

Abstract
New evidence indicates that neural mechanisms can down-regulate acute inflammation. In these studies, we tested the potential role of the α7 nicotinic acetylcholine receptor (α7 nAChR) in a rodent model of acid-induced acute lung injury. We first determined that the α7 nAChR was expressed by alveolar macrophages and lung epithelial cells. Then, using an acid-induced acute lung injury mouse model, we found that nicotine, choline, and PNU-282987 (a specific α7 nAChR agonist) decreased excess lung water and lung vascular permeability, and reduced protein concentration in the bronchoalveolar lavage (BAL). Deficiency of α7 nAChR resulted in a 2-fold increase in excess lung water and lung vascular permeability. The reduction of proinflammatory cytokines (macrophage inflammatory protein-2 and TNF-α) in the BAL with nicotine probably resulted from the suppression of NF-κB activation in alveolar macrophages. The beneficial effect of nicotine was also tested in rat model of acid-induced acute lung injury in which BAL protein and receptor for advanced glycation end products (RAGE), a marker of type I cell injury, were reduced by nicotine treatment. These results indicate that activation of α7 nAChR may provide a new therapeutic pathway for the treatment of acute lung injury.
Keywords: α7 nicotinic acetylcholine receptor, acute lung injury, alveolar macrophage, nicotine, proinflammatory cytokines
CLINICAL RELEVANCE
Activation of α7 nAChR down-regulates proinflammatory responses. Stimulation of pathway may provide a new therapeutic pathway for the treatment of acute lung injury.
There is new evidence that neural mechanisms can down-regulate inflammation in vivo by decreasing the release of TNF-α by endotoxin-stimulated macrophages (1). This anti-inflammatory pathway is mediated by an interaction between acetylcholine, the principal neurotransmitter of the vagus nerve, and cholinergic nicotinic acetylcholine receptors on macrophages (2). Nicotine pretreatment decreases inflammatory cell influx, proinflammatory cytokine levels, and liver injury in septic peritonitis (3). Stimulation of α7 nicotinic acetylcholine receptor (α7 nAChR) prevents activation of the NF-κB pathway and thereby improves survival in experimental models of sepsis (4, 5).
The overall aim of these experimental studies was to test the hypothesis that activation of the α7 nAChR–mediated cholinergic anti-inflammatory pathway could reduce the severity of acid aspiration induced acute lung injury by attenuating proinflammatory responses. The first objective was to determine whether alveolar macrophages, activated neutrophils, and (lung) epithelial cells expressed α7 nAChR. The secondary objective was to determine whether activation of the α7 nAChR by administration of the cholinergic agonist (nicotine, choline, and PNU-282987) would protect acid-induced acute lung injury. The third objective was to test whether knockout of α7 nAChR would worsen acid-induced acute lung injury in mice.
MATERIALS AND METHODS
Chemicals
(-)-nicotine, choline, and methyllycaconitine (MLA) were purchased from Sigma (St. Louis, MO). PNU-282987, a specific agonist of α7 nAChR, was purchased from Tocris Bioscience (Ellisville, MO). They were dissolved in 0.9% saline before each experiment. H-302, an anti–α7 nAChR antibody (used to detect α7 AChR of mouse and human origin), was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA)
Animals
Wild-type mice (C57BL/6J, 8 wk old) and mice deficient for the α7 nAChR (C57BL/6 background, B6.129S7-Chrna7tm1Bay, number 003232) were purchased from Jackson Laboratory (Bar Harbor, ME). Anesthesia was induced with an intraperitoneal injection of a mixture of ketamine (90 mg/kg) and xylazine (10 mg/kg). Sprague-Dawley rats weighing 350–400 g were anesthetized with an intraperitoneal injection of pentobarbital (75 mg/kg). The committee on animal research of the University of California, San Francisco approved the protocols.
Direct Visualized Instillation
As previously described, mice were anesthetized by an intraperitoneal injection of ketamine (90 mg/kg) and xylazine (10 mg/kg). The mice were suspended with their incisors attached to a ∼ 60° wood support by 3/0 suture. A cold-light source (Dolan-Jenner Industries Inc., Lawrence, MA) with two 25-inch flexible fiber-optic arms allowed transillumination to visualize the glottis and vocal cords to deliver the acid into the airspaces (6).
Acid-Induced Acute Lung Injury in Mice
Mice were intratracheally instilled with hydrochloride acid (pH 1.0, prepared in 1/3 saline, 1.25 ml/kg) by the direct visualized instillation (DVI) method. Before exposure to acid, mice were given an injection of 0.05 μCi 125I-albumin via right jugular vein. Mice were monitored for 4 h and killed to measure excess lung water and lung vascular permeability or to carry out bronchoalveolar lavage (BAL).
Acid-Induced Acute Lung Injury in a Ventilated Rat Model
A ventilated rat model was used to compare with the experimental setting in our prior studies (7). Rats were placed supine, and a tracheotomy tube (15-gauge luer stub adapter; Becton Dickinson, Sparks, MD) was inserted. The rats were treated with either saline or nicotine (3.5 mg/kg, intraperitoneally) 5–10 min before intratracheal instillation with pH 1.0 hydrochloric acid (3 ml/kg), and then ventilated with a tidal volume of 7 ml/kg, a rate of 60 times/min, and 5 cm H2O PEEP. Two hours later, rats were killed and the lungs were lavaged with 3 ml cold PBS. We then measured protein concentration and soluble receptor for advanced glycation end products (RAGE; a marker of alveolar type I cell injury) levels in the BAL.
Excess Lung Water and Lung Extravascular Plasma Equivalent
As previously described (8), the lungs were removed, counted in a γ-counter (Packard, Meriden, CT), weighed, and homogenized (after addition of 1 ml distilled water). The blood was collected through right ventricle puncture. The homogenate was weighed and a fraction was centrifuged (12, 000 rpm, 8 min) for assay of hemoglobin concentration in the supernatant. Another fraction of homogenate, supernatant, and blood were weighed and then desiccated in an oven (60°C for 24 h). We used the following formula to calculate ELW:
- 1. Measurement of the water fraction separately in the lung homogenate (WFH), the supernatant of the homogenate (WFS), and the blood (WFB) by:
where Wwet is the wet weight and Wdry is the dry weight.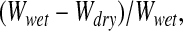
- 2. Calculation of blood volume in the lung:
where 1.039 is the density of blood, QH is the weight of lung homogenate (whole lung wet weight + weight of the distilled water), WFH is the water fraction of lung homogenate, HbS is the hemoglobin concentration of supernatant of lung homogenate, WFS is the water fraction in supernatant of lung homogenate, and HbB is the hemoglobin concentration of blood.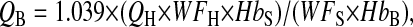
- 3. Calculation of water volume in the lung:
where Ww is the weight of the distilled water added when the lung is homogenized.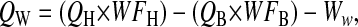
- 4. Calculation of whole lung dry weight (Qd):
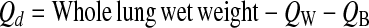
- 5. Calculation of the excess lung water (ELW):
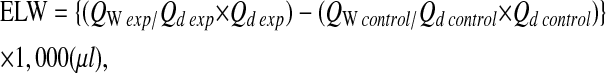
where QW exp equals water volume of the lung in the experimental group; Qd exp equals dry weight of the lung in the experimental group. The controls were the normal mice with the same age as the experimental group.
Lung extravascular plasma equivalents (EPE) (index of lung vascular permeability to protein) were calculated as the counts of 125I-albumin in the blood free lung tissue divided by the counts of 125I-albumin in the plasma.
BAL, Protein Levels, and Leukocyte Count
We used the methods as previously described (8). White blood cells were counted by a counter (Beckman Coulter, Inc., Fullerton, CA). Protein concentration in the BAL was determined by a Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA). The white blood cells were counted by a counter (Beckman Coulter). Blood cell smear was made using cytospin (Shandon Inc, Pittsburgh, PA). The slides were visualized using Wright-Giemsa staining (Fisher Scientific Co., Middletown, VA). Neutrophils were identified by a certified laboratory technologist in a blinded fashion.
Enzyme-Linked Immunosorbent Assay Measurements
The levels of TNF-α and macrophage inflammatory protein (MIP)-2 (analog of IL-8) in the BAL were measured by enzyme-linked immunosorbent assay (R&D Systems Inc., Minneapolis, MN). The soluble RAGE levels in the BAL were measured as previously described (7).
Histology
After mice were killed by inhalation of halothane, the chest and abdomen were rapidly opened and the base of the heart was clamped to prevent escape of the pulmonary blood volume. The thoracic organs were removed en bloc, and 10% formalin was instilled through the trachea at a pressure of 25 cm H2O. After 72 h fixation, lungs were embedded in paraffin, and 5-μm sections were cut and stained with hematoxylin and eosin (H&E), as in our prior studies (8).
Detection of α7 nAChR Expression in the Lung Section by Immunochemistry
Lung sections (5 μm) were deparafinized with xyline, hydrated, and boiled in citrate/citric acid buffer (pH 7.4) for antigen retrieval. Endogenous peroxidase was quenched by H2O2. Rabbit polyclonal IgG α7 nAChR antibody H-302 (1:200 diluted) was added on the sections as the primary antibodies. Histostain SP rabbit primary (AEC) kit (Zymed Laboratories, Inc., San Francisco, CA) was used for antigen detection based on the suggested staining protocol.
Detection of α7 nAChR Expression in Alveolar Macrophages and Neutrophils, Cultured Human Alveolar Type II Cells by Immunofluorescence and Immunohistochemistry
Mice were killed and the lungs of control mice or mice with acid-induced acute lung injury (at 4 h after intratracheal acid challenge) were lavaged with 3 ml PBS to harvest alveolar macrophages and neutrophils. The isolation protocol of human alveolar type II cells (ATII) was previously described by Fang and coworkers (9). Briefly, AT II cells were isolated from cadaveric human lungs that were declined by the Northern California Transplant Donor Network. As described in our prior studies (10, 11), these lungs are in relatively normal physiologic conditions. Using cytospin, alveolar macrophages, neutrophils, and alveolar type II cells were prepared on slides and fixed in 4% paraformaldehyde. The smear was permeablized in 0.1–0.2% triton and incubated with rabbit anti-mouse 1:200 diluted α7 nAChR antibody (H-302). Then, goat-anti-rabbit fluorescein isothiocyanate (FITC)-labeled secondary antibody (Santa Cruz Biotechnology, Inc.) was added on the smear and incubated for 1 h. After washing with hypertonic PBS (2.7% NaCl), the slides were mounted with Vectashield Mounting Medium (Vector Laboratories Inc. Burlingame, CA). For immunohistochemistry, Histostain SP rabbit primary (AEC) kit (Zymed Laboratories, Inc.) was used for antigen detection based on the suggested staining protocol.
NF-κB p65 Western Blot for BAL Proinflammatory Cells
As previously described (12), cytoplasm and nuclear proteins were extracted from BAL proinflammatory cells from saline + saline, saline + acid, and nicotine + acid treated groups (Nuclear Extract Kit; Active Motif, Carlsbad, CA). Denatured protein samples were run on a 4–12% gradient Bis-Tris gel (Invitrogen, Carlsbad, CA). Equal amounts of protein (10–20 μg, each time) per lane was loaded. The gel was run at 100 V for 1.5 h using a MOPS SDS running buffer (Invitrogen). The proteins were then transferred to a nitrocellulose membrane and blocked with blocking buffer for 1 h. The membrane was exposed to NF-κB p65 rabbit polyclonal antibody (H-286) at 1:100 dilution overnight at 4°C. The blot was washed and then exposed to a horseradish peroxidase–labeled antibody at 1:5,000 dilution for 45 min. The protein bands were developed with ECL kit (Amersham, Piscataway, NJ).
Statistical Analysis
Statistics were done by SPSS software (SPSS Inc., Chicago, IL). An unpaired t test was used unless there were multiple comparisons in which case we used ANOVA with post hoc Bonferroni test (significance level set at P < 0.05). The results are shown as mean ± SD.
RESULTS
Expression of α7 nAChR
In the normal mouse lung, α7 nAChR immunoreactivity was present in the bronchial epithelia (Figures 1A and 1B). Because we could not isolate a sufficiently pure population of mouse type II cells, we studied human alveolar epithelial type II cells and found that there was strong α7 nAChR immunoreactivity in these human alveolar type II cells (Figures 1C and 1D). In the normal mouse alveolar macrophage, intense α7 nAChR immunoreactivity was present on the membrane and cytoplasm (Figures 1E and 1F). To study α7 nAChR immunoreactivity in alveolar proinflammatory cells, mice were intratracheally instilled with either saline or acid, and the lungs were lavaged 4 h later. The proinflammatory cells in the airspaces of the lung were collected by performing BAL. In the injured lung, intense α7 nAChR immunoreactivity was found on alveolar neutrophils adjacent to alveolar macrophages (Figures 1G and 1H). The intensity of α7 nAChR immunostaining in proinflammatory cells in the injured lung was increased 3-fold compared with the control (Figure 1I).
Figure 1.

Expression of α7 nAChR in mouse airway epithelial cells, human alveolar type II cells, mouse alveolar macrophages, and neutrophils. (A and B) Representative immunohistochemistry of α7 nAChR in the lung section in mice. (A) Without H-302 antibody; (B) With H-302 antibody. In the inset, red-brownish staining indicates α7 nAChR–positive bronchial epithelial cells. (C and D) Immunofluorescence of α7 nAChR in isolated alveolar epithelial type II cells in human. (C) Without H-302 antibody; (D) With H-302 antibody (the second antibody labeled with FITC). (E and F) Immunofluorescence of α7 nAChR in alveolar macrophages in mice (the second antibody labeled with Cy3). (E) Without H-302 antibody; (F) With H-302 antibody. (G and H) Immunohistochemistry of α7 nAChR in BAL proinflammatory cells. (G) The control group was instilled with saline. (H) The acid-instilled group. Cells were treated with H-302 antibody and Histostain SP rabbit primary (AEC) kit (Zymed Laboratories). Green arrows indicate α7 nAChR–positive alveolar macrophages. Red arrows indicate α7 nAChR–positive neutrophils. (I) The intensity of immunostaining of α7 nAChR in G and H measured by NIH Image-J software.
Nicotine Attenuated Acid-Induced Acute Lung Injury in Mice and Rats
To test whether activation of α7 nAChR would reduce excess lung water and lung vascular permeability in acid-induced acute lung injury, we divided mice into four groups: (1) saline + saline group: the mice were pretreated with saline and then given intratracheal (IT) saline; (ii) nicotine + saline group: the mice were treated with nicotine (3.5 mg/kg, iv) 5 ∼ 10 min before IT saline; (iii) saline + acid group: the mice were pretreated with saline and then intratracheal acid; (iv) nicotine + acid group, the mice were pretreated with nicotine and then received IT acid. Four hours later, blood was withdrawn and the lungs were removed to measure excess lung water and lung vascular permeability. Excess lung water and extravascular plasma equivalents in the saline + acid group was increased 5- to 6-fold compared with the saline + saline group. Excess lung water and extravascular plasma equivalents in the nicotine + acid group were reduced by 60% compared with the saline + acid group (Figures 2A and 2B). Histologically, there was less pulmonary edema and neutrophil infiltration in the nicotine + acid mice compared with the saline + acid mice (Figures 2C and 2D). We also tested the effect of nicotine in acid-induced acute lung injury in rats to verify that the effect of nicotine was not just specific to mice. Also, we tested whether nicotine would affect the release of RAGE, a type I epithelial cell injury marker in the rat acid-induced acute lung injury. Nicotine reduced the BAL protein concentration (a measure of lung vascular permeability) and the levels of RAGE in the BAL compared with the saline therapy (Figures 2E and 2F). To determine whether the protective effect of nicotine on acid-induced acute lung injury is α7 nAChR dependent, mice were separately treated with saline, nicotine (3.5 mg/kg, intravenously), or nicotine (3.5 mg/kg, intravenously) plus MLA (a specific α7 nAChR antagonist which can induce a functional α7 nAChR deficiency, 4 mg/kg, intravenously) (10) 5–10 min before intratracheal instillation of acid. Mice were killed at 4 h. Nicotine decreased excess lung water and lung extravascular plasma equivalents. MLA counteracted the effects of nicotine and restored excess lung water and extravascular plasma equivalents to the acid-induced effects (Figures 2G and 2H).
Figure 2.

(A and B) Nicotine reduced pulmonary edema and lung vascular permeability in mice. (A) Excess lung water. (B) Extravascular plasma equivalents. The mice were pretreated with nicotine 5–10 min before intratracheal instillation of acid and killed at 4 h. **P < 0.01 for saline + acid (n = 6) versus the saline + saline group (n = 5); *P < 0.05 for nicotine + acid (n = 6) versus the saline + acid group (n = 6). (C and D). Representative histologic change (objective magnification: ×20; scale bar: 50 μm) (H&E staining). (C) Saline + acid. (D) Nicotine + acid (n = 3, each). (E and F). Effects of nicotine on acid-induced acute lung injury in ventilated rats. (E) The protein concentration in the BAL. (F) Soluble RAGE levels in the BAL. n = 3, in the saline + acid group; n = 4 in the nicotine +acid group. (G and H). Specific blockade of α7 nAChR counteracted the protective effect of nicotine in acid-induced acute lung injury in mice. To antagonize the effects of nicotine, MLA was also pretreated 5–10 min before acid. (G) Excess lung water. (H) Extravascular plasma equivalents. *P < 0.05 versus acid only; #P < 0.05 for MLA + nicotine versus nicotine treatment only (n = 5 in each group). Data are mean ± SD.
Nicotine Reduced Protein, Neutrophil Differentiation, and Cytokine Levels in the BAL in Mice and Prevented Activation of NF-κB in Mouse Alveolar Macrophages
To determine whether activation of α7 nAChR would affect the protein concentration, neutrophil counts, and cytokine levels in the BAL in acid-induced acute lung injury, mice were divided into three groups: (1) saline + saline group: the mice were pretreated with saline and then given IT saline; (2) saline + acid group: the mice were pretreated with saline and then received IT acid; (3) nicotine + acid group, the mice were pretreated with nicotine (3.5 mg/kg, intravenously) 5–10 min before IT acid. After 4 h, nicotine reduced acid-induced increase in the protein concentration, leukocyte counts, and the number of neutrophils in the BAL compared with saline (Figures 3A–3C). Nicotine also reduced MIP-2 and TNF-α levels in the BAL compared with saline (Figures 3D and 3E).
Figure 3.

Effects of nicotine on protein concentration, neutrophil counts, and cytokine levels in the BAL and activation of NF-κB in the BAL proinflammatory cells in mice. The mice were pretreated with nicotine 5–10 min before intratracheal instillation of acid. (A) Protein concentration. (B) Leukocyte counts. (C) Neutrophil counts. **P < 0.01 for saline +acid (n = 5) versus the saline + saline group (n = 4); #P < 0.05 or ##P < 0.01 nicotine + acid (n = 5) versus the saline + acid group (n = 5). (D and E). Cytokine levels in the BAL. (D) TNF-α. (E) MIP-2. *P < 0.05 for nicotine + acid (n = 5) versus the saline + acid (n = 4) group. Data are mean ± SD. (F) Western blot for NF-κB p65 in the cytoplasm and nucleus in the BAL proinflammatory cells (n = 3 in each group).
To study whether activation of α7 nAChR would alter activation of NF-κB p65 in acid-induced acute lung injury, mice were divided into three groups: (1) the saline + saline group, the mice were pretreated with saline, and then given IT saline; (2) the saline + acid group, the mice were pretreated with saline, and then received IT acid; (3) the nicotine + acid group, the mice were pretreated with nicotine (3.5 mg/kg, intravenously) 5–10 min before IT acid. Four hours later, proinflammatory cells were lavaged to perform Western blot for NF-κB p65. Western blot displayed faint bands in the saline + acid and nicotine + acid group in the cytoplasm in the BAL proinflammatory cells. The intensity of 65 kD band was reduced in the nucleus in the BAL proinflammatory cells in the nicotine + acid group compared with the acid group (Figure 3F).
Choline Attenuated Acid-Induced Acute Lung Injury in Mice
To test whether activation of α7 nAChR by choline (EC50 of 1.6 mM, a full α7 nAChR agonist) (13) would alter the course of acid-induced acute lung injury, mice were pretreated with saline or choline (60 mg/kg, intravenously) 5–10 min ahead of intratracheal instillation of acid or saline. Mice were killed 4 h later, and the lungs were harvested or lavaged with PBS. Intratracheal instillation of acid increased excess lung water and extravascular plasma equivalents. Administration of choline decreased excess lung water and extravascular plasma equivalents by 2-fold compared with the saline-treated group (Figures 4A and 4B). Neutrophil accumulation in the airspaces was increased 4 h after intratracheal challenge with acid, while injection of choline decreased the neutrophil differentiation in BAL compared with the saline-treated group (Figures 4D and 4E).
Figure 4.

Effects of choline on excess lung water, extravascular plasma equivalents, and neutrophil differentiation in the BAL in mice. The mice were pretreated with choline 5–10 min before intratracheal instillation of saline or acid. (A) Excess lung water. (B) Extravascular plasma equivalents. **P < 0.01 for the saline + acid (n = 5) versus the saline + saline group (n = 4); #P < 0.05 for the nicotine + acid (n = 5) versus the saline + acid group. (C) Neutrophil differentiation in the BAL. **P < 0.01 versus the saline + saline group; ##P < 0.01 versus the saline + acid group (n = 3 in each group). (D and E). BAL cytological change (Wright-Giemsa staining). (D) The saline + acid group (n = 4). (E) The choline + acid group (n = 5). Arrows indicate neutrophils. Data are mean ± SD.
PNU-282987 Attenuated Acid-Induced Acute Lung Injury in Mice
To confirm whether activation of α7 nAChR would reduce acid-induced acute lung injury, PNU-282987 (a highly specific α7 nAChR agonist, Ki of 27 nM) (14) was given (2.4 mg/kg, intraperitoneally) 20 min before acid instillation. The control group received the same volume of saline. After 4 h, PNU-282987 reduced excess lung water and extravascular plasma equivalents compared with saline (Figures 5A and 5B). PNU-282987 also decreased neutrophil count and protein level in the BAL (Figures 5C and 5D). PNU-282987 reduced TNF-α and MIP-2 levels in the BAL (Figures 5E and 5F).
Figure 5.

Effects of PNU-282987 (a specific α7 nAChR agonist) on excess lung water, extravascular plasma equivalents, neutrophil accumulation, protein concentration, and cytokine levels in the BAL in the acid-induced acute lung injury in mice. (A) Excess lung water. (B) Extravascular plasma equivalents. **P < 0.01 for PNU282987 versus the saline group (n = 5 in each group). (C) Neutrophil counts in the BAL. (D) Protein concentration in the BAL. (E and F). TNF-α and MIP-2 levels in the BAL. *P < 0.05, **P < 0.01 for PNU282987 versus the saline group (n = 5 in each group). Data are mean ± SD.
Knockout of α7 nAChR Reduced Acid-Induced Acute Lung Injury in Mice
To test whether α7 nAChR deficiency would worsen acid-induced acute lung injury, wild-type (n = 5) and α7 nAChR–deficient mice (n = 4) were intratracheally challenged with acid. One mouse in the α7 nAChR–deficient group died at 2 h, and the lung was grossly edematous. The other three α7 nAChR knockout mice manifested more severe respiratory distress compared with the wild-type. No death occurred in the wild-type group. Four hours later, the excess lung water and lung vascular permeability in the α7 nAChR–deficient mice increased 2-fold compared with the wild-type (Figures 6A and 6B).
Figure 6.

Deficiency of α7 nAChR worsened acid induced acute lung injury in mice. (A) Excess lung water. (B) Extravascular plasma equivalents. *P < 0.05,**P < 0.01 for wild-type versus knockout mice (n = 5 wild-type, n = 4 α7 nAChR knockout). Data are mean ± SD.
DISCUSSION
Acute lung injury in association with gastric aspiration carries a mortality of up to 30% (15). NF-κB plays a key role in the molecular and cellular events in acid-induced acute lung injury (16). Depletion of alveolar macrophages results in decreased production of inflammatory mediators in acid aspiration (23–80%) (15). In addition, the neutrophil is an important mediator of acid-induced acute lung injury (17). Thus, with an animal model of acid-induced lung injury, it is possible to test the hypothesis that activation of α7 nAChR may regulate pulmonary edema and proinflammatory responses in acid-induced acute lung injury.
The importance of the cholinergic anti-inflammatory pathway has recently been recognized (1, 2). Acetylcholine released by efferent vagus nerves inhibits macrophage activation (18). α7 nAChR is required for acetylcholine inhibition of macrophage TNF release (2). Nicotine, as the prototype agonist of nAChR, can protect mice from lethal sepsis (4) by inhibiting HMGB1 production. A selective α7 nAChR agonist (2.4-dimethoxybenzylidene anabaseine dihydrochloride, GTS-21) has been used to attenuate experimental pancreatitis (19). In this study, to the best of our knowledge, we first demonstrated that activation of α7 nAChR with its agonists (nicotine, choline, and PNU-282987) protected against acid-induced acute lung injury as reflected by a marked reduction in excess lung water, lung vascular permeability, proinflammatory cytokines and protein in the BAL, neutrophil infiltration, and epithelial cell injury (decreased RAGE level in the BAL). Conversely, blockade of α7 nAChR with MLA (an antagonist) counteracted the protective effect of nicotine and deficiency of α7 nAChR worsened acid-induced acute lung injury.
Activation of α7 nAChR by its endogenous agonists (acetylcholine, or choline, a metabolite of acetylcholine) is a key pathway to limit lung inflammation. This notion was confirmed by deficiency of α7 nAChR resulted in a 2-fold increase in excess lung water and lung vascular permeability in the acid-induced acute lung injury. Alveolar macrophages and neutrophils expressed α7 nAChR, which possessed acting sites for endogenous and exogenous α7 nAChR agonists. In the injured lung, α7 nAChR expression in alveolar macrophages and neutrophils was increased. Up-regulation of α7 nAChR expression in these proinflammatory cells may probably compensate for the inflammation and also may provide more binding sites for exogenous α7 nAChR agonists. Activation of α7 nAChR inhibited NF-κB activity, which was demonstrated by reduction of NF-κB p65 translocation. As a result of reduced NF-κB activation, activation of α7 nAChR reduced proinflammatory cytokine (TNF-α, MIP-2) levels in the airspaces of the lung. Decreased MIP-2 production from alveolar macrophages may also reduce neutrophil chemotaxis to the airspaces and further reduce the severity of lung inflammation and injury.
Lung airway epithelia are innervated by the vagus nerve, where alveolar epithelial cells have access to endogenous α7 nAChR agonists (acetylcholine and choline). Also, alveolar epithelial II type cells express α7 nAChR. The lung epithelium is an important locus of NF-κB activation during inflammation (20). It is still unclear whether stimulation of α7 nAChR can suppress NF-κB activity and reduce proinflammatory cytokine production. Thus, we cannot rule out an inhibitory effect on inflammation of activation of α7 nAChR by lung epithelial cells in this study.
In addition to human bronchial cells, the endothelial cells express α7 nAChR (21). It is possible that activation of α7 nAChR in the endothelial cells reduces neutrophil adhesion and infiltration. As expected, neutrophils were the main cell population in the airspaces of the lung 4 h after acid instillation. Administration of nicotine, choline, and PNU-282987 reduced neutrophil accumulation in the airspaces. This result can be explained by reduction of MIP-2 and neutrophil chemotaxis. Another explanation is that activation of α7 nAChR in the endothelial cells may reduce endothelial cell activation and leukocyte binding (22).
In the endotoxemia mouse model, activation of α7 nAChR by vagus nerve stimulation reduced the levels of TNF-α in the plasma, liver, and spleen, but not in the lung (23, 24). There may be several reasons to explain why their findings were different from ours: (1) The endotoxemia model is different from our acid-induced acute lung injury model. (2) The TNF-α was measured only in the lung homogenate. Thus, we cannot rule out that there was a difference in the TNF-α level in the BAL because alveolar macrophages are the major sources of TNF-α. (3) LPS was given by intravenous injection rather than intratracheal instillation. Even with intratracheal instillation of LPS, 12–24 h are required to develop peak pulmonary edema and inflammation. Thus, we believe that the lungs were probably not injured sufficiently at 90 and 180 min after intravenous injection of LPS to demonstrate the beneficial effect of vagus nerve stimulation.
Taken together, activation of α7 nAChR can inhibit NF-κB activation in alveolar macrophages and reduces the production of proinflammatory cytokines (MIP-2 and TNF-α). Thus, administration of α7 nAChR agonist can reduce neutrophil accumulation in the airspaces of the lung as well as decrease pulmonary edema and lung inflammation. These results suggest that activation of α7 nAChR may be useful as a novel anti-inflammatory therapy to treat acute lung injury.
Acknowledgments
The authors appreciate the helpful suggestions and technical assistance of Mark Looney, M.D., Yuanlin Song, M.D., and Sandra Brady.
This work was supported by NIH Grant NHLBI HL51845 and HL51856 (to M.A.M.).
Originally Published in Press as DOI: 10.1165/rcmb.2006-0240OC on April 12, 2007
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000;405:458–462. [DOI] [PubMed] [Google Scholar]
- 2.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421:384–388. [DOI] [PubMed] [Google Scholar]
- 3.van Westerloo DJ, Giebelen IA, Florquin S, Daalhuisen J, Bruno MJ, de Vos AF, Tracey KJ, van der Poll T. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. J Infect Dis 2005;191:2138–2148. [DOI] [PubMed] [Google Scholar]
- 4.Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al-Abed Y, Wang H, Metz C, Miller EJ, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004;10:1216–1221. [DOI] [PubMed] [Google Scholar]
- 5.Matthay MA, Ware LB. Can nicotine treat sepsis? Nat Med 2004;10:1161–1162. [DOI] [PubMed] [Google Scholar]
- 6.Su X, Looney M, Robriquet L, Fang X, Matthay MA. Direct visual instillation as a method for efficient delivery of fluid into the distal airspaces of anesthetized mice. Exp Lung Res 2004;30:479–493. [DOI] [PubMed] [Google Scholar]
- 7.Uchida T, Shirasawa M, Ware LB, Kojima K, Hata Y, Makita K, Mednick G, Matthay ZA, Matthay MA. Receptor for advanced glycation end-products is a marker of type I cell injury in acute lung injury. Am J Respir Crit Care Med 2006;173:1008–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su X, Camerer E, Hamilton JR, Coughlin SR, Matthay MA. Protease-activated receptor-2 activation induces acute lung inflammation by neuropeptide-dependent mechanisms. J Immunol 2005;175:2598–2605. [DOI] [PubMed] [Google Scholar]
- 9.Fang X, Song Y, Hirsch J, Galietta LJ, Pedemonte N, Zemans RL, Dolganov G, Verkman AS, Matthay MA. Contribution of CFTR to apical-basolateral fluid transport in cultured human alveolar epithelial type II cells. Am J Physiol Lung Cell Mol Physiol 2006;290:L242–L249. [DOI] [PubMed] [Google Scholar]
- 10.Ware LB, Wang Y, Fang X, Warnock M, Sakuma T, Hall TS, Matthay MA. Assessment of lungs rejected for transplantation and implications for donor selection. Lancet 2002;360:619–620. [DOI] [PubMed] [Google Scholar]
- 11.Frank JA, Briot R, Lee JW, Ishizaka A, Uchida T, Matthay MA. Physiological and biochemical markers of alveolar epithelial barrier dysfunction in perfused human lungs. Am J Physiol Lung Cell Mol Physiol. Epub 2007, Mar 9. [DOI] [PMC free article] [PubMed]
- 12.Chilton M, Mastropaolo J, Rosse RB, Bellack AS, Deutsch SI. Behavioral consequences of methyllycaconitine in mice: a model of alpha7 nicotinic acetylcholine receptor deficiency. Life Sci 2004;74:3133–3139. [DOI] [PubMed] [Google Scholar]
- 13.Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX. Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci 1997;9:2734–2742. [DOI] [PubMed] [Google Scholar]
- 14.Bodnar AL, Cortes-Burgos LA, Cook KK, Dinh DM, Groppi VE, Hajos M, Higdon NR, Hoffmann WE, Hurst RS, Myers JK, et al. Discovery and structure-activity relationship of quinuclidine benzamides as agonists of alpha7 nicotinic acetylcholine receptors. J Med Chem 2005;48:905–908. [DOI] [PubMed] [Google Scholar]
- 15.Beck-Schimmer B, Rosenberger DS, Neff SB, Jamnicki M, Suter D, Fuhrer T, Schwendener R, Booy C, Reyes L, Pasch T, et al. Pulmonary aspiration: new therapeutic approaches in the experimental model. Anesthesiology 2005;103:556–566. [DOI] [PubMed] [Google Scholar]
- 16.Madjdpour L, Kneller S, Booy C, Pasch T, Schimmer RC, Beck-Schimmer B. Acid-induced lung injury: role of nuclear factor-kappaB. Anesthesiology 2003;99:1323–1332. [DOI] [PubMed] [Google Scholar]
- 17.Folkesson HG, Matthay MA, Hebert CA, Broaddus VC. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest 1995;96:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 2005;6:844–851. [DOI] [PubMed] [Google Scholar]
- 19.van Westerloo DJ, Giebelen IA, Florquin S, Bruno MJ, Larosa GJ, Ulloa L, Tracey KJ, van der Poll T. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology 2006;130:1822–1830. [DOI] [PubMed] [Google Scholar]
- 20.Quinton LJ, Jones MR, Simms BT, Kogan MS, Robson BE, Skerrett SJ, Mizgerd JP. Functions and regulation of NF-κB RelA during pneumococcal pneumonia. J Immunol 2007;178:1896–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Pereira EF, Maus AD, Ostlie NS, Navaneetham D, Lei S, Albuquerque EX, Conti-Fine BM. Human bronchial epithelial and endothelial cells express alpha7 nicotinic acetylcholine receptors. Mol Pharmacol 2001;60:1201–1209. [DOI] [PubMed] [Google Scholar]
- 22.Saeed RW, Varma S, Peng-Nemeroff T, Sherry B, Balakhaneh D, Huston J, Tracey KJ, Al-Abed Y, Metz CN. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med 2005;201:1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa L, Yang H, Sudan S, Czura CJ, Ivanova SM, Tracey KJ. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med 2002;195:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, Gallowitsch-Puerta M, Ashok M, Czura CJ, Foxwell B, et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 2006;203:1623–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]