

Abstract
The proto-oncogene AKT (also known as PKB) is activated in many human cancers, mostly owing to loss of the PTEN tumour suppressor1. In such tumours, AKT becomes enriched at cell membranes where it is activated by phosphorylation. Yet many targets inhibited by phosphorylated AKT (for example, the FOXO transcription factors) are nuclear; it has remained unclear how relevant nuclear phosphorylated AKT (pAKT) function is for tumorigenesis. Here we show that the PML tumour suppressor prevents cancer by inactivating pAKT inside the nucleus. We find in a mouse model that Pml loss markedly accelerates tumour onset, incidence and progression in Pten-heterozygous mutants, and leads to female sterility with features that recapitulate the phenotype of Foxo3a knockout mice2. We show that Pml deficiency on its own leads to tumorigenesis in the prostate, a tissue that is exquisitely sensitive to pAkt levels, and demonstrate that Pml specifically recruits the Akt phosphatase PP2a as well as pAkt into Pml nuclear bodies. Notably, we find that Pml-null cells are impaired in PP2a phosphatase activity towards Akt, and thus accumulate nuclear pAkt. As a consequence, the progressive reduction in Pml dose leads to inactivation of Foxo3a-mediated transcription of proapoptotic Bim and the cell cycle inhibitor p27kip1. Our results demonstrate that Pml orchestrates a nuclear tumour suppressor network for inactivation of nuclear pAkt, and thus highlight the importance of AKT compartmentalization in human cancer pathogenesis and treatment.
Pml nuclear bodies represent distinct yet dynamic intranuclear structures involved in apoptosis, proliferation and senescence3. Nuclear bodies are absent from Pml-deficient cells, causing aberrant nuclear patterns of nuclear body resident proteins. We have recently reported the high correlation between reduction in nuclear body number and prostate and colon cancer4, both tumours showing frequent PTEN loss5,6. Because we and others have established faithful mouse models for the role of PTEN in cancer7–9, we sought to determine whether Pml loss would affect the signature of Pten deficiency. After crossing Pten-heterozygous mice (Pten+/−) with Pml-null mice (Pml−/−) to produce six genotypes of interest, we found a marked Pml-dependent reduction in lifespan of Pten+/− mice (Supplementary Fig. S1a). Pten+/− female lethality, dictated by an autoimmune disorder10, was not precipitated by Pml loss. In contrast, the male cohort displayed an exquisite, Pml dose dependence in survival (the autoimmune disorder is minor in males10), thus exposing a novel lethal phenotype in compound mutants with Pml haploinsufficiency for its repression. Magnetic resonance imaging (MRI) visualized intestinal and prostate anomalies (not shown), and post-mortem analysis confirmed large intestinal lesions in Pten+/−Pml+/− and Pten+/−Pml−/− mice, suggesting obstruction as a probable (males) or additional (females) cause of death. Intestines of Pten+/−Pml−/− mice presented invasive adenocarcinoma of the colon (Fig. 1a, colon), whereas Pten+/− mice displayed only pre-cancerous polyps (Supplementary Fig. S1b, colon). Disease-free survival analysis summarizes onset of the Pten+/−Pml−/−-specific colon carcinoma (Fig. 1b). Notably, Pml levels also dictated polyp numbers in Pten+/− animals: Pml dose reduction resulted in a 3–5-fold increase in the number of polyps per mouse (Fig. 1b, top inset). Average polyp diameter also correlated with Pml status (Fig. 1b, bottom inset). Taken together, in Pten+/− mice, Pml loss causes the appearance of polyposis and colon cancer, which reduces lifespan severely.
Figure 1. Pml status dictates carcinogenesis in Pten+/− mice.
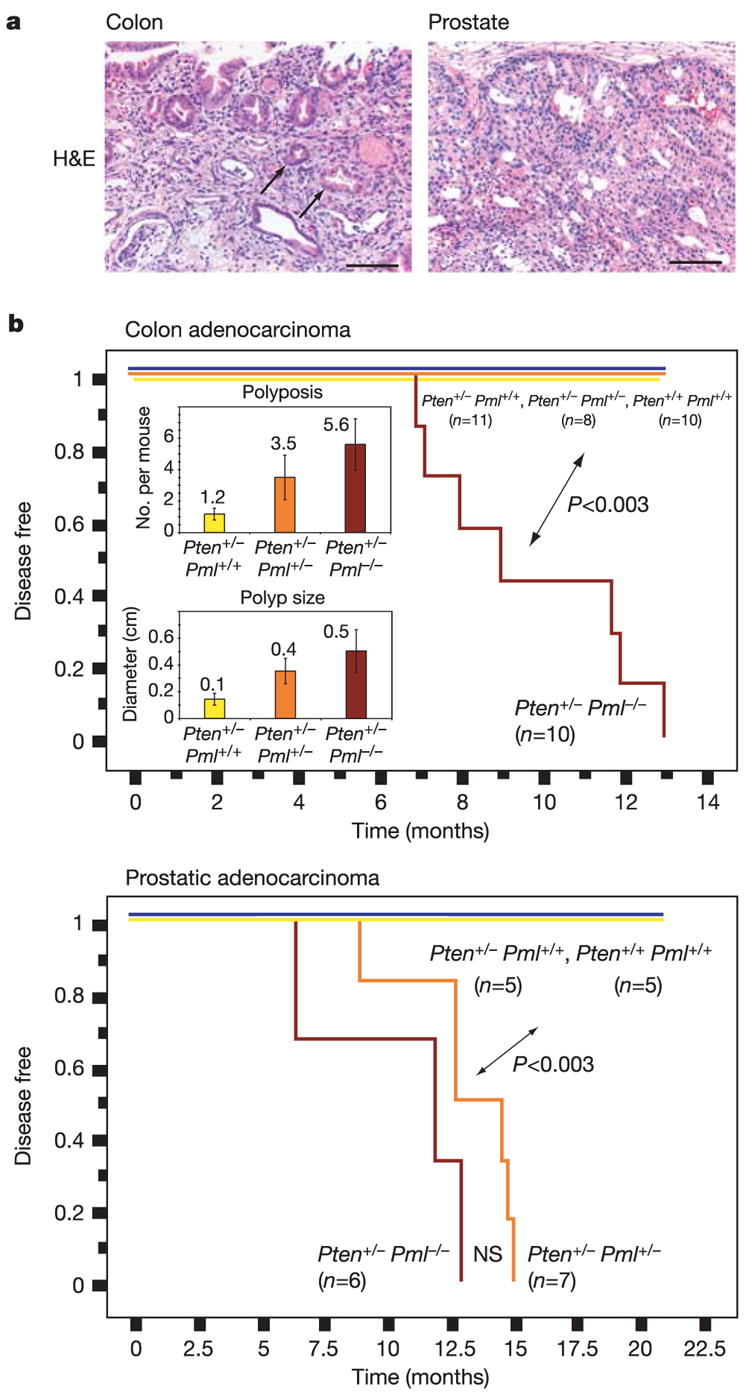
a, Haematoxylin and eosin (H&E) analysis of colon and prostate tissue reveals invasive adenocarcinoma in Pten+/− Pml−/− mice. Arrows show invasive glands in colonic submucosa. Scale bars, 100μm. b, Kaplan–Meier plots for disease-free survival. Average polyps per mouse (top inset) and average polyp size (bottom inset) are shown. Error bars are s.d. (see also Methods).
Pten activity regulates prostate cancer, as by 8 months some Pten+/− animals develop prostatic intraepithelial neoplasia (the in situ form of prostate cancer). Invasive cancer is observed on further lowering of Pten activity, as shown in Pten-hypomorphic or prostate-specific null mice11,12. Haematoxylin and eosin staining and prostate cancer-free survival analysis revealed that Pten+/−Pml−/− and Pten+/−Pml+/− males (but not Pten+/− mice) developed highly invasive cancers, suggesting that prostate epithelia are more sensitive to Pml status than intestine (Fig. 1 and Supplementary Fig. S1b, prostate). We also found a characteristic increased cell proliferation marker staining13 (even in tumour-free areas) in Pten+/−Pml+/−and Pten+/−Pml−/− intestines as well as in Pten+/−Pml−/− prostate glands (Supplementary Fig. S1c).
The degree of Pten deficiency closely correlates with activation of the oncogenic kinase Akt11. Akt activation by membrane recruitment is inhibited by Pten14,15 and leads to phosphorylation of Akt at Thr 308 and Ser 473 (refs 16, 17). To address whether Pml loss affects Akt activation we performed immunohistochemistry (IHC) staining and quantified western blotting of tissues. Staining of pAkt at Ser 473 was clearly increased in polyps, normal colon and in prostate lesions of Pten+/−Pml−/− mice when compared with Pten+/− mice (Fig. 2a). Importantly, both colon and prostate presented dominant nuclear pAkt accumulation, indicating a qualitative change in kinase localization (Fig. 2a, insets). Quantification of pAkt levels in Pten+/− mice confirmed a 50% increase in normal colon and 2-month-old prostate on Pml loss (Supplementary Fig. S1d, compare Pten+/−Pml−/− (en) and Pten+/−Pml+/+ (ew)), comparable to the reported Pten+/+ to Pten+/−transition11. Loss of the remaining Pten allele was excluded through western blot (not shown), IHC and Southern blot analysis of the macro-dissected cancers (Supplementary Fig. S1e). Taken together, our data suggest that in Pten+/− mice, loss of Pml leads to further activation of Akt but not because of complete Pten loss.
Figure 2. Pml loss leads to an increase in pAkt in vivo.
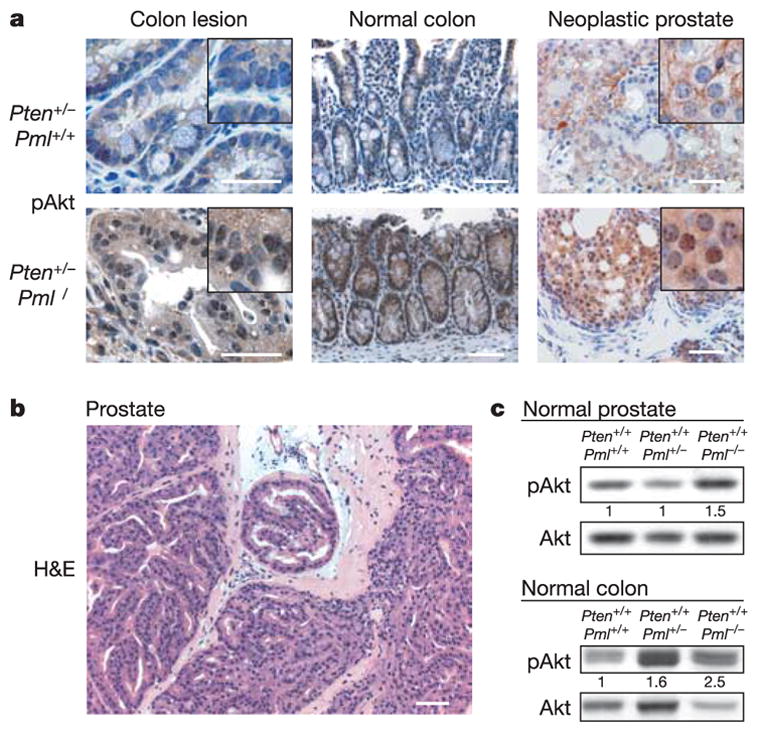
a, IHC staining comparing pAkt levels and localization in Pten+/− Pml+/+ and Pten+/− Pml−/− mice. b, Haematoxylin and eosin (H&E) staining of Pml−/− prostate with high-grade prostatic intraepithelial neoplasia. Scale bars (a, b), 100μm. c, Pml status affects pAkt levels in pre-neoplastic Pten+/+ tissues.
When analysing Pten+/+ prostates we found that Pml−/− animals presented high-grade prostatic intraepithelial neoplasia and areas of focally invasive cancer in their anterior prostates around 12 months of age (Fig. 2b and Supplementary Fig. S2a, b). Normal colon and prostate in Pml-deficient mice showed increased levels of pAkt (Fig. 2c) but no signs of colon neoplasia/dysplasia, consistent with the above finding that prostate is most sensitive to Pml loss (Fig. 1b). We frequently found enlarged prostates in Pml-null mice, yet larger organ size was also due to large luminal non-cellular areas (see Supplementary Fig. S2c, asterisk in the Pten+/−Pml+/+ panel). To measure size effects we determined average individual cell (not organ) sizes. Pml−/− and Pten+/− glands were mostly hypercellular but filled with cells of only half the size of their wild-type counterparts (Supplementary Fig. S2c). Thus, Pml deficiency leads to Akt activation in prostate and colon and triggers initiation of prostate cancer.
To test whether the qualitative difference in pAkt localization was an inherent feature of Pml loss we used mouse embryonic fibroblasts (MEFs). First, we determined Akt status in primary littermate Pml+/+ and Pml−/− MEFs by subjecting them to serum starvation and stimulation (see Methods). These cells had little and comparable pAkt activation at steady state and during serum starvation (Supplementary Fig. S2d, top panels). But serum stimulation for 10 min gave stronger Akt activation in Pml−/−than in Pml+/+ MEFs, as confirmed by Akt kinase assays (Supplementary Fig. S2e). At 3 h after stimulation, Akt activation was slightly higher in null MEFs, an effect more readily observed at later passage (Supplementary Fig. S2f, top panels). To test whether transgenic expression of human PML also affects Akt activation we used stably PML-transduced Pml−/− cells, which showed decreased Akt activation and faster inactivation after stimulation (Supplementary Fig. S2d, bottom panels). PML over-expression in Pml+/+ MEFs had almost no effect (not shown), similar to overexpression in PML+/+ human cells (Supplementary Fig. S2f, middle panels). Thus, Pml is able to suppress Akt activation after a membrane stimulus and it is sufficient to remedy this defect in Pml−/− cells. To exclude effects on Akt caused by cell cycle perturbations after PML add back, cell cycle profiles of Pml−/− and add-back cells were compared but no differences found, especially after starvation/stimulation (Supplementary Fig. S2g).
To account for qualitative and quantitative differences by confocal laser scanning microscopy (CLSM), primary Pml+/+ and Pml−/− MEFs were seeded on the same coverslips. Minor differences in pAkt staining were observed between wild-type and null cells at steady state and starvation (Fig. 3a). In contrast, Pml−/− cells showed not only stronger but notably nuclear pAkt accumulation after 10 min or 3 h of serum stimulation (see also Supplementary Fig. S3a). This localization was in marked contrast to that observed in Pten−/−cells, where pAkt is concentrated at cell membranes (Fig. 3a, bottom row). This membrane-associated pAkt pool was not antagonized by overexpression of PML in these MEFs (not shown), or in human PTEN−/− cells (Supplementary Fig. S2f, bottom panels), consistent with inhibition of nuclear, not cytoplasmic, pAkt by PML. Cell fractionation confirmed that a distinct nuclear population of pAkt was present in Pml−/− but not Pml+/+ cells after stimulation (Supplementary Fig. S3b). We next compared pAkt localization in three invasive prostate cancer models. The prostate-specific Pten-null and Pten-hypomorphic models showed strong membrane accumulation of pAkt, as previously described11 (Fig. 3b). In contrast, the invasive prostate cancer of Pten+/− Pml−/− mice showed dominant nuclear and cytoplasmic staining (see also Fig. 2a, insets). Therefore, we next tested whether Pml loss would enhance nuclear pAkt function.
Figure 3. Pml deficiency leads to increased nuclear pAkt localization and function in vitro and in vivo.
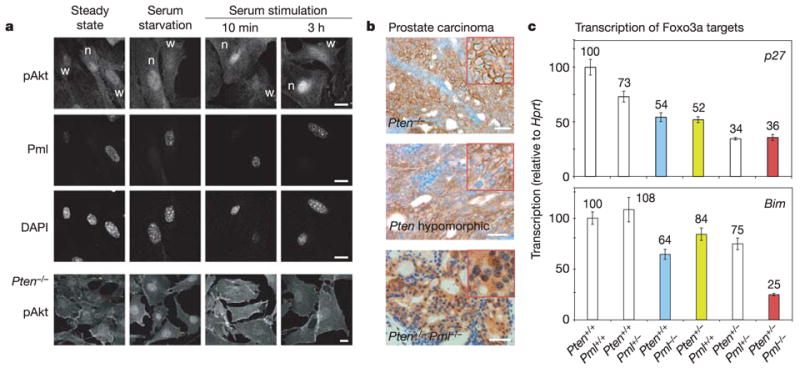
a, Immunofluorescence CLSM on primary littermate Pml+/+ (w) and Pml−/−(n) MEFs seeded on the same coverslips. pAkt in Pten−/− MEFs is shown for comparison. Scale bars, 10μm. b, pAkt in immunohistochemistry staining of prostate cancers from indicated mouse models. Scale bars, 50μm. c, Foxo3a activity measured by quantitative real-time PCR of prostates of the indicated genotypes. Error bars are s.d. of triplicates.
The forkhead transcription factor FOXO3A is a well-studied nuclear target of pAKT18, as AKT-mediated phosphorylation causes FOXO3A inactivation and export19. We quantified nuclear Foxo3a amounts by western blotting and found that Pml−/−cells, in contrast to wild-type cells, had decreased exogenous and endogenous nuclear Foxo3a after serum stimulation (Supplementary Fig. S3c). To quantify Foxo3a distribution by non-disruptive means, GFP–FOXO3A (human FOXO3A tagged with green fluorescent protein) localization in fixed Pml+/+ and Pml−/− MEFs was scored. At 10 min after stimulation, 80% of Pml−/− cells showed cytoplasmic FOXO3A whereas 70% of wild-type cells still retained FOXO3A in the nucleus (Supplementary Fig. S3d). Importantly, an Akt-insensitive FOXO3A mutant retained its nuclear localization even after a 3 h stimulation in Pml−/− cells. In agreement, a twofold increase in Akt-mediated FOXO3A phosphorylation was observed after serum stimulation when comparing Pml-deficient cells with add-back cells (Supplementary Fig. S3e).
Foxo3a exerts some of its tumour-suppressive functions by inducing transcription of p27kip1 (see ref. 18), which inhibits prostate cancer after Pten loss13. As quantified by real-time polymerase chain reaction (PCR), Pml status effectively dictates p27kip1 messenger RNA levels in prostates of both Pten+/+ and Pten+/− mice (Fig. 3c). Two additional Foxo3a target genes—the proapoptotic factor Bim and DNA repair protein Gadd45 (ref. 18)—responded similarly, consistent with Foxo3a inactivation (see also Supplementary Fig. S3f).
Foxo3a-deficient females display early infertility due to premature ovarian failure20,21, an event normally associated with ageing2. To determine the effect of Pml loss on fertility we set up mating pairs using fertile wild-type males. Pml−/− and Pten+/− females accumulated litters (Supplementary Fig. S4a) similar to wild-type mice (not shown). In contrast, although Pten+/−Pml−/− females had near-normal first litter sizes, they were invariably infertile by 5 months of age and their ovaries showed signs of follicular degeneration at 2 months and near-complete follicle depletion by 5 months (Supplementary Fig. S4b). Taken together, our results demonstrate that Pml loss enhances Foxo3a inactivation, especially under conditions of elevated Akt (Pten loss in vivo, serum stimulation in vitro).
Mechanistically, Pml loss could either stimulate Akt activation or antagonize pAkt inactivation. To distinguish between the two, we used okadaic acid, which selectively inactivates PP2a, the only known Akt-Thr 308 phosphatase22,23. If Pml deficiency favours Akt activation mechanisms (independent of PP2a), okadaic acid treatment should increase Akt phosphorylation at Thr 308 in Pml−/− cells. Alternatively, if Pml deficiency antagonizes PP2a, Pml−/− cells should be insensitive to okadaic acid. Our results suggest the latter: Pml−/− MEFs, in contrast to wild type, showed no response in the levels of pAkt at Thr 308 on okadaic acid addition in fresh serum (Fig. 4a and Supplementary Fig. S4c). Thus, after this stimulation in the presence of okadaic acid, Pml+/+ cells were finally able to match the pAkt levels of Pml−/− cells (Fig. 4b). Importantly, Pml−/− cell extracts retained normal PP2a levels and general activity, suggesting that Pml specifically cooperates with PP2a in pAkt inactivation (Supplementary Fig. S4d, e).
Figure 4. Pml affects PP2a activity towards pAkt.
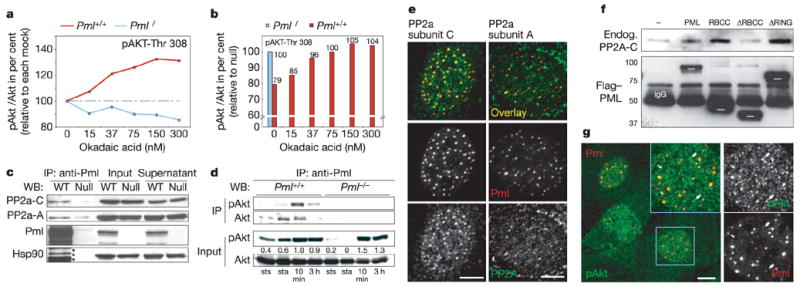
a, pAkt-Thr 308/Akt ratios after okadaic acid treatment of MEFs (relative to mock, see Methods). b, Ratios from a expressed relative to untreated Pml−/− cells. Note the broken ordinate axis for clarity. c, Endogenous co-immunoprecipitation of PP2a with Pml in MEFs. Asterisks indicate leftover Pml staining, not Hsp90. d, Endogenous co-immunoprecipitation of Akt with Pml at steady state (sts), starvation (sta) and serum stimulation for times indicated. e, CLSM co-localization of endogenous Pml and PP2a in MEFs. Scale bars, 5μm. f, Anti-Flag co-immunoprecipitation of PP2a-C with Flag-tagged PML/PML mutants. White bars indicate PML/mutant migration. IgG, anti-Flag IgG heavy chains. Molecular weights are in kDa. g, CLSM co-localization of pAkt and Pml. Arrowheads indicate pAkt-deficient nuclear bodies and arrows indicate pAkt accumulation outside nuclear bodies. Scale bar, 10μm.
PP2a is a heterotrimer of A, B and catalytic (C) subunits, which can function alone or in a ‘core dimer’ with A. Whereas the two known A and C subunit isoforms are ubiquitous, the B subunit families can determine tissue/substrate specificity and give rise to over 70 holoenzyme variations24. We found the C and to some extent the A subunits co-precipitating with Pml (Fig. 4c), which prompted us to test whether PP2a could act in nuclear bodies. Co-immunoprecipitation experiments showed that Akt is able to associate with Pml both when activated or not (Fig. 4d); using CLSM of primary MEFs, significant nuclear body co-localization of PP2a-C and PP2a-A with Pml as well as nuclear body enrichment was evident (Fig. 4e), and PP2a-C was also found enriched in many Pml+/+ (but not Pml−/−) nuclei (Supplementary Fig. S4f). No Pml co-localization was found in the randomly tested PP2a-B PR-55 family members (Supplementary Fig. S4g).
As a result of probing PML deletion mutants for their ability to bind endogenous PP2a-C, we found that deletion of the RBCC motif abolished binding whereas the RING finger motif was dispensable (Fig. 4f), thus delineating the necessary interaction domains (Supplementary Fig. S5a). Similarly, Δ RBCC was unable to antagonize pAkt activation (Supplementary Fig. S5b). PP2a might also affect Akt indirectly via PDK1 (ref. 25), but we found unaltered steady-state phosphorylated PDK1 levels in tissues and MEFs of various genotypes (not shown) and similar steady-state levels of pAkt in Pml+/+ and Pml−/− cells (Supplementary Figs S3b, cytoplasm, and S5c, nucleus). Specific enrichment of pAkt in Pml nuclear bodies was found in wild-type MEFs when visualizing subnuclear pAkt localization by CLSM (Fig. 4g). Taken together, our data suggest that Pml can markedly increase effective concentrations of PP2a and its target, pAkt, in nuclear bodies, consistent with a nuclear pAkt clearing deficiency of Pml−/− cells.
Characterization of the molecular pathways leading to cancer is a major step towards understanding and combating the disease26. Here we have used mouse genetics to gather insights into AKT-driven tumorigenesis and established a mouse model for epithelial cancers triggered by Pml loss.
First, we have demonstrated the importance of Pml gene dose in prostate and colon carcinoma especially after Pten loss. For comparison, Trp53−/− prostates never show neoplasia27, and Pten+/−Trp53−/− prostates (ref. 27) or Pten+/−pRb+/− mice show no invasive prostate cancer (M. Niki and P.P.P., unpublished data). Second, PML loss highlights nuclear AKT function, a novel quality in PTEN-loss-driven tumorigenesis for which no model system exists so far. Colon, male prostate glands and female ovaries appear especially sensitive to increases in nuclear Akt and decreased Foxo3a levels. As we have shown that p27kip1 becomes a prostate and colon cancer inhibitor in conditions of Pten heterozygosity13, these results highlight the decisive role of FOXO3A function in human tumours with loss of one PTEN copy. Finally, we have identified a Pten–Pml–PP2a tumour-suppressive network for the inactivation of nuclear pAkt (see model, Supplementary Fig. S5d). Thus, PML regulates the activity of major opponents of AKT signalling in epithelia, which allows us to envision PML-stabilizing approaches for therapy of AKT-driven cancers.
METHODS
Mice
Pml−/− (ref. 28) and Pten+/− (ref. 7) mutants were crossed as described in Supplementary Information. The six cohort genotypes were: (1) Pten+/+ Pml+/+; (2) Pten+/+ Pml+/−; (3) Pten+/+ Pml−/−; (4) Pten+/− Pml+/+; (5) Pten+/− Pml+/−; and, (6) Pten+/− Pml−/−.
MRI
For initial tumour assessment, mice of all genotypes were subjected to monthly MRI screening similar to the protocol described previously11 (see also Supplementary Information).
Western blotting, immunoprecipitation and in vitro kinase assays
Tissue and cell lysates were prepared as previously described11 and processed as outlined in Supplementary Information. In vitro kinase assays were done using a non-radioactive kinase kit (see Supplementary Information).
Cells, plasmids and immunofluorescence
Pten−/− Trp53−/− MEFs were prepared as described27. Plasmids for FOXO3A and a FOXO3A mutant19 were gifts of M. Greenberg. The Flag–PML IV-derived plasmids were done as previously described29. See Supplementary Information for further information.
IHC and quantifications
For IHC, tissues were fixed and embedded in paraffin according to standard procedures (see Supplementary Information) and determination of average prostate cell size was calculated as outlined in Supplementary Information.
Quantitative real time PCR
Tissue RNA was isolated using the Trizol method and quantitative real-time PCR performed as described30. See Supplementary Information for all primer sets and detailed methods.
A detailed Methods section including antibodies used is available as online Supplementary Information.
Supplementary Material
Acknowledgments
We thank R. Bernardi, B. Carver, Z. Chen, S. Clohessy, T. Maeda and T. Yung for discussion, help with reagents and data analysis; W. Golden and M. S. Jiao for help with pathology analysis; and C. Le, M. Lupu and C. Matei for help with MR imaging. This work was supported by NIH grants to P.P.P. and P.P.S. as well as grants to J.A.K. and the Memorial Sloan-Kettering Cancer Center.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions The experiments were conceived and designed by L.C.T., A.A., P.P.S., J.A.K., C.C.-C. and P.P.P. Experiments were performed by L.C.T., A.A., P.P.S. and J.A.K. Data were analysed by L.C.T., A.A., P.P.S., J.A.K., C.C.-C. and P.P.P. The paper was written by L.C.T. and P.P.P.
References
- 1.Luo J, Manning BD, Cantley LC. Targeting the PI3K–Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 2.Brenkman AB, Burgering BM. FoxO3a eggs on fertility and aging. Trends Mol Med. 2003;9:464–467. doi: 10.1016/j.molmed.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108:165–170. doi: 10.1016/s0092-8674(02)00626-8. [DOI] [PubMed] [Google Scholar]
- 4.Gurrieri C, et al. Loss of the tumour suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst. 2004;96:269–279. doi: 10.1093/jnci/djh043. [DOI] [PubMed] [Google Scholar]
- 5.Goel A, et al. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res. 2004;64:3014–3021. doi: 10.1158/0008-5472.can-2401-2. [DOI] [PubMed] [Google Scholar]
- 6.Porkka KP, Visakorpi T. Molecular mechanisms of prostate cancer. Eur Urol. 2004;45:683–691. doi: 10.1016/j.eururo.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 7.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nature Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 8.Podsypanina K, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci USA. 1999;96:1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki A, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 10.Di Cristofano A, et al. Impaired Fas response and autoimmunity in Pten+/− mice. Science. 1999;285:2122–2125. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]
- 11.Trotman LC, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang S, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 13.Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nature Genet. 2001;27:222–224. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- 14.Maehama T, Taylor GS, Dixon JE. PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem. 2001;70:247–279. doi: 10.1146/annurev.biochem.70.1.247. [DOI] [PubMed] [Google Scholar]
- 15.Stambolic V, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 16.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor–mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 17.Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004;29:233–242. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Tran H, Brunet A, Griffith EC, Greenberg ME. The many forks in FOXO’s road. Sci STKE. 2003:RE5. doi: 10.1126/stke.2003.172.re5. 2003. [DOI] [PubMed] [Google Scholar]
- 19.Brunet A, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 20.Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301:215–218. doi: 10.1126/science.1086336. [DOI] [PubMed] [Google Scholar]
- 21.Hosaka T, et al. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci USA. 2004;101:2975–2980. doi: 10.1073/pnas.0400093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24:186–191. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- 23.Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 24.Janssens V, Goris J, Van Hoof C. PP2A: the expected tumor suppressor. Curr Opin Genet Dev. 2005;15:34–41. doi: 10.1016/j.gde.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Ren CH, Tahir SA, Ren C, Thompson TC. Caveolin-1 maintains activated Akt in prostate cancer cells through scaffolding domain binding site interactions with and inhibition of serine/threonine protein phosphatases PP1 and PP2A. Mol Cell Biol. 2003;23:9389–9404. doi: 10.1128/MCB.23.24.9389-9404.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang ZG, et al. PML is essential for multiple apoptotic pathways. Nature Genet. 1998;20:266–272. doi: 10.1038/3073. [DOI] [PubMed] [Google Scholar]
- 29.Guo A, et al. The function of PML in p53-dependent apoptosis. Nature Cell Biol. 2000;2:730–736. doi: 10.1038/35036365. [DOI] [PubMed] [Google Scholar]
- 30.Brunet A, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.