2004;101:1315–20. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rocca SA, Herbert RJ, Crooke H, Drew TW, Wileman TE, Powell PP. Loss of interferon regulatory factor 3 in cells infected with classical swine fever virus involves the N-terminal protease, Npro. J Virol. 2005;79:239–47. doi: 10.1128/JVI.79.11.7239-7247.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMorte VJ, Dyck JA, Ochs RL, Evans RM. Localization of nascent RNA and CREB binding protein with the PML-containing nuclear body. Proc Nat Acad Sci USA. 1998;28:4991–6. doi: 10.1073/pnas.95.9.4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, Spencer CA. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J Virol. 1996;70:7471–7477. doi: 10.1128/jvi.70.11.7471-7477.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale J,M, Lemon SM. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Nat Acad Sci USA. 2005;102:2992–7. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol. 1998;18:2986–2996. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Mamane Y, Hiscott J. Structural and functional analysis of interferon regulatory factor 3: Localization of the transactivation and autoinhibitory domains. Mol Cell Biol. 1999;19:2465–2474. doi: 10.1128/mcb.19.4.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J Virol. 2004;78:1675–84. doi: 10.1128/JVI.78.4.1675-1684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonte P, Thomas J, Texier P, Caron C, Khochbin S, Epstein AL. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J Virol. 2004;78:6744–57. doi: 10.1128/JVI.78.13.6744-6757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9- mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. 2003;198:513–20. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews JM, sunce M. Ziinc fingers-folds for many occasions. IUBMB Life. 2002;54:351–5. doi: 10.1080/15216540216035. [DOI] [PubMed] [Google Scholar]
- Maul GG, Everett RD. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICPO. Journal of General Virology. 1994;75:1223–1233. doi: 10.1099/0022-1317-75-6-1223. [DOI] [PubMed] [Google Scholar]
- Maul GG, Guldner HH, Spivack JG. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0) Journal of General Virology. 1993;74:2679–2690. doi: 10.1099/0022-1317-74-12-2679. [DOI] [PubMed] [Google Scholar]
- Melroe G, DeLuca N, Knipe DM. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. Journal of Virology. 2004;78:8411–20. doi: 10.1128/JVI.78.16.8411-8420.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merika M, Thanos D. Enhancesosomes. Curr Opin Genet Dev. 2001;11:205–8. doi: 10.1016/s0959-437x(00)00180-5. [DOI] [PubMed] [Google Scholar]
- Mittnacht S, Straub P, Kirchner H, Jacobsen H. Interferon treatment inhibits onset of herpes simplex virus immediate-early transcription. Virology. 1988;164:201–210. doi: 10.1016/0042-6822(88)90637-x. [DOI] [PubMed] [Google Scholar]
- Morency E, Coute Y, Thomas J, Texier P, Lomonte P. The protein ICP0 of herpes simplex virus type 1 is targeted to nucleoli of infected cells. Arch Virol. 2005 doi: 10.1007/s00705-005-0546-5. [DOI] [PubMed] [Google Scholar]
- Mossman KL, Macgregor PF, Rozmus JJ, Goryachev AB, Edwards AM, Smiley JR. Herpes simplex virus triggers and then disarms a host antiviral response. J Virol. 2001;75:750–758. doi: 10.1128/JVI.75.2.750-758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman KL, Saffran HA, Smiley JR. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J Virol. 2000;74:2052–2056. doi: 10.1128/jvi.74.4.2052-2056.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional roles of type I and type II interferons in antivrial defense. Science. 1994;264:1918–21. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Nabel GJ, Rice SA, Knipe DM, Baltimore D. Alternative mechanisms for activation of human immunodeficiency virus enhancer in T cells. Science. 1988;239:1299–1302. doi: 10.1126/science.2830675. [DOI] [PubMed] [Google Scholar]
- Nicholl MJ, Robinson LH, Preston CM. Activation of cellular interferon-responsive genes after infection of human cells with herpes simplex virus type 1. J Gen Virol. 2000;81:2215–2218. doi: 10.1099/0022-1317-81-9-2215. [DOI] [PubMed] [Google Scholar]
- O’Hare P, Hayward GS. Evidence for a direct role for both the 175,000- and 110,000-molecular-weight immediate-early proteins of herpes simplex virus in the transactivation of delayed-early promoters. J Virol. 1985;53(3):751–760. doi: 10.1128/jvi.53.3.751-760.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberman F, Panet A. Inhibition of transcription of herpes simplex virus immediate early genes in interferon-treated human cells. J Gen Virol. 1988;69:1167–1177. doi: 10.1099/0022-1317-69-6-1167. [DOI] [PubMed] [Google Scholar]
- Oberman F, Panet A. Characterization of the early steps of herpes simplex virus replication in interferon-treated human cells. Journal of Interferon Research. 1989;9:563–571. doi: 10.1089/jir.1989.9.563. [DOI] [PubMed] [Google Scholar]
- Parkinson J, Lees-Miller SP, Everett RD. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol. 1999;73:650–657. doi: 10.1128/jvi.73.1.650-657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston CM, Harman AN, Nicholl MJ. Activation of interferon response factor-3 in human cells infected with herpes simplex virus type 1 or human cytomegalovirus. J Virol. 2001;75:8909–8916. doi: 10.1128/JVI.75.19.8909-8916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan MP, Knipe DM. Stimulation of expression of a herpes simplex virus DNA-binding protein by two viral functions. Molecular & Cellular Biology. 1985;5:957–963. doi: 10.1128/mcb.5.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Tun-Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T, Akira S, Yamamoto N, Lu KP, Yamaoka S. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol. 2006;7:598–605. doi: 10.1038/ni1347. [DOI] [PubMed] [Google Scholar]
- Samaniego LA, Neiderhiser L, DeLuca NA. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J Virol. 1998;72:3307–3320. doi: 10.1128/jvi.72.4.3307-3320.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, T. T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFNalpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- Sato M, Tanaka N, Hata N, Oda E, Taniguchi T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett. 1998;425:112–6. doi: 10.1016/s0014-5793(98)00210-5. [DOI] [PubMed] [Google Scholar]
- Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- Spear PG, Roizman B. Proteins specified by herpes simplex virus. V. Purification and structural proteins of the herpesvirion. J Virol. 1972;9:143–159. doi: 10.1128/jvi.9.1.143-159.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack J, Haga IR, Schroder M, Bartlett NW, Maloney G, Reading PC, Fitzgerald KA, Smith GL, Bowie AG. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J Exp Med. 2005;201:1007–18. doi: 10.1084/jem.20041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelow LI, Leib DA. Role of the virion host shutoff (vhs) of herpes simplex virus type 1 in latency and pathogenesis. J Virol. 1995;69:6779–6786. doi: 10.1128/jvi.69.11.6779-6786.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterstab G, Ludwig S, Anton A, Planz O, Dauber B, Krappmann D, Heins G, Ehrhardt C, Wolff T. Viral targeting of the interferon-{beta}- inducing Traf family member-associated NF-{kappa}B activator (TANK)- binding kinase-1. Proc Nat Acad Sci USA. 2005;102:13640–5. doi: 10.1073/pnas.0502883102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wathelet MG, Clauss IM, Content J, Huez GA. Regulation of two interferon-inducible human genes by interferon, poly(rI).poly(rC) and viruses. Eur J Biochem. 1988;174:323–9. doi: 10.1111/j.1432-1033.1988.tb14101.x. [DOI] [PubMed] [Google Scholar]
- Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Molecular Cell. 1998;1:507–518. doi: 10.1016/s1097-2765(00)80051-9. [DOI] [PubMed] [Google Scholar]
- Yang H, Lin CH, Ma G, Orr M, Baffi MO, Wathelet MG. Transcriptional activity of interferon regulatory factor (IRF)-3 depends on multiple protein-protein interactions. Eur J Biochem. 2002;269:6142–51. doi: 10.1046/j.1432-1033.2002.03330.x. [DOI] [PubMed] [Google Scholar]
- Yang H, Ma G, Lin CH, Orr M, Wathelet MG. Mechanism for transcriptional synergy between interferon regulatory factor (IRF)-3 and IRF-7 in activation of the interferon-beta gene promoter. Eur J Biochem. 2004;271:3693–703. doi: 10.1111/j.1432-1033.2004.04310.x. [DOI] [PubMed] [Google Scholar]
- Yeow WS, Au WC, Lowther WJ, Pitha PM. Downregulation of IRF-3 levels by ribozyme modulates the profile of IFNA subtypes expressed in infected human cells. Journal of Virology. 2001;75:3021–7. doi: 10.1128/JVI.75.6.3021-3027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. Embo J. 1998;17:1087–95. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jiang Y, Geiser V, Zhou J, Jones C. Bovine herpesvirus 1 immediate-early protein (bICP0) interacts with the histone acetyltransferase p300, which stimulates productive infection and gC promoter activity. J Gen Virol. 2006;87:1843–51. doi: 10.1099/vir.0.81766-0. [DOI] [PubMed] [Google Scholar]