


Abstract
The 2′-deoxyguanosine-3′,5′-diphosphate, 2′-deoxyadenosine-3′,5′-diphosphate, 2′-deoxycytidine-3′,5′-diphosphate and 2′-deoxythymidine-3′,5′-diphosphate systems are the smallest units of a DNA single strand. Exploring these comprehensive subunits with reliable density functional methods enables one to approach reasonable predictions of the properties of DNA single strands. With these models, DNA single strands are found to have a strong tendency to capture low-energy electrons. The vertical attachment energies (VEAs) predicted for 3′,5′-dTDP (0.17 eV) and 3′,5′-dGDP (0.14 eV) indicate that both the thymine-rich and the guanine-rich DNA single strands have the ability to capture electrons. The adiabatic electron affinities (AEAs) of the nucleotides considered here range from 0.22 to 0.52 eV and follow the order 3′,5′-dTDP > 3′,5′-dCDP > 3′,5′-dGDP > 3′,5′-dADP. A substantial increase in the AEA is observed compared to that of the corresponding nucleic acid bases and the corresponding nucleosides. Furthermore, aqueous solution simulations dramatically increase the electron attracting properties of the DNA single strands. The present investigation illustrates that in the gas phase, the excess electron is situated both on the nucleobase and on the phosphate moiety for DNA single strands. However, the distribution of the extra negative charge is uneven. The attached electron favors the base moiety for the pyrimidine, while it prefers the 3′-phosphate subunit for the purine DNA single strands. In contrast, the attached electron is tightly bound to the base fragment for the cytidine, thymidine and adenosine nucleotides, while it almost exclusively resides in the vicinity of the 3′-phosphate group for the guanosine nucleotides due to the solvent effects. The comparatively low vertical detachment energies (VDEs) predicted for 3′,5′-dADP− (0.26 eV) and 3′,5′-dGDP− (0.32 eV) indicate that electron detachment might compete with reactions having high activation barriers such as glycosidic bond breakage. However, the radical anions of the pyrimidine nucleotides with high VDE are expected to be electronically stable. Thus the base-centered radical anions of the pyrimidine nucleotides might be the possible intermediates for DNA single-strand breakage.
INTRODUCTION
The knowledge of the distribution of excess electron sites for DNA single strands is attracting increasing attention. (1,2) The formation of anions in DNA fragments has been found to be related to important biochemical processes such as DNA damage and repair (3–10), charge transfer along DNA (11–15), and the initiation of reactions leading to mutation (3,5,16).
Experimentally based investigations suggest that the nucleobases have small electron affinities (EAs), ∼0.1 eV for thymine (T), cytosine (C) and uracil (U) (17). Negative EA values have been determined for adenine (A) and cytosine in gas-phase experiments (18,19). The identification of the existence of two types of anions (dipole bound and covalent) (20–22) partly explains the differences among the different experimental EA values (23). Recent experiments on the electron-capturing efficiencies of short DNA oligomers provide the only estimate of the relative order of the vertical attachment energies (VAEs) for DNA single strands (1). However, the direct experimental determination of the EAs of nucleosides and nucleotides has proven difficult. Large DNA fragments, such as nucleotides, are non-volatile and the requirement for the vaporization of the species without thermal degradation makes it difficult to carry out reliable experimental studies in the gas phase.
Theoretical investigations at various levels of sophistication have complemented the experimental studies. While second-order Møller–Plesset perturbation theory (MP2) underestimates the adiabatic electron affinities (AEAs) for the five bases (24,25), density functional theory (DFT) approaches yield experimentally consistent AEA values for the individual bases (26–28). Theoretical studies of excess charge in DNA have been extended to the prediction of the EAs of subunits such as nucleosides and nucleotides. Semi-empirical methods have been applied to evaluate the EAs of nucleosides and nucleotides. (29,30) With the reliably calibrated B3LYP/DZP++ approach (31), meaningful predictions of the EAs of the 2′-deoxyribonucleosides have been completed (2). Recently, the EAs of the pyrimidine nucleotides (3′-dCMP, 3′-dTMP, 5′-dCMP, 5′-dTMP) have also been predicted at the B3LYP/DZP++ level of theory (10,32–35).
To understand the effects of excess electrons on single-strand DNA, the backbone of DNA should be realistically simulated in the model study. Previous studies reveal that the phosphate group at either the 3′ or 5′ position increases the electron acquisition ability of the pyrimidines (10,32,33). Thus, properly modeled systems representing single-strand DNA should include the phosphate group at both the 3′ and 5′ positions of the nucleotides. Moreover, the knowledge of the distribution of the excess electron sites and the availability of reliable EAs for the purine nucleotides are of equal importance. The vertical attachment of electrons to guanine-rich DNA single strands appears to predominate over entrapment by the cytosine-rich strands, as reported in the important 2005 experiment by Ray et al. (1). Although previous studies of the pyrimidine nucleosides and nucleotides suggest that the excess electron is mainly located on the bases in the electron-attached radical anions, the small AEA values and the large dipole moments of the purine bases (2,26) suggest that the situation for the purine nucleotides might be different. Here, we report a theoretical investigation of electron attachment to reasonable models of DNA single strands. The 2′-deoxyguanosine-3′,5′-diphosphate (3′,5′-dGDP), 2′-deoxyadenosine-3′,5′-diphosphate (3′,5′-dADP), 2′-deoxycytidine-3′,5′-diphosphate (3′,5′-dCDP) and 2′-deoxythymidine-3′,5′-diphosphate (3′,5′-dTDP) systems in their protonated forms have been selected as models (see Scheme 1). For a better description of the influence of the 3′–5′ phosphodiester linkage in DNA, the -OPO3H moiety at the 5′ position was terminated with a methyl group. These systems represent the most complete descriptions to date of the minimal DNA subunits and are expected to provide reliable information concerning electron attachment to single-strand DNA.
Scheme 1.

Models of the DNA single strands: 2′-deoxyguanosine-3′,5′-diphosphate (3′,5′-dGDP), 2′-deoxyadenosine-3′,5′-diphosphate (3′,5′-dADP), 2′-deoxycytidine-3′,5′-diphosphate (3′,5′-dCDP) and 2′-deoxythymidine-3′,5′-diphosphate (3′,5′-dTDP).
THEORETICAL METHODS
The B3LYP functional approach (36,37) with basis sets of double-ζ quality plus polarization and diffuse functions (denoted DZP++) was used to obtain optimized geometries, vibrationally zero-point corrected energies and natural charges for the model molecules in both neutral and anionic forms. The DZP++ basis sets were constructed by augmenting the Huzinaga–Dunning (38,39) set of contracted double-ζ Gaussian functions with one set of p-type polarization functions for each H atom and one set of five d-type polarization functions for each C, N, O and P atom (αp(H) = 0.75, αd(C) = 0.75, αd(N) = 0.80, αd(O) = 0.85, αd(P) = 0.60). To complete the DZP++ basis, one even-tempered diffuse s function was added to each H atom, while sets of even-tempered diffuse s and p functions were centered on each heavy atom. The even-tempered orbital exponents were determined according to the prescription of Lee and Schaefer (40). Each adiabatic electron affinity was computed as the difference between the absolute energies of the appropriate neutral and anion species at their respective optimized geometries AEA = Eneut − Eanion.
To evaluate the electron-capture abilities of DNA single strands in aqueous solution, the polarizable continuum model (PCM) (41) with a dielectric constant of water (ε = 78.39) was used to simulate the solvated environment of an aqueous solution. To analyze the distributions of each unpaired electron, molecular orbital and the spin-density plots were constructed from the corresponding B3LYP/DZP++ densities. Natural population analysis (NPA) was determined using the B3LYP functional and the DZP++ basis set with the Natural bond orbital (NBO) analysis of Reed and Weinhold (42,43). The GAUSSIAN 98 system of DFT programs (44) was used for all computations.
RESULTS AND DISCUSSION
Electron affinities
The positive EAs (Table 1) of the model systems suggest that the DNA single strands have a tendency to capture low-energy electrons and to form electronically stable radical anions.
Table 1.
Electron affinities of nucleic acid bases, nucleosides and nucleotides (in eV)
| Process | AEA | VEAa | VDEb |
|---|---|---|---|
| 3′,5′-dADP → 3′,5′-dADP− | 0.10 (0.22) | 0.02 | 0.26 |
| 3′,5′-dGDP → 3′,5′-dGDP− | 0.24 (0.36) | 0.14 | 0.32 |
| 3′,5′-dCDP → 3′,5′-dCDP− | 0.27 (0.44) | 0.03 | 0.71 |
| 3′,5′-dTDP → 3′,5′-dTDP− | 0.35 (0.52) | 0.17 | 0.67 |
| 3′-dCMP → 3′-dCMP− | 0.33 (0.44)c | 0.15c | 1.28c |
| 3′-dTMP → 3′-dTMP− | 0.44 (0.56)c | 0.26c | 1.53c |
| 5′-dCMP → 5′-dCMP− | 0.20 (0.34)d | −0.11d | 0.85d |
| 5′-dTMP → 5′-dTMP− | 0.28 (0.44)d | 0.01d | 0.99d |
| dA → dA− | −0.05 (0.06)e | 0.91e | |
| dG → dG− | 0.01 (0.09)e | 0.05e | |
| dC → dC− | 0.21 (0.33)e | 0.72e | |
| dT → dT− | 0.31 (0.44)e | 0.94e | |
| A → A− | −0.37 (−0.28)f | ||
| G → G− | −0.14 (−0.07)f | ||
| C → C− | −0.09 (0.03)f | ||
| T → T− | 0.06 (0.20)f |
Values with zero-point vibrational corrections are reported in parentheses.
aVAE = E(neutral) − E(anion), the energies are evaluated using the optimized neutral structures.
bVDE = E(neutral) − E(anion), the energies are evaluated using the optimized anion structures.
dReference (10), where 5′-dCMP and 5′-dTMP were labeled as 5′-dCMPH and 5′-dTMPH, respectively.
eReference (2).
fReference (26).
To understand the electron-capturing ability of DNA single strands at the nascent stage of electron attachment, it is necessary to estimate the vertical electron attachment energies (VEAs). Relatively large VEAs are predicted for 3′,5′-dTDP (0.17 eV) and 3′,5′-dGDP (0.14 eV) in this investigation. This indicates that both the thymine-rich and the guanine-rich DNA single strands are prepared to capture low-energy electrons. Conversely, the near zero VEA values for 3′,5′-dCDP (0.03 eV) and 3′,5′-dADP (0.02 eV) suggest less effective electron capturing abilities for cytosine and adenine-derived DNA single strands. It must be noted that the high electron-capturing ability of guanine-rich DNA single strands and the low electron-capturing ability for cytosine-rich and adenine-rich DNA single strands have been observed in the experiments of Ray et al. (1). Although the VEA value is not the sole factor related to the electron-capture ability, our predictions for the VEAs of the nucleoside-3′,5′-diphosphate systems allow us to understand these experimental observations from the viewpoint of energy.
The AEAs of the nucleotides follow the order 3′,5′-dTDP > 3′,5′-dCDP > 3′,5′-dGDP > 3′,5′-dADP, which is consistent with that for the nucleobases and the nucleosides. However, substantial increases in the AEAs are predicted compared to those for the corresponding nucleic acid bases (0.52 eV versus 0.20 eV for T, 0.44 eV versus 0.03 eV for C, 0.36 eV versus −0.07 eV for G and 0.22 eV versus −0.28 eV for A, respectively; see Table 1). In addition, the increase in the AEAs from the nucleosides to the corresponding nucleotides amounts to ∼0.08–0.27 eV, signifying the importance of the phosphate groups in the stabilization of the radical anions of the DNA components. It is interesting to note that the AEA of 3′,5′-dTDP is almost the same as that of 3′-dTMP (2′-deoxythymidine-3′-monophosphate). The influence of the OH group at the 5′ position on the AEA of 3′-dTMP is equivalent to that of the 5′-phosphate group in 3′,5′-dTDP. The similar AEA value of 3′,5′-dCDP and that of 3′-dCMP (2′-deoxycytidine-3′-monophosphate) suggest the same equivalency.
To evaluate the electronic stability of the radical anions, the vertical detachment energies of the anions have been predicted. The VDEs of the radical anions of the purines are found to be about 0.3 eV, significantly smaller than those of the radical anions of the pyrimidines (∼0.7 eV). Consequently, reactions with activation barriers higher than 0.3 eV (∼7 kcal/mol), such as that for N-glycosidic bond breakage (35), are not expected for the radical anions of 3′,5′-dGDP and 3′,5′-dADP. As will be shown below, the low VDE values for 3′,5′-dGDP and 3′,5′-dADP may be traced to the nature of the phosphate group anion, while the high VDE values for 3′,5′-dTDP and 3′,5′-dCDP are concordant with the characteristics of the base-centered anions.
Molecular orbital analysis
Examination of the molecular orbital that the ‘last’ electron occupies provides an electronic structure-based rationale for the electron-attracting capabilities of the nucleotides. Plots of the singly occupied molecular orbitals (SOMOs) for the radical anions are shown in Figure 1. The most striking feature revealed by the SOMOs of these nucleoside-3′,5′-diphosphates molecules is that the excess electron density resides in part in the vicinity of the phosphate moiety, at the 3′ position. This phenomenon has not been found for either the pyrimidine nucleoside-3′-monophophate radical anions (32,33) or the corresponding 5′-monophophate radical anions (10). Previous investigations have shown that the excess electron density is not located around the -PO4H2 moiety of the stable radical anions of the pyrimidine nucleoside monophosphate (10,32,33). The present results seem to suggest that introducing a phosphate group at the 3′ or 5′ position of the ribose improves the electron-capturing ability of the 3′-phosphate fragment. The partial electron distribution on the -PO4H2 moiety might have its origin in the relatively low VDEs of the nucleoside–diphosphate complexes. However, guanine is different, in that the excess electron is partly covalent bonded to the 3′-phosphate in 3′,5′-dGDP− while it appears to be dipole-bound in the nucleoside (dG−) (2). The VDE of 3′,5′-dGDP− is thus significantly higher than that of dG− (0.32 eV versus 0.05 eV). Examination of the SOMOs depicted in Figure 1 enables one to conclude that the excess electron largely resides in the vicinity of the phosphate moiety in the purine radical anions, while it is mainly located near the nucleobase unit in the pyrimidines. Recall that the AEA of the sugar-phosphate-sugar model is close to zero (∼0.00–0.03 eV) (45). In this light, the difference in electron distributions between the purines and the pyrimidines is expected.
Figure 1.

Plots of the SOMOs of the radical anions of 3′,5′-dADP, 3′,5′-dGDP, 3′,5′-dCDP and 3′,5′-dTDP.
It should be noted that the excess electron at the base fragment in 3′,5′-dADP−, 3′,5′-dCDP− and 3′,5′-dTDP− is valence-bound (the π* anti-bonding orbital of the base is partly occupied, as shown in the SOMOs), while it is typically dipole-bound in the guanine moiety in 3′,5′-dGDP−. This feature can also be seen in electron attachment to the nucleosides (2). Compared to the nucleosides, the net result of the introduction of phosphate groups at the 3′ and 5′ positions is to reduce the population near the base moiety by increasing the electron density around the phosphate groups.
Charge distributions
The location of negative charge on the constituent parts of the nucleoside pair also provides some insight into the overall electronic effect of the charge. Table 2 summarizes the charge distributions among the bases, ribose and phosphates for both the neutral and anionic complexes. The analysis of the NPA charge differences between the neutral and anionic nucleotides supports the conclusion that the excess electron mainly resides in the vicinity of the nucleobase moiety in the pyrimidine radical anions, while it is largely located at the 3′-phosphate group in the purine. The NPA charge differences suggest that there is 0.63 a.u. of negative charge located on the thymine and 0.29 a.u. on the 3′-phosphate for 3′,5′-dTDP−; with 0.63 a.u. of negative charge located on the cytosine and 0.21 a.u. on the 3′-phosphate for 3′,5′-dCDP−. Conversely, the charge distribution differences of the purine nucleotides show significant increases in the negative charge resident on the 3′-phosphate group (0.46 a.u. for both 3′,5′-dADP− and 3′,5′-dGDP−) and remarkable decreases in the negative charge populations on the base moieties (0.42 a.u. for 3′,5′-dADP− and 0.46 a.u. for 3′,5′-dGDP−). The NPA analyses for the pyrimidine nucleoside-5′-monophosphate (10) and nucleoside-3′-monophosphate (31) indicate that the excess electron does not qualitatively reside on the phosphate group for the nucleoside-monophosphate. Approximately 20% of the negative charge distribution on the 3′-phosphate group for the pyrimidine nucleoside-3′,5′-diphosphates thus originates from cooperative effects owing to the coexistence of these two phosphate groups. The distribution of the ‘last’ electron amongst the constituent parts seems directly correlated to the VDEs discussed above.
Table 2.
NPA charge distributions of the neutrals and radical anions for the nucleoside-3′,5′-diphosphates
| Component | Neutral | Anion | Δa |
|---|---|---|---|
| 3′,5′-dADP | |||
| A | −0.263 | −0.683 | −0.42 |
| Ribose | 0.999 | 0.921 | −0.08 |
| 5′-Phosphate | −0.363 | −0.405 | −0.04 |
| 3′-Phosphate | −0.372 | −0.833 | −0.46 |
| 3′,5′-dGDP | |||
| G | −0.266 | −0.726 | −0.46 |
| Ribose | 1.007 | 0.955 | −0.05 |
| 5′-Phosphate | −0.369 | −0.391 | −0.02 |
| 3′-Phosphate | −0.372 | −0.837 | −0.46 |
| 3′,5′-dCDP | |||
| C | −0.287 | −0.917 | −0.63 (−0.86b; −0.82c) |
| Ribose | 1.011 | 0.861 | −0.15 (−0.10b; −0.17c) |
| 5′-Phosphate | −0.366 | −0.378 | −0.01 (−0.01c) |
| 3′-Phosphate | −0.358 | −0.565 | −0.21(−0.04b) |
| 3′,5′-dTDP | |||
| T | −0.288 | −0.919 | −0.63 (−0.86b; −0.85c) |
| Ribose | 1.008 | 0.938 | −0.07 (−0.10b; −0.14c) |
| 5′-Phosphate | −0.363 | −0.371 | −0.01 (−0.01c) |
| 3′-Phosphate | −0.357 | −0.647 | −0.29 (−0.04b) |
Geometries
The fully optimized geometries of the neutral and anionic nucleoside-3′,5′-diphosphates are depicted in Figure 2. The most striking finding is that pyramidization of atom C8 in the radical anion dA− and atom C6 of dC− and dT− (also in the pyrimidine nucleoside-monophosphates) (2,10,32,33) is barely detected in the anionic nucleoside-3′,5′-diphosphates. The dihedral angle DN1-C5-H6-C6 is less than 3° in 3′,5′-dCDP− and 3′,5′-dTDP− [it is ∼10–20° in the corresponding nucleosides and their monophosphate complexes (2,10,33)], and the dihedral DN9-N7-H8-C8 in 3′,5′-dADP− is less than 1° (in contrast, it is about 30° in dA−) (2). Compared to the nucleosides and the nucleoside-monophosphates, the geometric variations due to electron attachment are less obvious for the bases of the nucleoside-3′,5′-diphosphates. Typically, the C5–C6 and C6–N1 bond distances in 3′,5′-dCDP− are 0.03 and 0.04 Å longer than those for the neutral species. These differences are about 0.02 Å less than the corresponding bond-length elongations found for the formation of 5′-dCMP− (the C5–C6 and C6–N1 bond increases amount to 0.05 and 0.06 Å for in the radical anion of 5′-dCMP, and 0.05 and 0.06 Å in the radical anion of dC, respectively) (2,10). Similarly, the C5–C6 and C6–N1 bond distance increases are 0.03 and 0.03 Å in 3′,5′-dTDP−, while they are 0.05 and 0.06 Å in dT− and 5′-dTMP−, respectively. (2,10) Moreover, the bond elongations in N7–C8 and C8–N9 due to electron attachment to 3′,5′-dADP are significantly less than those (2) for the related nucleoside (0.01 Å for 3′,5′-dADP versus 0.07–0.09 Å for dA). The reduction of the glycosidic bond distance in the radical anions of the nucleoside-3′,5′-diphosphates is about 0.01 Å less than that for the corresponding nucleosides and nucleoside-monophosphates. The less significant geometric alterations of the base moiety of the radical anions of 3′,5′-dADP, 3′,5′-dCDP and 3′,5′-dTDP might originate from the less negative charge distribution near the base moiety, consistent with the molecular orbital analysis and the charge distribution analysis discussed above. Due to the nature of the dipole-bound anion, it is not unexpected that the geometry of the guanine base of 3′,5′-dGDP is little changed by electron attachment.
Figure 2.

Optimized geometrical structures for the neutrals and radical anions of 3′,5′-dADP, 3′,5′-dGDP, 3′,5′-dCDP and 3′,5′-dTDP. Bond distances are in Å. Color representations: orange for P, gray for C, red for O, blue for N and white for H.
Effects of solvation
Analogous to the pyrimidine-monophosphates, interaction with water greatly improves the electron-capture ability of DNA single strands. Note that when we speak of the ‘electron affinity’ of a solvated molecule M, the physical situation described is a microsolvated M(H2O)n system, in which the water uniformly enclose M and n becomes arbitrarily large. In this sense, the ‘AEA’ values are 1.59 eV for 3′,5′-dADP, 0.95 eV for 3′,5′-dGDP, 1.99 eV for 3′,5′-dCDP and 1.98 eV for 3′,5′-dTDP in aqueous solution. The increases in the AEAs with solvation range from 0.6 eV (3′,5′-dGDP) to 1.5 eV (3′,5′-dCDP), similar to those for the pyrimidine-monophosphates (10,32–34).
Table 3.
Dihedral angles of the pyramidized base atoms for the radical anions of nucleotides
| Species | Dihedrala | Comparisons |
|---|---|---|
| 3′,5′-dADP− | 0.6° | (30.7°)b |
| 3′,5′-dGDP− | 0.2° | (0.4°)b |
| 3′,5′-dCDP− | 2.8° | (8.7°)b, (20.2°)c, (11.8°)d |
| 3′,5′-dTDP− | 2.9° | (16.5°)b, (23.7°),c (16.8°)d |
Table 4.
‘Electron affinities’ of nucleotides (in eV) in aqueous solution
It is even more important to notice that the polar solvent surroundings associated with aqueous solution alter the distribution of the excess electron in each DNA single strand dramatically (see Figure 3). Due to the solvation effects, the excess electron resides almost completely near the base for 3′,5′-dCDP−, 3′,5′-dTDP− and even for 3′,5′-dADP−, for which case the base fragment excess electron density is low in the gas phase. Conversely, the excess electron distribution in aqueous solution is largely limited to the vicinity of the 3′-phosphate group in 3′,5′-dGDP−, resulting in a phosphate-located radical anion. Although there is a hypothesis (45) that the excess electron might reside on the phosphate group of DNA single strands, the present study provides the sound theoretical evidence for only the guanine-related nucleotides. However, the present study also reveals that this hypothesis might be applicable for the purine nucleotides in the gas phase and only valid for the guanine nucleotide in aqueous solution.
Figure 3.

Plots of the SOMOs of the radical anions of 3′,5′-dADP, 3′,5′-dGDP, 3′,5′-dCDP and 3′,5′-dTDP in aqueous solution.
It is well known that the phosphate groups of the nucleotides are deprotonated in aqueous solution. Recent studies demonstrate that the EAs of the pyrimidine nucleotides are nearly independent of the existence of counterions in aqueous solution (32,34). Accordingly, the deprotonation of the phosphate groups in aqueous solution should not affect the EAs of 3′,5′-dCDP and 3′,5′-dTDP. Note that the excess electron density in 3′,5′-dADP− is distributed mainly on the base moiety in aqueous solution. Thus, one might expect that deprotonation of the phosphate in dADP would influence its electron affinity to a minor degree. However, since the unpaired electron is primarily located on the phosphate moiety of 3′,5′-dGDP− in aqueous solution, the deprotonation of the phosphates might reduce the electron affinity of dGDP considerably.
CONCLUSIONS: IMPACT ON LEE-INDUCED DNA SINGLE-STRAND BREAKAGE (SSB)
The 2′-deoxyguanosine-3′,5′-diphosphate, 2′-deoxyadenosine-3′,5′-diphosphate, 2′-deoxycytidine-3′,5′-diphosphate and 2′-deoxythymidine-3′,5′-diphosphate systems are the simplest fragments of DNA single strands that might be considered representative. Exploring electron attachment to these archetypal units of DNA single strands enables one to approach reliable predictions of the EAs of DNA single strands.
DNA single strands have a strong tendency to capture low-energy electrons and to form electronically stable radical anions. The relatively large VEAs predicted for 3′,5′-dTDP (0.17 eV) and 3′,5′-dGDP (0.14 eV) in this investigation indicate that both the thymine-rich and the guanine-rich DNA single strands have the ability to capture low-energy electrons. Conversely, the small VEA values of 3′,5′-dCDP (0.03 eV) and 3′,5′-dADP (0.02 eV) imply that the electron-capture efficiency of the cytosine and adenine-rich DNA single strands is lower than those for guanine and thymine.
The AEAs of the nucleotides range from 0.22 to 0.52 eV and follow the order 3′,5′-dTDP > 3′,5′-dCDP > 3′,5′-dGDP > 3′,5′-dADP. A substantial increase in the AEA is predicted compared to that of the corresponding nucleic acid bases and the corresponding nucleosides. The effect of the phosphate groups in stabilizing the radical anions of the DNA components is crucial.
The coexistence of the phosphate groups at the 3′ and 5′ positions of the ribose results in a more delocalized electron distribution around both the base and the phosphate moieties. About 20–30% of the negative charge is located at the phosphate group for the pyrimidine nucleotides, while the analogous distribution is about 50% for the purine nucleotides.
Aqueous solution dramatically increases the electron-capturing ability of the DNA single strands, by up to 1–2 eV. Moreover, the solvent effect localize the excess electron on either the base (for 3′,5′-dADP, 3′,5′-dCDP and 3′,5′-dTDP) or the phosphate (for 3′,5′-dGDP) subunit.
It is worthwhile to point out that base stacking seems unlikely to affect the EAs of single strands of DNA, as shown in the computational study by Simons’ group (46). The primary results of our own study of the solvated dGdC base-stacking model suggest that this is also true for the stacked bases in aqueous solution.
Low energy electron (LEE) attachment induced DNA SSB has attracted great interest from both experiment and theory (6–10,33,35,45,47–49). Electron attachment to DNA single strands may lead to either N–C glycosidic bond rupture or C3′–O3′ and C5′–O5′ σ-bond breakage. Distinct electron attachment positions have been proposed based on different model studies (7–10,32–37). The present investigation demonstrates that in the gas phase, the excess electron is located both on the nucleobase and the phosphate moiety for DNA single strands. However, the distribution of the negative charge is not even. The attached electron favors the base moiety for the pyrimidine DNA oligomers, and it prefers the 3′-phosphate subunit for the purine DNA single strands. In contrast, the distribution of the extra negative charge is more localized in aqueous solution. The attached electron is tightly bound to the base fragment for the cytidine, thymidine and adenosine nucleotides; for the guanosine nucleotides, this ‘last electron’ resides primarily in the vicinity of the 3′-phosphate group due to the solvation effects.
The comparatively low VDE values predicted for 3′,5′-dADP− and 3′,5′-dGDP− indicate that both are electronically less favorable, and electron detachment might compete with reactions involving relatively high activation barriers such as glycosidic bond breakages. However, the radical anions of the pyrimidine nucleotides with high VDE values are expected to be electronically stable. Thus the base-centered radical anions of the pyrimidine nucleotides might be the possible intermediates for DNA SSB.
ACKNOWLEDGEMENTS
This research was supported by the U.S. National Science Foundation, Grant CHE-0451445. Funding to pay the Open Access publication charge was provided by the same NSF source.
Conflict of interest statement. None declared.
REFERENCES
- 1.Ray SG, Daube SS, Naaman R. On the capturing of low-energy electrons by DNA. J. Proc. Natl Acad. Sci. USA. 2005;102:15–19. doi: 10.1073/pnas.0407020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richardson NA, Gu J, Wang S, Xie Y, Schaefer HF. DNA nucleosides and their radical anions: molecular structures and electron affinities. J. Am. Chem. Soc. 2004;126:4404–4411. doi: 10.1021/ja030487m. [DOI] [PubMed] [Google Scholar]
- 3.Becker D, Sevilla MD. The chemical consequences of radiation damage to DNA. In: Lett J, editor. Advances in Radiation Biology. Vol. 17. Academic Press, New York; 1993. pp. 121–180. [Google Scholar]
- 4.Kelley SO, Barton JK. Electron transfer between bases in double helical DNA. Science. 1999;283:375–381. doi: 10.1126/science.283.5400.375. [DOI] [PubMed] [Google Scholar]
- 5.Ratner M. Photochemistry – electronic motion in DNA. Nature. 1999;397:480–481. doi: 10.1038/17232. [DOI] [PubMed] [Google Scholar]
- 6.Boudaiffa B, Cloutier P, Hunting D, Huels MA, Sanche L. Resonant formation of DNA strand breaks by low-energy (3 to 20 eV) electrons. Science. 2000;287:1658–1659. doi: 10.1126/science.287.5458.1658. [DOI] [PubMed] [Google Scholar]
- 7.Pan X, Cloutier P, Hunting D, Sanche L. Dissociative electron attachment to DNA. Phys. Rev. Lett. 2003;90:208102. doi: 10.1103/PhysRevLett.90.208102. [DOI] [PubMed] [Google Scholar]
- 8.Caron LG, Sanche L. Low-energy electron diffraction and resonances in DNA and other helical macromolecules. Phys. Rev. Lett. 2003;91:113201. doi: 10.1103/PhysRevLett.91.113201. [DOI] [PubMed] [Google Scholar]
- 9.Zheng Y, Cloutier P, Hunting D, Wagner JR, Sanche L. Glycosidic bond cleavage of thymidine by low-energy electrons. J. Am. Chem. Soc. 2004;126:1002–1003. doi: 10.1021/ja0388562. [DOI] [PubMed] [Google Scholar]
- 10.Bao X, Wang J, Gu J, Leszczynski J. DNA strand breaks induced by near-zero-electron volt electron attachment to pyrimidine nucleotides. Proc. Natl Acad. Sci. USA. 2006;103:5658–5663. doi: 10.1073/pnas.0510406103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall DB, Holmlin RE, Barton JK. Oxidative DNA damage through long-range electron transfer. Nature. 1996;382:731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 12.Steenken S. Electron transfer in DNA? Competition by ultra-fast proton transfer? Biol. Chem. 1997;378:1293–1297. [PubMed] [Google Scholar]
- 13.Taubes G. Double helix chemistry at a distance – but how? Science. 1997;275:1420–1421. doi: 10.1126/science.275.5305.1420. [DOI] [PubMed] [Google Scholar]
- 14.Berlin YA, Burin AL, Ratner MA. Charge hopping in DNA. J. Am. Chem. Soc. 2001;123:260–268. doi: 10.1021/ja001496n. [DOI] [PubMed] [Google Scholar]
- 15.Beljonne D, Pourtois G, Ratner MA, Bredas JL. Pathways for photoinduced charge separation in DNA hairpins. J. Am. Chem. Soc. 2003;125:14510–14517. doi: 10.1021/ja035596f. [DOI] [PubMed] [Google Scholar]
- 16.Huels MA, Hahndorf I, Illenberger E, Sanche L. Resonant dissociation of DNA bases by subionization electrons. J. Chem. Phys. 1998;108:1309–1312. [Google Scholar]
- 17.Schiedt J, Weinkauf R, Neumark DM, Schlag EW. Anion spectroscopy of uracil, thymine and the amino-oxo and amino-hydroxy tautomers of cytosine and their water clusters. Chem. Phys. 1998;239:511–524. [Google Scholar]
- 18.Desfrancois C, Abdoul-Carime H, Schermann JP. Electron attachment to isolated nucleic acid bases. J. Chem. Phys. 1996;104:7792–7794. [Google Scholar]
- 19.Periquet V, Moreau A, Carles S, Schermann JP, Desfrancois C. Cluster size effects upon anion solvation of N-heterocyclic molecules and nucleic acid bases. J. Electron Spectrosc. Relat. Phenom. 2000;106:141–151. [Google Scholar]
- 20.Oyler NA, Adamowicz L. Electron-attachment to uracil – theoretical ab-initio study. J. Phys. Chem. 1993;97:11122–11123. [Google Scholar]
- 21.Hendricks JH, Lyzpustina SA, de Clercq HL, Bowen KH. Dipole bound, nucleic acid base anions studied via negative ion photoelectron spectroscopy. J. Chem. Phys. 1996;104:7788–7794. [Google Scholar]
- 22.Hendricks JH, Lyzpustina SA, de Clercq HL, Bowen KH. The dipole bound-to-covalent anion transformation in uracil. J. Chem. Phys. 1998;108:8–11. [Google Scholar]
- 23.Chen ECM, Chen ESD. Negative ion mass spectra, electron affinities, gas phase acidities, bond dissociation energies, and negative ion states of cytosine and thymine. J. Phys. Chem. B. 2000;104:7835–7844. [Google Scholar]
- 24.Wetmore SD, Boyd RJ, Eriksson LA. Electron affinities and ionization potentials of nucleotide bases. Chem. Phys. Lett. 2000;322:129–135. [Google Scholar]
- 25.Russo N, Roscano M, Grand A. Theoretical determination of electron affinity and ionization potential of DNA and RNA bases. J. Comput. Chem. 2000;21:1243–1250. [Google Scholar]
- 26.Wesolowski SS, Leininger ML, Pentchev PN, Schaefer HF. Electron affinities of the DNA and RNA bases. J. Am. Chem. Soc. 2001;123:4023–4028. doi: 10.1021/ja003814o. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Sevilla MD, Sanche L. DFT investigation of dehalogenation of adenine-halouracil base pairs upon low-energy electron attachment. J. Am. Chem. Soc. 2003;125:8916–8920. doi: 10.1021/ja034286u. [DOI] [PubMed] [Google Scholar]
- 28.Ban F, Lundqvist MJ, Boyd RJ, Eriksson LA. Theoretical studies of the cross-linking mechanisms between cytosine and tyrosine. J. Am. Chem. Soc. 2002;124:2753–2761. doi: 10.1021/ja011528m. [DOI] [PubMed] [Google Scholar]
- 29.Harris DG, Shao J, Anderson JM, Marx DP, Zimmerman SS. Procedure for selecting starting conformations for energy minimization of nucleosides and nucleotides. Nucleosides Nucleotides Nucleic Acids. 2002;21:803–812. doi: 10.1081/NCN-120016482. [DOI] [PubMed] [Google Scholar]
- 30.Voityuk AA, Michel-Beyerle M, Rosch N. Energetics of excess electron transfer in DNA. Chem. Phys. Lett. 2001;342:231–238. [Google Scholar]
- 31.Rienstra-Kiracofe JC, Tschumper GS, Schaefer HF, Nandi S, Ellison GB. Atomic and molecular electron affinities: photoelectron experiments and theoretical computations. Chem. Rev. 2002;102:231–282. doi: 10.1021/cr990044u. [DOI] [PubMed] [Google Scholar]
- 32.Gu J, Xie Y, Schaefer HF. Near 0 eV electrons attach to nucleotides. J. Am. Chem. Soc. 2006;128:1250–1252. doi: 10.1021/ja055615g. [DOI] [PubMed] [Google Scholar]
- 33.Gu J, Wang J, Leszczynski J. Electron attachment-induced DNA single strand breaks: C-3′-O-3′ sigma-bond breaking of pyrimidine nucleotides predominates. J. Am. Chem. Soc. 2006;128:9322–9323. doi: 10.1021/ja063309c. [DOI] [PubMed] [Google Scholar]
- 34.Gu J, Xie Y, Schaefer HF. Electron attachment to nucleotides in aqueous solution. Chem. Phys. Chem. 2006;7:1885–1887. doi: 10.1002/cphc.200600294. [DOI] [PubMed] [Google Scholar]
- 35.Gu J, Xie Y, Schaefer HF. Glycosidic bond cleavage of pyrimidine nucleosides by low-energy electrons: a theoretical rationale. J. Am. Chem. Soc. 2005;127:1053–1057. doi: 10.1021/ja0400990. [DOI] [PubMed] [Google Scholar]
- 36.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 37.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 38.Huzinaga S. Gaussian-type functions for polyatomic systems I. J. Chem. Phys. 1965;42:1293. [Google Scholar]
- 39.Dunning TH. Gaussian basis functions for use in molecular calculations: I. Contraction of (9S5P) atomic basis sets for first-row atoms. J. Chem. Phys. 1970;53:2823. [Google Scholar]
-
40.Lee TJ, Schaefer HF. Systematic study of molecular anion within the self-consistent-field approximation: OH−, CN−, C2H−,
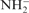 and
and 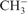 . J. Chem. Phys. 1985;83:1784–1794. [Google Scholar]
. J. Chem. Phys. 1985;83:1784–1794. [Google Scholar] - 41.Cossi M, Barone V, Cammi R, Tomasi J. Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996;255:327–335. [Google Scholar]
- 42.Reed AE, Schleyer PR. Chemical bonding in hypervalent molecules: the dominance of ionic bonding and negative hyperconjugation over d-orbital participation. J. Am. Chem. Soc. 1990;112:1434–1445. [Google Scholar]
- 43.Reed AE, Curtiss LA, Weinhold F. Intermolecular interactions from a natural bond orbital donor-acceptor viewpoint. Chem. Rev. 1988;88:899–926. [Google Scholar]
- 44.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Stratmann RE, et al. Pittsburgh, PA: Gaussian, Inc; 2001. Gaussian 98 (revision a.10) [Google Scholar]
- 45.Li X, Sevilla MD, Sanche L. Density functional theory studies of electron interaction with DNA: can zero eV electrons induce strand breaks? J. Am. Chem. Soc. 2003;125:13668–13669. doi: 10.1021/ja036509m. [DOI] [PubMed] [Google Scholar]
- 46.Berdys J, Ansiewicz I, Skurski P, Simons J. Damage to model DNA fragments from very low-energy (<1 eV) electrons. J. Am. Chem. Soc. 2004;126:6441–6447. doi: 10.1021/ja049876m. [DOI] [PubMed] [Google Scholar]
- 47.Simons J. How do low-energy (0.1-2 eV) electrons cause DNA-strand breaks? Acc. Chem. Res. 2006;39:772. doi: 10.1021/ar0680769. [DOI] [PubMed] [Google Scholar]
- 48.Sanche L. Low energy electron-driven damage in biomolecules. Eur. Phys. J. D. 2005;35:367–390. [Google Scholar]
- 49.Anusiewicz I, Berdys J, Sobczy M, Skurski P, Simons J. Effects of base p-stacking on damage to DNA by low-energy electrons. J. Phys. Chem. A. 2004;108:11381–1138. [Google Scholar]