

Abstract
Virus-specific CD4+ T cells contribute to effective virus control through a multiplicity of mechanisms including direct effector functions as well as “help” for B cell and CD8+ T cell responses. Here, we have used the lymphocytic choriomeningitis virus (LCMV) system to assess the minimal constraints of a dominant antiviral CD4+ T cell response. We report that the core epitope derived from the LCMV glycoprotein (GP) is 11 amino acids in length and provides optimal recognition by epitope-specific CD4+ T cells. Surprisingly, this epitope is also recognized by LCMV-specific CD8+ T cells and thus constitutes a unique viral determinant with dual MHC class I- and II-restriction.
Keywords: Virus, LCMV, T cell response, CD4, CD8, MHC class I, MHC class II
Introduction
Effective control of microbial infections, in particular viruses, relies on the concerted activity of pathogen-specific T and B cells. While the relative contribution of individual immune cell subsets depends on the precise determinants of host-pathogen interaction (nature of the pathogen, route of pathogen entry, pathogen dosage and host immune status), CD8+ T cells and B cells constitute the main effector cell populations in most viral infections (Burton, 2002; Whitton and Oldstone, 2001; Zinkernagel, 2002). In contrast, antiviral CD4+ T cell responses are for the most part significantly smaller than CD8+ T cell responses and may wane over time (Seder and Ahmed, 2003; Sprent and Surh, 2002). Nevertheless, CD4+ T cells play a critical and multifaceted role at all stages of the adaptive immune response by serving as effectors, helpers and/or regulators/supressors.
Direct antiviral effector functions elaborated in vivo by specific CD4+ T cells include the production of cytokines and chemokines and possibly cytotoxic T cell activity (Appay, 2004; Jenkins et al., 2001; Seder and Paul, 1994). The traditional designation of CD4+ T cells as “helper T cells” is based on their importance for enhancement and modulation of specific antibody responses (McHeyzer-Williams et al., 2003), but work over the past few years has made increasingly clear that additional help is also provided to CD8+ T cell responses (reviewed, Bevan, 2004). Here, the relevant molecular interactions remain to be elucidated in detail but it appears that, depending on the in vivo model system under study, induction of primary CD8+ T cell responses, generation and maintenance of CD8+ T cell memory as well as secondary CD8+ T cell immunity are all improved by the activity of specific CD4+ T cells (Bevan, 2004; Homann, 2002). The complex function of antiviral CD4+ T cell immunity is further supported by studies on relevant human diseases such as HIV, EBV, CMV, HBV and HCV infections (Norris and Rosenberg, 2002). Altogether, it may be said that the relative importance of antigen-specific CD4+ T cell responses increases with the extent of viral disease (viral load and associated inflammatory alterations) (Homann, 2002). Finally, in their incarnation as “regulatory T cells”, CD4+ T cells are actively engaged in curtailing immune responses which may have beneficial (limitation of immunopathology) or adversarial (incapacitation of CD8+ T cell responses) effects (Belkaid and Rouse, 2005; Dittmer et al., 2004; Mittrucker and Kaufmann, 2004).
In the present study, we have employed the lymphocytic choriomeningitis virus (LCMV) system to further define the nature of antiviral CD4+ T cell responses (Homann et al., 2001; Kamperschroer and Quinn, 1999; Lenz et al., 2004; Oxenius et al., 1995; Varga and Welsh, 1998a, 1998b, 2000; Whitmire et al., 1998). Originally identified by Oxenius et al. (1995) the immunodominant population of LCMV-specific CD4+ T cells in C57Bl/6J mice targets an epitope derived from the viral glycoprotein (GP61–80) and restricted by I-Ab. Here, we have used several GP61–80-specific CD4+ T cell lines and clones, a battery of truncated peptides from the GP61–80 epitope and surface plasmon resonance to determine the minimal constraints and avidities of a dominant, virus-specific T cell receptor (TCR)–MHC interaction.
Results
Generation of LCMV-specific CD4+ T cell lines and clones
To generate CD4+ T cell lines and clones specific for the I-A b-restricted LCMV GP61–80 epitope, a C57Bl6/J mouse was infected with LCMV Armstrong clone 53b (1.5×105pfu i.p.) to induce LCMV-specific T cell responses and virus clearance. Thirteen days later, spleen cells were isolated, depleted of CD8+ T cells and cultured in the presence of GP61–80 peptide as detailed in Materials and methods. Forty CD4+ T cell lines and/or clones were established and tested for TCR usage and IFNγ production, a hallmark of LCMV-specific CD4+ T cells (Homann et al., 2001; Kamperschroer and Quinn, 1999; Varga and Welsh, 1998a; Whitmire et al., 1998). As about 1/3 of GP61–80-specific effector (data not shown) and memory (Homann et al., 2001) CD4+ T cells utilize Vβ8.1/8.2 TCRs, we selected 6 cell lines/clones for further analysis based on Vβ8.1/8.2 and Vα2 TCR expression as well as efficient IFNγ induction after resting and restimulation with GP61–80 peptide (Figs. 1 and 2B).
Fig. 1. Establishment and functional characterization of GP61–80-specific CD4+ T cell lines and clones.
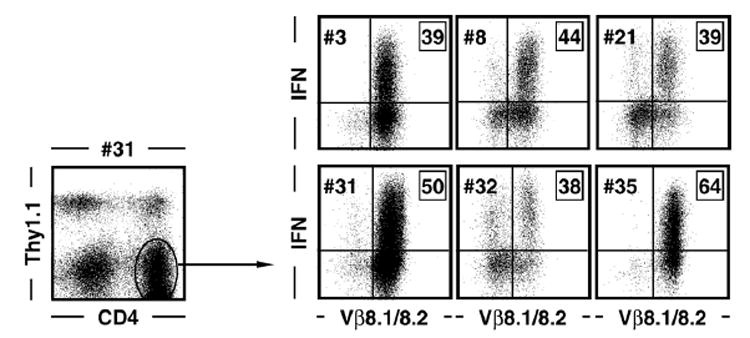
CD4+ T cell lines and clones were generated as detailed in Materials and methods. To evaluate antigen-specific T cell function and TCR usage, cells were rested by antigen withdrawal, supplemented with congenic (Thy1.1+) spleen cells to provide a source for MHC class II-restricted antigen presentation and stimulated for 5 h with GP61–80 peptide. Cells were subsequently stained for CD4, Thy1.1 and TCR Vβ8.1/8.2 as well as intracellular IFNγ. Dot plots on the right are gated on Thy1.1-negative CD4+ T cells as indicated by the exemplary dot plot on the left; boxed values indicate percentages of IFNγ+ gated CD4+ T cells.
Fig. 2. T cell receptor usage by GP61–80-specific CD4+ T cell clones/lines.
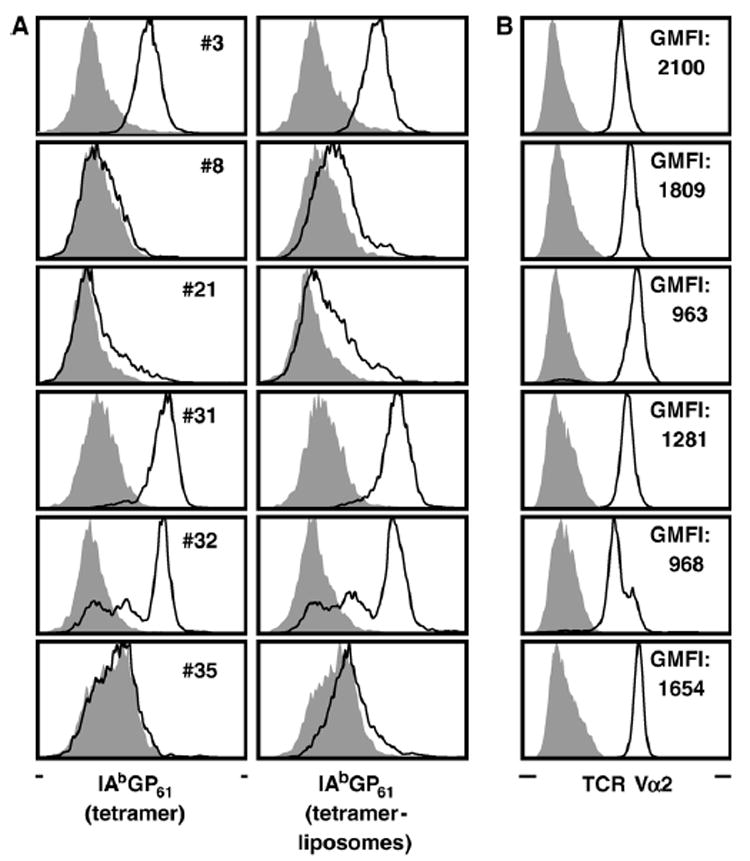
(A) CD4+ T cell clones/lines were rested in the absence of antigen and stained for CD4 as well as tetramers (left column) or tetramer-liposomes (right column) as detailed in Materials and methods. Black tracings indicate staining with I-AbGP61–80 tetramers or tetramer-liposomes; gray histograms indicate control stains with I-AbOVA323–339 tetramers or tetramer-liposomes. (B) In parallel stains, TCR expression levels were determined by staining for TCR Vα2 (black tracing); for control stains (gray histograms) cells were stained for TCR Vα8. Values represent the geometric mean of fluorescence intensity (GMFI) of Vα2 stains.
Differential I-AbGP61–80 binding by CD4+ T cell lines and clones
We subsequently evaluated the selected cell lines/clones for their ability to bind I-AbGP61–80 tetramers. Here, three distinct staining patterns emerged: (1) efficient tetramer binding (clones #3 and #31), (2) partial tetramer binding indicative of more than one TCR specificity present in the cell preparation (cell lines #21 and #32) and (3) absence of tetramer binding (clones/cell lines #8 and #35) (Fig. 2A). To evaluate if effective tetramer staining could be “rescued”, we used a more sensitive staining technique, tetramers deployed on fluorescent liposomes (Mallet-Designé et al., 2003). Although this approach reliably improved the “signal-to-noise ratio” for clones #3 and #31, it demonstrated only slight staining above background levels for clones/cell lines #8, #21 and #35 (Fig. 2A). Furthermore, tetramer binding did not demonstrate any appreciable correlation with the extent of TCR or CD4 co-receptor expression on the cell surface as determined by measuring the mean fluorescent intensity of Vβ8.1/8.2, Vα2 and CD4 staining (Fig. 2B and data not shown).
Identification of an 11 amino acid core epitope (GP67–77)
In spite of their differential ability to bind MHC class II tetramers, the prompt induction of IFNγ production in the selected cell lines/clones (#3, #8, #21, #31, #32, #35) made this functional assay suitable to determine the minimal constraints of I-Ab-restricted GP61–80 epitope presentation and TCR reactivity. Using a panel of truncated peptides and comparing the extent of IFNγ induction to that elicited by the native GP61–80 peptide, we identified a core motif of 11 amino acids (GP67–77) that on average induces 2/3 of the maximal IFNγ response in the 6 cell lines/clones (Fig. 3A). When tested on primary LCMV-specific CD4+ T cells (8 days after LCMV infection), the GP67–77-specific response was consistently of slightly greater magnitude as compared to GP61–80- or GP64–80-specific responses (Fig. 3B). These results indicate that flanking residues of the core epitope present in the GP61–80 and GP64–80 peptides may preclude recognition of the epitope by a subset of virus-specific CD4+ T cells and identify GP67–77 as the immunodominant LCMV-specific, I-Ab-restricted CD4+ T cell core epitope.
Fig. 3. Identification of the minimal GP61–80 binding constraints.
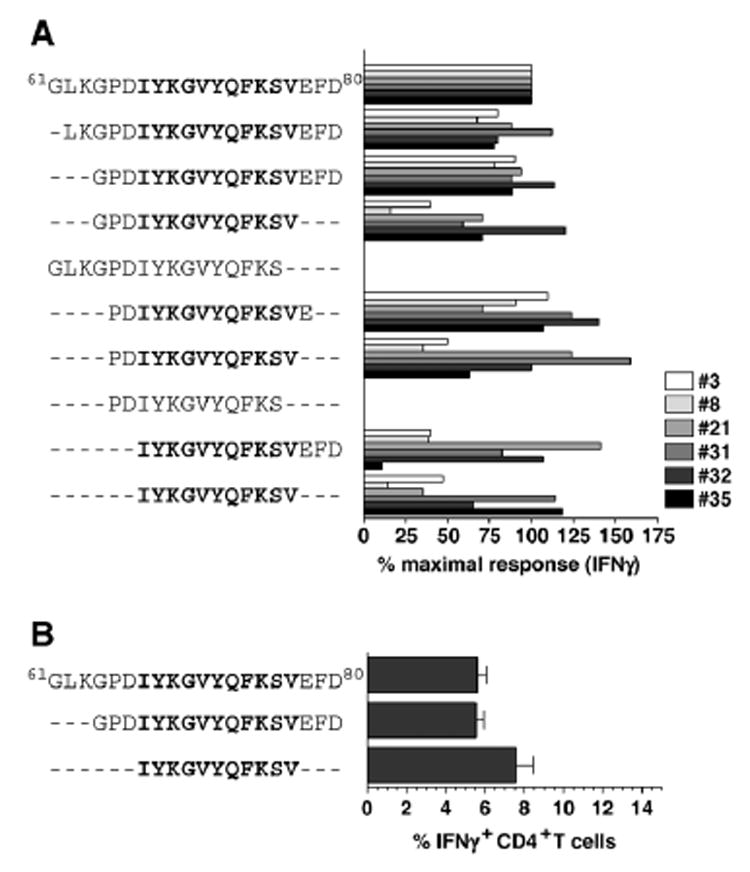
(A) CD4+ T cell clones and lines #3, #8, #21, #31, #32 and #35 were rested by antigen withdrawal, restimulated with indicated peptides and stained for intracellular IFN. The percentage of IFNγ+ cells after stimulation with the native GP61–80 peptide of individual cell lines or clones was set at 100% and the values after stimulation with truncated peptides were calculated accordingly. All clones/cell lines (#3, #8, #21, #31, #32 and #35) were tested in parallel. Data for individual clones/cell lines obtained in two separate experiments were combined according to peptide sequences used for stimulation; no statistical analysis was performed as each value consists of only 2 data points. (B) Eight days after LCMV infection of B6 mice, spleen cells were restimulated with indicated peptides and frequencies of epitope-specific CD4+ T cells were determined by intracellular IFNγ staining (3 mice/experiment; values indicate SEM of one out of four independent experiments).
Restriction of the core epitope GP67–77 by both I-Ab and Db
During our analyses of peptide-induced IFNγ production by primary murine effector CD4+ T cells (Fig. 3A), we noted unusually high “background staining” in CD4-negative cells obtained from cultures stimulated with GP67 but not GP64 or NP309 peptides (Fig. 4A). We therefore evaluated the possibility that the GP67–77 epitope may also be recognized in an MHC-I-restricted fashion by LCMV-specific CD8+ T cells. In B6 mice, the majority of specific CD8+ T cells recognize two co-dominant epitopes restricted by Db (GP33–41 and NP396–404) as well as two subdominant epitopes restricted by Db and Kb, respectively (GP276–286 and GP34–43) (Gairin et al., 1995; Hudrisier et al., 1997; Klavinskis et al., 1990; Oldstone et al., 1988; Schulz et al., 1989). More recently, several additional subdominant epitopes have been identified and include the epitopes GP92–101 and NP166–175 (Db-restricted) as well as GP118–125, and NP205–212 (Kb-restricted) (van der Most et al., 2003; van der Most et al., 1998). Indeed, we found that the GP67–77 epitope is recognized by both LCMV-specific CD4+ and CD8+ T cells (Fig. 4B) and that Db is the restriction element for specific CD8+ T cell responses (Fig. 4C) Thus, the GP67–77 epitope constitutes a rather unique viral determinant as it is targeted, by virtue of its dual restriction, by both CD4+ and CD8+ T cell populations. Furthermore, since stimulation with the GP64–80 peptide was not associated with IFNγ induction in CD8+ T cells unless provided at excessively high concentrations (50 μg/ml, data not shown), the presence of the 3 additional carboxy-terminal (GP64–66) and amino-terminal(GP78–80) residues apparently interferes with effective Db-restricted presentation.
Fig. 4. Recognition of the GP67–77 core epitope by both virus-specific CD4+ and CD8+ T cells.
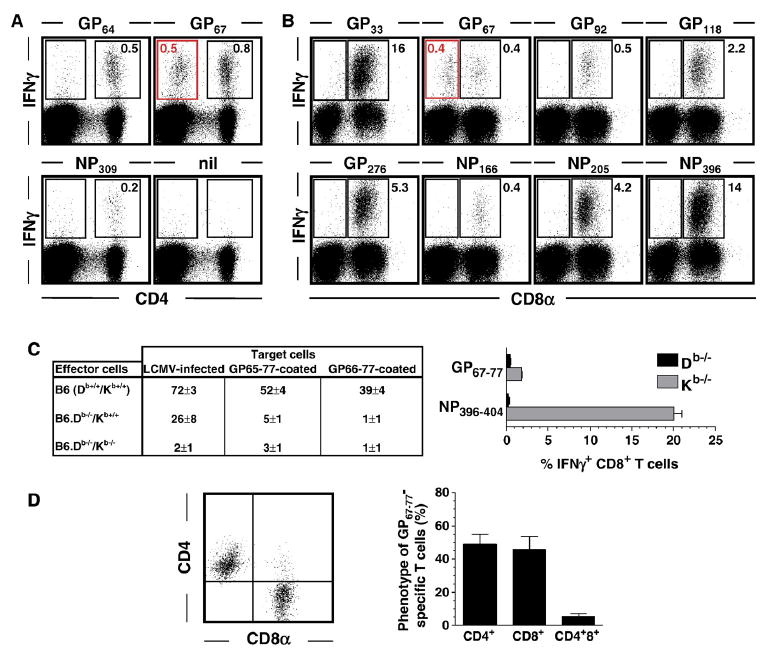
Spleen cells harvested from B6 mice infected with 1.5×105 pfu LCMV Armstrong i.p. 8 days earlier were stimulated for 5 h with the indicated peptides (nil: no peptide) and analyzed for induced IFNγ expression (Plots are gated on spleen cells according to FSC/SSC properties. Values indicate percentages of IFNγ+ cells in indicated rectangular gates; absence of values in some gates refers to background staining levels of <0.1%). (A) Analysis of specific CD4+ T cell responses demonstrates IFNγ induction in CD4-negative cells only after stimulation with GP67 (red gate) but not GP61 or NP309 peptides. (B) Parallel analyses of IFNγ production by CD8+ T cells revealed induction of IFNγ in CD8-negative cells only after stimulation with GP67 peptide (red gate). (C) Left panel: Seven days after LCMV infection of B6 (Db+/+/Kb+/+), B6.Db−/−/Kb+/+ and B6.Db−/−/Kb−/− mice, spleen cell suspensions were tested for specific epitope-specific CTL activity using virus-infected or peptide-coated target cells in a conventional 5 h ex vivo CTL assay. Specific lysis for effector:target cell cultures at a ratio of 50:1 is shown; values indicate the percentage (SEM) of 51Cr release from target cells. Note that lack of Db expression abolishes killing of GP65–77 and GP67–77 coated target cells. Right panel: Eight days after infection of B6.Db−/−/Kb+/+ and B6.Db+/+/Kb−/− mice, epitope-specific CD8+T cell responses were quantified by peptide restimulation and intracellular IFNg staining; displayed are the percentages of epitope-specific CD8+T cells (3 mice/group, SEM). Note that lack of Db expression abolishes GP67–77-induced IFNg production. The NP396–404-specific IFNg response has been included as a control for a bona fide Db-restricted CD8+T cell response. (D) Phenotype of GP67–77-specific T cells. Eight days after LCMV infection of B6 mice, spleen cells were restimulated with GP67–77 peptide and stained for CD8α, CD4 and intracellular IFNγ. The dot plot is gated on IFNγ+T cells indicating the distribution of GP67–77-specific T cells among CD8+ and CD4+ subsets. The adjacent bar diagram summarizes these findings (3 mice/experiment; 3 independent experiments).
Finally, the distribution of GP67–77-specific T cells among CD4+ and CD8+ T cell subsets was found to be about similar with 45–50% of GP67–77-reactive T cells expressing either CD4 or CD8 and a minority of T cells specific for this epitope (<10%) expressing both co-receptors (Fig. 4D).
TCR Vβ usage and functional avidities of primary I-Ab and Db-restricted GP67–77-specific T cells
To assess the distribution of TCR usage by GP67–77-restricted CD4+ and CD8+ T cells, we utilized a panel of TCR Vβ-specific antibodies in conjunction with intracellular IFNγ staining. GP67–77-specific CD4+ T cells were preferentially recruited from just two TCR Vβ families (8.1/8.2 and 14) with several other TCR Vβ families contributing less than 5% each to the overall GP67–77-restricted CD4+ T cell response. In contrast, GP67–77-specific CD8+ T cells appeared more diversified as indicated by five TCR Vβ families (2, 8.1/8.2, 8.3, 9, 10) contributing >5% to the overall specific response as well as larger error bars in the bar diagram reflective of differential private TCR Vβ usage among individual mice (Fig. 5A).
Fig. 5. TCRVβ usage and functional avidities of GP67–77-specific CD4+ and CD8+ effector Tcells.

(A) Effector spleen cells (8 days after LCMV) were restimulated for 5 h with GP67–77 peptide and stained for surface markers (CD8, CD4, TCRVβ subfamily) and intracellular IFNγ. Bar diagrams display relative distribution of TCRVβ usage by GP67–77-specific (IFNγ+) CD4+ (black) and CD8+ (gray) Tcells. The dotted line indicates the level of non-specific background staining. Bars indicate mean± SE from 2 independent experiments using 2–3 mice each. (B) Functional avidities were determined by culture of effector spleen cell suspension with graded dosages of GP61–80 or GP67–77 peptide and plotting of IFNγ+ effector T cell frequencies (mean±SE) against the peptide concentration used for restimulation (data from 1 of 3 independent experiments using 2–3 mice each). Values listed below the graphs represent the EC50 for indicated peptide specificities and specific Tcells, i.e. the peptide concentration required to elicit detectable IFNγ induction in half of the GP61–80- (left) and GP67–77- specific (right) CD4+ or CD8+ Tcell populations (mean±SE, 1 of 3 independent experiments). Although EC50 values of GP67–77- specific CD4+ and CD8+ T cells as well as of GP61–80- and GP67–77-specific CD4+ T cells were not significantly different, GP67–77-specific CD8+ T cells exhibited a lower EC50 when compared to GP61–80-specific CD4+ T cells (p=0.0085).
We and others have previously reported that the functional avidities of LCMV-specific CD4+ T cells (measured by determining the reciprocal peptide concentration required for IFNγ induction in half of an epitope-specific T cell population) are considerably lower than those of LCMV-specific CD8+ T cells (Homann et al., 2002; 2001; Whitmire et al., 2006). However, a careful interpretation of such data has to consider that induction of effector functions in this assay is dependent on both T cell-intrinsic properties (“functional avidities”) as well as differential binding affinities of synthetic peptides to MHC class I or II molecules (“structural aviditities”). Our observation of virus-specific CD4+ and CD8+ T cells responses specific for the same peptide epitope now allows for a more direct comparison of functional avidities between these cell lineages and indicates comparable functional avidities of GP 67–77-specific CD4+ and CD8+ T cell populations (Fig. 5B).
Fitting of the core epitope GP67–77 to I-Ab motifs
Based on the identity of naturally processed I-Ab peptides and the recently solved of I-Ab crystal structure (Dongre et al., 2001; Zhu et al., 2003), we proposed a common I-Ab-restricted peptide-binding motif. When applied to the LCMV-GP sequences, we found that no optimal alignment could be detected for the 11-mer GP67–77 peptide or longer variants of the same peptide (Fig. 6A). Attempts to optimize the two most likely sequence alignments (K-Y-F-V and Y-V-Q-S for P1-P4-P6-P9, respectively) by substituting non-optimal residues with canonical residues at the same position failed to produce better agonistic peptides (Fig. 6B and LT/MBAO, unpublished). The absence of an optimum register for the GP peptide suggests that like in the case of I-Ad and I-Ag7, peptides bound to I-Ab can adopt different alternate registers of binding (Corper et al., 2000; Scott et al., 1998). This capacity to use alternate registers has two practical consequences: (i) lower peptide affinity for the MHC and (ii) increase the repertoire of peptides from any given pathogen and therefore increase T cell repertoire against this pathogen. In the case of LCMV, the usage of this strategy to increase the efficiency of the anti-viral response appears very appropriate.
Fig. 6. Fitting of dominant LCMV-GP epitope sequences to I-Ab.
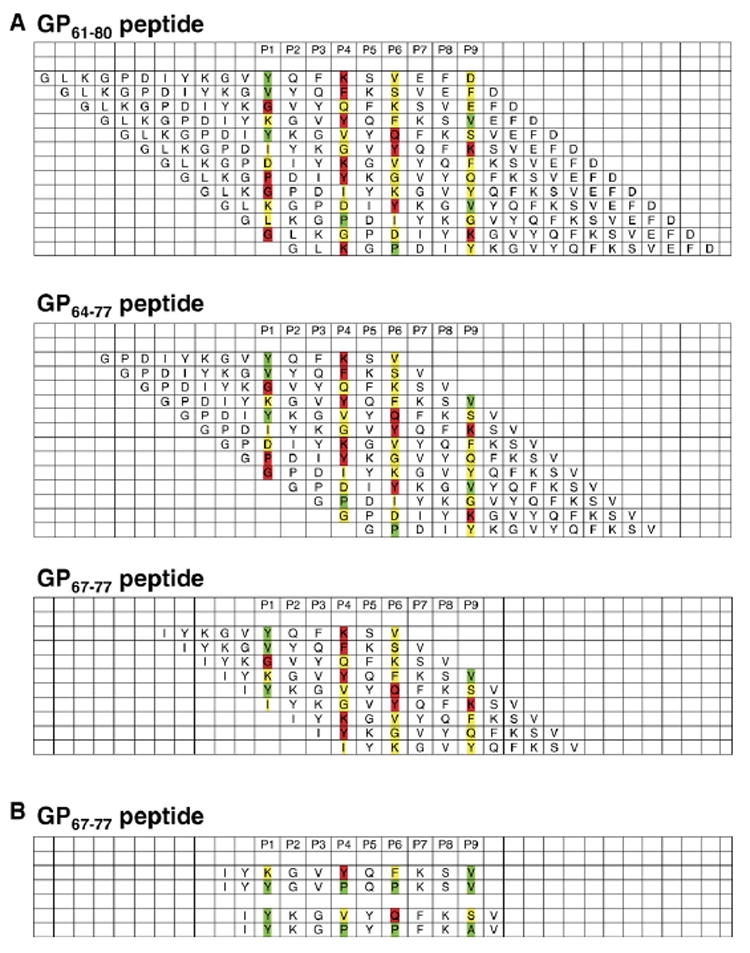
P1 to P9 signify I-Ab binding pockets. The interactions of individual amino acids with I-Ab binding pockets are differentiated according to optimal (green), neutral (yellow) and unfavorable (red) binding properties. (A) Potential alignments and registers for GP61–80, GP64–77 and GP67–77. The shortening of the original peptide was done first using each alignment. (B) Then, the minimal peptide, GP67–77, was modified in 2 registers to try to increase affinity and T cell response by introducing optimal anchor residues.
Binding affinity of LCMV-specific CD4+ T cell receptors
Having determined the minimal requirements for GP61–80 epitope binding and recognition, we proceeded to investigate in more detail the binding affinity of selected CD4+ T cell clones. To this extent, PCR products from clones #3 and #31 were inserted into the pCR2.1-TOPO cloning vector and sequenced. Both I-Ab tetramer-binding CD4+ T cell clones exhibited identical CDR3 amino acid sequences although we noted small differences at the level of nucleotide sequences (not shown). For direct measurement of TCR binding affinity, we generated recombinant TCRs from clone #3 and evaluated binding to I-AbGP61–80 complexes by surface plasmon resonance (SPR) as detailed in Materials and methods (Fig. 7). SPR measurements at equilibrium demonstrated a KD of 4.2 μM indicating a relatively high binding affinity as compared to other published CD4+ TCR affinities (Krummel et al., 2000).
Fig. 7. Determining the affinity of TCR #3.

Recombinant T cell receptors based on sequences derived from clone #3 were constructed as detailed in Materials and methods and tested for binding to I-AbGP61–80 complexes by plasmon surface resonance. TCR3: recombinant GP61–80-specific TCR obtained from clone #3; Ctl: control.
Discussion
In this study, we have defined the minimal constraints for the recognition of a dominant, MHC class II-restricted viral determinant. Naturally processed peptides selected by MHC class II molecules demonstrate considerable variability of length (usually 12–26 amino acids) but tend to cluster into families that share a 9 amino acid core segment that interacts with the four major binding pockets of the MHC II moleculev (p1, p4, p6 and p9). Flanking residues of natural peptides can modulate binding affinities and also serve as direct contact residues for TCR recognition (Suri et al., 2006). A recent study has discerned 128 naturally processed I-Ab-restricted peptides that are 11–21 amino acids in length and derived from both endocytic and cytosolic compartments. Although some studies have begun to employ similar techniques based on tandem mass-spectrometry for the identification of microbial determinants that are processed and presented by MHC II-bearing cells in vivo (Meiring et al., 2005; Ovsyannikova et al., 2003), the majority of studies performed to date have relied on measuring the binding affinity of synthetic microbial peptides to recombinant MHC molecules as well as the induction of effector functions in specific responder T cells. The latter approach has been guided, in particular for the mapping of MHC I-restricted epitopes, by the identification of common binding motifs and is usually less effective for prediction of MHC II-restricted determinants. Nevertheless, information about the naturally processed I-Ab-restricted peptide sequences together with the recently solved crystal structure of I-Ab has allowed for the development of a simple scoring matrix (Dongre et al., 2001; Zhu et al., 2003). However, retroactive application of this scoring matrix to the core and related LCMV-GP epitopes identified in this study failed to reveal optimal binding sequences. These observations emphasize that in spite of the comparatively high binding affinities of the dominant LCMV-GP epitope (Krummel et al., 2000), any attempts to predict optimal MHC class II-binding epitope sequences are impaired by the likely usage of alternating peptide binding registers.
The LCMV GP61–80 epitope was originally identified by Oxenius et al. (1995) as an immunodominant, I-Ab-restricted determinant derived from the LCMV WE strain. Although LCMV Armstrong differs from LCMV WE by a single N → K substitution in position GP63 of this determinant, subsequent work by other groups established that the GP61–80 epitope is also dominant for CD4+ T cell responses directed against LCMV Armstrong (Homann et al., 2001; Kamperschroer and Quinn, 1999; Varga and Welsh, 1998a; Varga and Welsh, 1998b; Whitmire et al., 1998). Indeed, we and others have estimated that more than half of the I-Ab-restricted, LCMV Armstrong-specific CD4+ T cell response is directed against this epitope (Homann et al., 2001; Kamperschroer and Quinn, 1999). More recently, Oxenius’ group has utilized a truncated GP peptide that lacks the first three C-terminal amino acids (GP64–80) to effectively stimulate TCR transgenic CD4+ T cells originally derived from a clone specific for the GP61–80 epitope (Wolint et al., 2004). This finding is in agreement with our observation that residues GP61–63 are completely dispensable for recognition by GP61–80 specific CD4+ T cell clones and also supported by unpublished findings that the GP-specific CD4+ T cell response is unaltered after infection with a variant virus lacking GP residues 59–63 (virus obtained, cloned and sequenced from persistently LCMV Armstrong-infected Db−/−×Kb−/− mice).
Interestingly, our analyses of primary LCMV-specific T cell responses directed against the GP67–77 epitope demonstrated the presence of CD8+ T cells that recognize this epitope in the context of Db. Although a previous publication, using an in vitro peptide binding assay, has described a truncated version of the GP67–77 epitope (GP70–77) as a peptide with high binding affinity to Kb in (van der Most et al., 1998), the use of Kb as a restriction element for the shorter GP70–77 epitope has, to the best of our knowledge, not been validated in ex vivo experiments. It thus remains possible that the GP67–77 and GP70–77 epitopes indeed demonstrate restriction by different MHC molecules (Db vs. Kb, respectively) or that GP70–77, similar to the GP33–41 epitope, is restricted by both Db and Kb. Several features are noteworthy in considering binding of GP67–77 to H-2Db, a restriction element that has been investigated in great detail. The signature motif has an Asn at P5 and an hydrophobic amino acid at P9 (Meth, Ileu or Leu) (Falk et al., 1991). In the present case, GP67–77 is missing the P5 anchor and will have to bind to Db in a non-classical way (Ostrov et al., 2002). It is likely that Valine77 anchors the C-terminus of the peptide, whereas the middle part bulges out, like most 11 mer peptides bound to MHC class I do (Apostolopoulos et al., 2002; Ostrov et al., 2002), and is exposed for T cell recognition. The recognition of one and the same viral determinant by both CD8+ and CD4+ T cells constitutes a unique feature within the LCMV system and should be taken into consideration for the interpretation of differential CD4+ and CD8+ T cell activities in this model system. For example, CD8+ T cells that recognize the I-Ab-restricted GP61–80 epitope have been described in CD4-deficient mice (Pearce et al., 2004; Tyznik et al., 2004). However, this CD8+ T cell response is restricted by I-Ab and its existence was attributed to abnormal T cell development in CD4+ T cell-deficient mice since further experiments indicated that neither wild-type nor acutely CD4-depleted mice mounted an MHC II-restricted CD8+ T cell response following infection with recombinant L. monocytogenes expressing a secreted form of chicken ovalbumin (Tyznik et al., 2004).
Finally, recognition of the same viral determinant by CD4+ and CD8+ T cells may offer an opportunity to optimize immunization strategies utilizing small, defined peptide sequences. As shown by van der Most and colleagues, protection of mice against chronic infection can be achieved even through induction of subdominant CD8+ T cell responses (van der Most et al., 1998) that constitute only a very small fraction of the antiviral CD8+ Tcell population in a natural infection (~2%). To improve the overall effectiveness of these CD8+ T cell responses, the authors utilized a lipidated LCMV GP92–101 peptide conjugated to an ovalbumin “helper” epitope (van der Most et al., 1998), a strategy that may be simplified by the use of the GP67–77 core peptide and the concurrent induction of genuine virus-specific CD8+ and CD4+ T cell responses.
Materials and methods
Mice and virus
C57BL/6 (H-2b, Thy1.2+) and congenic B6.PL (Thy1.1+) mice were purchased from the closed vivarium breeding colony at The Scripps Research Institute. Mice lacking Db, Kb or Db ×Kb expression were obtained from Taconic, Hudson NY. Origin, growth and titration of lymphocytic choriomeningitis virus (LCMV) Armstrong clone 53b have been described (Homann et al., 1998). Eight to ten week old C57BL/6 mice were infected with a single intraperitoneal dose of 105 plaque forming units LCMV Armstrong.
CD4+ T cell lines and clones
LCMV GP61–80-specific, I-Ab-restricted CD4+ T cell lines and clones were obtained from C57BL/6 mice 13 days after infection with 1.5×105 pfu LCMVArmstrong i.p. Spleens were harvested, single cell suspensions prepared, red blood cell lysed and CD8+ T cells depleted with antibody-coated magnetic beads as described (Berger et al., 2000). Cell counts were adjusted to 5×106 cells/ml in cloning medium (RPMI 1640 supplemented with 8% FCS, 1% penicillin/streptomycin, 1 mM L-glutamine and 50 units of human IL-2) supplemented with 1 μg/ml GP61–80 peptide, and plated in 24-well tissue culture plates. After 1 week, cultures were supplemented regularly with “feeder cells” (irradiated C57BL/6 spleen cells) as needed, intermittently rested by withdrawal of LCMV GP61–80 peptide and specific CD4+ T cell lines and clones established by limiting dilution techniques (Anderson et al., 1985).
Flow cytometry
For detection of intracellular IFNγ, rested cell lines and clones as well as primary T cells were restimulated for 5 h with peptides (1–5 μg/ml) prior to surface and intracellular stains as detailed elsewhere (Homann et al., 2001). Preparation and use of I-Ab tetramers and tetramer-liposomes have been described (Homann et al., 2001; Mallet-Designé et al., 2003). In this study, tetramers (20 μg/ml; 40 μl volume) were incubated with cells at room temperature for 45–75 min prior to addition of antibody reagents, further incubation (20 min), washes and immediate acquisition on a BD FACSCalibur flow cytometer. For quantitation of expression levels, we used the normalized geometric mean of fluorescence intensity (GMFI, calculated by dividing GMFI of the experimental stain by GMFI of corresponding control stain). Functional avidities were determined as described (Homann et al., 2006; Homann et al., 2002). For determination of TCR Vβ usage, we employed a kit comprising a panel of available TCR Vβ-specific antibodies (BDPharmingen).
CTL assay
Direct ex vivo CTL activity was analyzed 7 days after LCMV infection in a standard 51Cr release assay as described (Homann et al., 1998).
Sequencing and recombinant expression of TCRs
The cDNA for the α and β chain of TCR for CD4 T cell clones #3 and #31 was obtained by RT-PCR using Ready-To-Go RT-PCR beads (Amersham Pharmacia Biotech, Piscataway, NJ) according to the manufacturer’s instructions. In brief, RNA isolated from clones was used for cDNA synthesis and PCR amplification. PCR products were subcloned into pCR2.1-TOPO cloning vector (Invitrogen, Carlsbad, CA), sequenced and subcloned into the metallothionein promoter-based fly expression vector pRMHa3 (Fremont et al., 1992). The final constructs coded for the α1α2 and β1β2 domains, respectively, followed by a linker sequence (SSADL), a thrombin site (LVPRGS), a leucine zipper (acidic for the α chain, basic for the β chain) (Scott et al., 1996) and a hexahistidine tag. Vectors were transfected into SC2 cells and stable cell lines and clones were established. Soluble TCRs were purified from culture supernatants as described (Garcia et al., 1997; Matsumura et al., 1992; Scott et al., 1998).
Surface plasmon resonance
A BIACORE 2000 instrument was used to determine interactions between purified pMHC (peptide:MHC) complexes and recombinant TCR molecules as described (Mallet-Designé et al., 2003). In brief, pMHC complexes or TCRs were immobilized by amine coupling chemistry on a CM5 research grade sensor chip. Successive injections of pMHC complexes or TCRs were performed in filtered and degassed PBS buffer at a flow rate of 20 μl/min at concentrations of 20, 10, 5, 2.5, and 1.25 μM. In all experiments, irrelevant TCR molecules or irrelevant pMHC (I-Ab/ova) were used as negative control and subtracted from experimental sensorgrams. On- and off- rates were obtained by non-linear curve fitting of subtracted curves using the 1:1 Langmuir binding model using the BIAevaluation program (version 3.0.2).
Statistical analysis
Data handling, analysis and graphic representation were performed using Prism 4.0a (GraphPad Software, San Diego, California); p values were calculated by Student’s t-test.
Acknowledgments
This is publication number 17545-NP from the Molecular and Integrative Neurosciences Department and Department of Infectology, The Scripps Research Institute, La Jolla, CA. This work was supported in part by NIH 1R21 AG026518-01 (D.H), AI009484 (M.B.A.O.), NIH training grant 5T32 AI007244-23 (D.B.) and NIH DK055037 (L.T.).
References
- Anderson J, Byrne JA, Schreiber R, Patterson S, Oldstone MB. Biology of cloned cytotoxic T lymphocytes specific for lymphocytic choriomeningitis virus: clearance of virus and in vitro properties. J Virol. 1985;53(2):552–560. doi: 10.1128/jvi.53.2.552-560.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolopoulos V, Yu M, Corper AL, Li W, McKenzie IF, Teyton L, Wilson IA, Plebanski M. Crystal structure of a non-canonical high affinity peptide complexed with MHC class I: a novel use of alternative anchors. J Mol Biol. 2002;318(5):1307–1316. doi: 10.1016/s0022-2836(02)00198-5. [DOI] [PubMed] [Google Scholar]
- Appay V. The physiological role of cytotoxic CD4 (+) T-cells: the holy grail? Clin Exp Immunol. 2004;138(1):10–13. doi: 10.1111/j.1365-2249.2004.02605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nat Immunol. 2005;6(4):353–360. doi: 10.1038/ni1181. [DOI] [PubMed] [Google Scholar]
- Berger DP, Homann D, Oldstone MB. Defining parameters for successful immunocytotherapy of persistent viral infection. Virology. 2000;266(2):257–263. doi: 10.1006/viro.1999.0074. [DOI] [PubMed] [Google Scholar]
- Bevan MJ. Helping the CD8 (+) T-cell response. Nat Rev Immunol. 2004;4(8):595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- Burton DR. Opinion: antibodies, viruses and vaccines. Nat Rev Immunol. 2002;2(9):706–713. doi: 10.1038/nri891. [DOI] [PubMed] [Google Scholar]
- Corper AL, Stratmann T, Apostolopoulos V, Scott CA, Garcia KC, Kang AS, Wilson IA, Teyton L. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science. 2000;288(5465):505–511. doi: 10.1126/science.288.5465.505. [DOI] [PubMed] [Google Scholar]
- Dittmer U, He H, Messer RJ, Schimmer S, Olbrich AR, Ohlen C, Greenberg PD, Stromnes IM, Iwashiro M, Sakaguchi S, Evans LH, Peterson KE, Yang G, Hasenkrug KJ. Functional impairment of CD8 (+) T cells by regulatory T cells during persistent retroviral infection. Immunity. 2004;20(3):293–303. doi: 10.1016/s1074-7613(04)00054-8. [DOI] [PubMed] [Google Scholar]
- Dongre AR, Kovats S, deRoos P, McCormack AL, Nakagawa T, Paharkova-Vatchkova V, Eng J, Caldwell H, Yates JR, Rudensky AY. In vivo MHC class II presentation of cytosolic proteins revealed by rapid automated tandem mass spectrometry and functional analyses. Eur J Immunol. 2001;31(5):1485–1494. doi: 10.1002/1521-4141(200105)31:5<1485::AID-IMMU1485>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351(6324):290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- Fremont DH, Matsumura M, Stura EA, Peterson PA, Wilson IA. Crystal structures of two viral peptides in complex with murine MHC class I H-2Kb. Science. 1992;257(5072):919–927. doi: 10.1126/science.1323877. [DOI] [PubMed] [Google Scholar]
- Gairin JE, Mazarguil H, Hudrisier D, Oldstone MB. Optimal lymphocytic choriomeningitis virus sequences restricted by H-2Db major histocompatibility complex class I molecules and presented to cytotoxic T lymphocytes. J Virol. 1995;69(4):2297–2305. doi: 10.1128/jvi.69.4.2297-2305.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia KC, Tallquist MD, Pease LR, Brunmark A, Scott CA, Degano M, Stura EA, Peterson PA, Wilson IA, Teyton L. Alphabeta T cell receptor interactions with syngeneic and allogeneic ligands: affinity measurements and crystallization. Proc Natl Acad Sci U S A. 1997;94(25):13838–13843. doi: 10.1073/pnas.94.25.13838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann D. Immunocytotherapy. Curr Top Microbiol Immunol. 2002;263:43–65. doi: 10.1007/978-3-642-56055-2_4. [DOI] [PubMed] [Google Scholar]
- Homann D, Tishon A, Berger DP, Weigle WO, von Herrath MG, Oldstone MB. Evidence for an underlying CD4 helper and CD8 T-cell defect in B-cell-deficient mice: failure to clear persistent virus infection after adoptive immunotherapy with virus-specific memory cells from muMT/muMT mice. J Virol. 1998;72(11):9208–9216. doi: 10.1128/jvi.72.11.9208-9216.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann D, Teyton L, Oldstone MB. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat Med. 2001;7(8):913–919. doi: 10.1038/90950. [DOI] [PubMed] [Google Scholar]
- Homann D, Jahreis A, Wolfe T, Hughes A, Coon B, van Stipdonk MJ, Prilliman KR, Schoenberger SP, von Herrath MG. CD40L blockade prevents autoimmune diabetes by induction of bitypic NK/DC regulatory cells. Immunity. 2002;16(3):403–415. doi: 10.1016/s1074-7613(02)00290-x. [DOI] [PubMed] [Google Scholar]
- Homann D, Dummer W, Wolfe T, Rodrigo E, Theofilopoulos AN, Oldstone MB, von Herrath MG. Lack of intrinsic ctla-4 expression has minimal effect on regulation of antiviral T-cell immunity. J Virol. 2006;80(1):270–280. doi: 10.1128/JVI.80.1.270-280.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudrisier D, Oldstone MB, Gairin JE. The signal sequence of lymphocytic choriomeningitis virus contains an immunodominant cytotoxic T cell epitope that is restricted by both H-2D (b) and H-2K (b) molecules. Virology. 1997;234(1):62–73. doi: 10.1006/viro.1997.8627. [DOI] [PubMed] [Google Scholar]
- Jenkins MK, Khoruts A, Ingulli E, Mueller DL, McSorley SJ, Reinhardt RL, Itano A, Pape KA. In vivo activation of antigen-specific CD4 T cells. Annu Rev Immunol. 2001;19:23–45. doi: 10.1146/annurev.immunol.19.1.23. [DOI] [PubMed] [Google Scholar]
- Kamperschroer C, Quinn DG. Quantification of epitope-specific MHC class-II-restricted T cells following lymphocytic choriomeningitis virus infection. Cell Immunol. 1999;193(2):134–146. doi: 10.1006/cimm.1999.1458. [DOI] [PubMed] [Google Scholar]
- Klavinskis LS, Whitton JL, Joly E, Oldstone MB. Vaccination and protection from a lethal viral infection: identification, incorporation, and use of a cytotoxic T lymphocyte glycoprotein epitope. Virology. 1990;178(2):393–400. doi: 10.1016/0042-6822(90)90336-p. [DOI] [PubMed] [Google Scholar]
- Krummel M, Wulfing C, Sumen C, Davis MM. Thirty-six views of T-cell recognition. Philos Trans R Soc London, Ser B Biol Sci. 2000;355(1400):1071–1076. doi: 10.1098/rstb.2000.0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz DC, Kurz SK, Lemmens E, Schoenberger SP, Sprent J, Oldstone MB, Homann D. IL-7 regulates basal homeostatic proliferation of antiviral CD4+T cell memory. Proc Natl Acad Sci U S A. 2004;101(25):9357–9362. doi: 10.1073/pnas.0400640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet-Designé VI, Stratmann T, Homann D, Carbone F, Oldstone MB, Teyton L. Detection of low-avidity CD4+ T cells using recombinant artificial APC: following the anti-ovalbumin immune response. J Immunol. 2003;170(1):123–131. doi: 10.4049/jimmunol.170.1.123. [DOI] [PubMed] [Google Scholar]
- Matsumura M, Saito Y, Jackson MR, Song ES, Peterson PA. In vitro peptide binding to soluble empty class I major histocompatibility complex molecules isolated from transfected Drosophila melanogaster cells. J Biol Chem. 1992;267(33):23589–23595. [PubMed] [Google Scholar]
- McHeyzer-Williams M, McHeyzer-Williams L, Panus J, Pogue-Caley R, Bikah G, Driver D, Eisenbraun M. Helper T-cell-regulated B-cell immunity. Microbes Infect. 2003;5(3):205–212. doi: 10.1016/s1286-4579(03)00012-1. [DOI] [PubMed] [Google Scholar]
- Meiring HD, Kuipers B, van Gaans-van den Brink JA, Poelen MC, Timmermans H, Baart G, Brugghe H, van Schie J, Boog CJ, de Jong AP, van Els CA. Mass tag-assisted identification of naturally processed HLA class II-presented meningococcal peptides recognized by CD4+ T lymphocytes. J Immunol. 2005;174(9):5636–5643. doi: 10.4049/jimmunol.174.9.5636. [DOI] [PubMed] [Google Scholar]
- Mittrucker HW, Kaufmann SH. Mini-review: regulatory T cells and infection: suppression revisited. Eur J Immunol. 2004;34(2):306–312. doi: 10.1002/eji.200324578. [DOI] [PubMed] [Google Scholar]
- Norris PJ, Rosenberg ES. CD4 (+) T helper cells and the role they play in viral control. J Mol Med. 2002;80(7):397–405. doi: 10.1007/s00109-002-0337-3. [DOI] [PubMed] [Google Scholar]
- Oldstone MB, Whitton JL, Lewicki H, Tishon A. Fine dissection of a nine amino acid glycoprotein epitope, a major determinant recognized by lymphocytic choriomeningitis virus-specific class I-restricted H-2Db cytotoxic T lymphocytes. J Exp Med. 1988;168(2):559–570. doi: 10.1084/jem.168.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrov DA, Roden MM, Shi W, Palmieri E, Christianson GJ, Mendoza L, Villaflor G, Tilley D, Shastri N, Grey H, Almo SC, Roopenian D, Nathenson SG. How H13 histocompatibility peptides differing by a single methyl group and lacking conventional MHC binding anchor motifs determine self-nonself discrimination. J Immunol. 2002;168(1):283–289. doi: 10.4049/jimmunol.168.1.283. [DOI] [PubMed] [Google Scholar]
- Ovsyannikova IG, Johnson KL, Naylor S, Muddiman DC, Poland GA. Naturally processed measles virus peptide eluted from class II HLA-DRB1*03 recognized by T lymphocytes from human blood. Virology. 2003;312(2):495–506. doi: 10.1016/s0042-6822(03)00281-2. [DOI] [PubMed] [Google Scholar]
- Oxenius A, Bachmann MF, Ashton-Rickardt PG, Tonegawa S, Zinkernagel RM, Hengartner H. Presentation of endogenous viral proteins in association with major histocompatibility complex class II: on the role of intracellular compartmentalization, invariant chain and the TAP transporter system. Eur J Immunol. 1995;25(12):3402–3411. doi: 10.1002/eji.1830251230. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Shedlock DJ, Shen H. Functional characterization of MHC class II-restricted CD8+CD4− and CD8−CD4− T cell responses to infection in CD4−/− mice. J Immunol. 2004;173(4):2494–2499. doi: 10.4049/jimmunol.173.4.2494. [DOI] [PubMed] [Google Scholar]
- Schulz M, Aichele P, Vollenweider M, Bobe FW, Cardinaux F, Hengartner H, Zinkernagel RM. Major histocompatibility complex-dependent T cell epitopes of lymphocytic choriomeningitis virus nucleoprotein and their protective capacity against viral disease. Eur J Immunol. 1989;19(9):1657–1667. doi: 10.1002/eji.1830190921. [DOI] [PubMed] [Google Scholar]
- Scott CA, Garcia KC, Carbone FR, Wilson IA, Teyton L. Role of chain pairing for the production of functional soluble IA major histo-compatibility complex class II molecules. J Exp Med. 1996;183(5):2087–2095. doi: 10.1084/jem.183.5.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CA, Peterson PA, Teyton L, Wilson IA. Crystal structures of two I-Ad-peptide complexes reveal that high affinity can be achieved without large anchor residues. Immunity. 1998;8(3):319–329. doi: 10.1016/s1074-7613(00)80537-3. [DOI] [PubMed] [Google Scholar]
- Seder RA, Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat Immunol. 2003;4(9):835–842. doi: 10.1038/ni969. [DOI] [PubMed] [Google Scholar]
- Seder RA, Paul WE. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu Rev Immunol. 1994;12:635–673. doi: 10.1146/annurev.iy.12.040194.003223. [DOI] [PubMed] [Google Scholar]
- Sprent J, Surh CD. T cell memory. Annu Rev Immunol. 2002;20:551–579. doi: 10.1146/annurev.immunol.20.100101.151926. [DOI] [PubMed] [Google Scholar]
- Suri A, Lovitch SB, Unanue ER. The wide diversity and complexity of peptides bound to class II MHC molecules. Curr Opin Immunol. 2006;18(1):70–77. doi: 10.1016/j.coi.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Tyznik AJ, Sun JC, Bevan MJ. The CD8 population in CD4-deficient mice is heavily contaminated with MHC class II-restricted T cells. J Exp Med. 2004;199(4):559–565. doi: 10.1084/jem.20031961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Most RG, Murali-Krishna K, Whitton JL, Oseroff C, Alexander J, Southwood S, Sidney J, Chesnut RW, Sette A, Ahmed R. Identification of Db- and Kb-restricted subdominant cytotoxic T-cell responses in lymphocytic choriomeningitis virus-infected mice. Virology. 1998;240(1):158–167. doi: 10.1006/viro.1997.8934. [DOI] [PubMed] [Google Scholar]
- van der Most RG, Murali-Krishna K, Lanier JG, Wherry EJ, Puglielli MT, Blattman JN, Sette A, Ahmed R. Changing immunodominance patterns in antiviral CD8 T-cell responses after loss of epitope presentation or chronic antigenic stimulation. Virology. 2003;315(1):93–102. doi: 10.1016/j.virol.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Varga SM, Welsh RM. Detection of a high frequency of virus-specific CD4+ T cells during acute infection with lymphocytic choriomeningitis virus. J Immunol. 1998a;161(7):3215–3218. [PubMed] [Google Scholar]
- Varga SM, Welsh RM. Stability of virus-specific CD4+ T cell frequencies from acute infection into long term memory. J Immunol. 1998b;161(1):367–374. [PubMed] [Google Scholar]
- Varga SM, Welsh RM. High frequency of virus-specific interleukin-2-producing CD4 (+) T cells and Th1 dominance during lymphocytic choriomeningitis virus infection. J Virol. 2000;74(9):4429–4432. doi: 10.1128/jvi.74.9.4429-4432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmire JK, Asano MS, Murali-Krishna K, Suresh M, Ahmed R. Long-term CD4 Th1 and Th2 memory following acute lymphocytic choriomeningitis virus infection. J Virol. 1998;72(10):8281–8288. doi: 10.1128/jvi.72.10.8281-8288.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmire JK, Benning N, Whitton JL. Precursor frequency, nonlinear proliferation, and functional maturation of virus-specific CD4+ T cells. J Immunol. 2006;176(5):3028–3036. doi: 10.4049/jimmunol.176.5.3028. [DOI] [PubMed] [Google Scholar]
- Whitton JL, Oldstone MB. The immune response to viruses. In: Knipe D, Howley P, Griffin D, Lamb R, Martin M, Strauss S, editors. Field’s Virology. 4. Lippincott Williams; Wilkins, Philadelphia: 2001. pp. 285–320. [Google Scholar]
- Wolint P, Betts MR, Koup RA, Oxenius A. Immediate cytotoxicity but not degranulation distinguishes effector and memory subsets of CD8+ T cells. J Exp Med. 2004;199(7):925–936. doi: 10.1084/jem.20031799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Rudensky AY, Corper AL, Teyton L, Wilson IA. Crystal structure of MHC class II I-Ab in complex with a human CLIP peptide: prediction of an I-Ab peptide-binding motif. J Mol Biol. 2003;326(4):1157–1174. doi: 10.1016/s0022-2836(02)01437-7. [DOI] [PubMed] [Google Scholar]
- Zinkernagel RM. On differences between immunity and immunological memory. Curr Opin Immunol. 2002;14(4):523–536. doi: 10.1016/s0952-7915(02)00367-9. [DOI] [PubMed] [Google Scholar]