

Abstract
Monitoring of mutagenesis by environmental agents for the purpose of preventing genetic disease including cancer must include quantitation of cell killing, sensitive measurement of mutation production by appropriate doses of each agent, and assessment of mutation repair effects in mammalian cells. A four-step procedure, in the presence and absence of a repair suppressor, is proposed: (i) determination of the survival curve; (ii) measurement of the mitotic index in cells collected after treatment with colcemid; (iii) construction of a mutagenesis yield curve in the presence and absence of a repair suppressor, like caffeine; and (iv) assessment of the effect of test agents on the repair of mutations produced by other mutagens. The procedure is quantitative, reproducible, and reasonably rapid. It involves measurement of mutations causing visible chromosomal aberrations. Numerical parameters are proposed defining quantitatively mutation, cell killing, and mutation repair capacity. The procedure is applied to γ-irradiation and can detect the effects of doses as low as 2–5 cGy. Theoretical analysis of the underlying processes is presented, using the concept of DE0, the effective dose of mutagen after repair mechanisms and neutralizing agents have acted. Microscopically visible chromosome aberrations are due to mutations that distort the process of mitotic chromosome condensation, with or without actual chromosome breakage.
While it has long been recognized that mutation is the first step in carcinogenesis, and although mutation monitoring has been carried out for ≈25 years, the results have not fulfilled expectations. Indeed, an alarming number of human cancers have increased in frequency (1). It has been demonstrated that methodologies based on single gene markers are usually insufficiently sensitive. Thus, methods that can detect mutagenic effects of an agent like x-rays only in doses far beyond the mean lethal dose for human cells cannot be expected to be effective in preventing human disease, since dead cells cannot produce cancer (2–4). Similar considerations would apply not only to cancer but to all mutation-caused human disease, so that the range of pathology involved is indeed vast.
To be effective in preventing human disease due to mutagenic agents, it appears necessary to measure three different effects in a sensitive, rapid, and convenient procedure: production of mutation, cell death, and action of cellular repair systems. A system of measurements to accomplish these ends is considered here. It involves measurement of several parameters including chromosomal aberrations in cells without and with caffeine treatment to inhibit repair. A large number of studies have contributed in very important ways to development of cytogenetic scoring of mutational events, the role of caffeine in mutation repair, and the relationship of these processes to human disease (5–22).
This paper develops the underlying theory and presents a set of procedures for evaluating several aspects of mutation, which it is hoped will be useful for prevention of human disease. X-irradiation is used as a model mutagen because of the ease and the accuracy of dose delivery and measurement, and because it produces an extremely wide range of mutational events.
Materials and Methods
A random culture of human immortalized lymphocytes is irradiated at time = 0 with the prescribed dose of γ-rays; caffeine at a concentration of 1 mg/ml is added to half of the cells; the cells are incubated at 37°C for 0.5 hr and then colcemid at a concentration of 0.05 μg/ml is added to each tube to collect mitotic figures. After a further 1.5 hr of incubation, the cells are prepared by standard methodology for cytogenetic analysis. Mitotic indices are determined on these same slides, counting at least 1000 cells. Chromosomal aberrations are scored on 20–80 cells for each treatment. Aberrations scored consist of chromatid and chromosome breaks and gaps in which the discontinuity was at least equal to the width of the chromatid. Smaller discontinuities were counted if they were accompanied by a displacement of the chromatid arm beyond the break. The identity of the slides was unknown to the scorer, and multiple scorers evaluated the same slides with good agreement. At the γ-ray doses and caffeine concentrations employed, translocations, dicentrics, and rings were rarely if ever seen and were not scored (6). Unirradiated cells, with or without caffeine, usually yielded about 1 aberration per 20 cells. Caffeine itself slightly increases the incidence of these aberrations. Any aberrations found in control slides, with or without caffeine, were subtracted from the corresponding aberrational yield in the test slides. Typical results are presented in Table 1. γ-Radiation was delivered at room temperature by a 137Cs source at a dose rate of 3.3–174 cGy/min as described (11). Human lymphocytes immortalized by Epstein–Barr virus (NIGMS Mutant Cell Repository, Camden, NJ) or fresh human lymphocytes activated by phytohemagglutinin were employed. Survival curves for the lymphocytes were constructed by means of the colorimetric cell proliferation assay (23–26), which has been shown by others and ourselves to yield essentially the same curves as the standard single cell plating assay for a variety of mammalian cells (27). Rapidly growing fibroblasts like those of the Chinese hamster ovary (CHO) cell can also be used for all of the procedures described.
Table 1.
Typical experimental results showing chromosome aberrational yield and mitotic index changes in human lymphocytes induced by γ-irradiation.
| Dose, cGy | Aberrations per 20 cells
|
Mitotic index
|
||
|---|---|---|---|---|
| Caffeine absent | Caffeine present | Caffeine absent, % | Caffeine present, % | |
| 0* | 1† | 2† | 3.7 | 4.7 |
| 10 | 5 | 22 | 2.6 | 6.9 |
| 25 | 17 | 38 | 0.8 | 4.5 |
| 50 | 23 | 87 | 0.2 | 4.7 |
| 100 | 39 | 171 | 0.1 | 4.7 |
| 250 | ND‡ | 451 | 0 | 4.8 |
| 500 | ND‡ | 874 | 0 | 4.6 |
*Control.
These two values were averaged from 600 cells in 20 experiments. In each experiment, the number of aberrations in the absence of irradiation was substracted from that in the irradiated cells, in the presence and absence of caffeine respectively.
ND, not determinable. Too few cells reached mitosis to permit scoring.
A Four-Step Methodology for Characterization of γ-Ray Mutagenesis
The approach proposed here envisages carrying out four experimental procedures: (i) determination of the single cell survival curve or its equivalent for the test agent in the presence and absence of caffeine, (ii) measurement of the mitotic index in colcemid-treated cells that have been incubated with the test agent in the presence and absence of caffeine, (iii) measurement of microscopically visible chromosomal aberrations in these same cells, and (iv) measurement of the effect of the test agent on the mutational yield and G2 delay produced by other mutagens.
The Single Cell Survival Curve.
Survival curves define the lethal effects on mammalian cell reproduction of radiation and chemical agents (27). The survival curve is a necessary part of mutagenesis determination for any particular agent to ascertain the dose ranges to be tested. We tentatively propose that the dose between 100 and 1.0% survival would encompass the range most important for human disease. This figure can be changed if experience dictates its necessity.
The survival curve also permits determination of the parameters DL0, the mean lethal dose, and DR0, the maximum instantaneous dose of mutagenic agent that can be neutralized by the cellular repair system. The standard single cell survival curve exhibits an initial shoulder followed by a transition to a linearly logarithmic decrease in cell survival (27) (Fig. 1). For most γ-irradiation of mammalian cells tested, DL0, the mean lethal dose for human cells defined as the dose needed to reduce survivors to the 37% level (i.e., by the fraction 1/e) as measured in the linear portion of the logarithmic curve, lies between 50 and 175 cGy (27–29). The exact value varies with the experimental conditions, but approximate values are sufficient for present purposes. We use an approximate value of 100 cGy as the DL0 value of human cells generally. DR0 is obtained by the intersection of the extrapolated linear portion of the survival curve with the ordinate representing 100% survival. Many investigators make a practice of using the 50% cell lethality dose to describe cell killing by an agent. The parameters DL0 and DR0 are preferred, however, because each describes a single process, whereas the 50% survival dose is a mixed function of both cell killing and repair. DR0 also shows variability depending on experimental conditions with a range between 40 and 200 cGy. The variability in these parameters is not important for purposes of the present paper. In further work where more precision may be required, analysis of conditions governing this variability will be presented.
Figure 1.

A typical x-ray survival curve for the human HeLa cell (top curve) and demonstration of removal of the shoulder by caffeine (lower curve) to produce a linear curve with approximately the same slope as the limiting slope of the curve without caffeine (28). DL0, the mean lethal dose is obtained from this slope. DR0, the maximum dose that can be repaired is obtained from intersection of the top curve with the ordinate (survival = 100%). It can also be approximated as the interval between the two curves in the region where they are parallel.
Caffeine in appropriate dose erases the shoulder on the survival curve, producing a single exponential or one-hit curve, whose slope is almost the same as the limiting slope of the survival curve in the absence of caffeine (Fig. 1) (28). Caffeine has been shown to inhibit repair of mutations and to eliminate the interruption in traverse of G2 by mutagenized cells (G2 lag) (5–26, 28, 30–32).
It may be noted that survival curves constructed by the recently described cell proliferation method (23–25) yield results similar to those obtained by single cell plating for a wide variety of cells, and so can be used for cells like activated human lymphocytes for which single cell plating is difficult, but which are particularly convenient for cytogenetic analysis. However, fibroblasts could be substituted for activated lymphocytes if desired (18–22).
We utilize the following conceptual model: cell killing occurs as a result of unrepaired mutations located widely throughout the genome. The capacity of the cell to repair simultaneous mutations has a maximum limit. Killing is due to apoptosis/necrosis triggered in cells by a single unrepaired mutation occurring in an exposed region of the genome (see Discussion). Thus, in understanding mutagenic effects of any agent, it becomes necessary to determine the mean lethal dose, DL0, the mean mutagenic dose, DM0, and DR0, the maximum dose that when given at a single point in time can still be repaired by the test cell.
DL0 is obtained from the limiting slope of the standard survival curve. DR0 can be calculated either from the survival curve as the intersection of the extrapolated linear part of the survival curve with the ordinate, 100% survival (Fig. 1), or somewhat less accurately from the difference between survival curves carried out in the presence or absence of caffeine in the region where the two curves are virtually parallel, as shown in Fig. 1. DM0 is obtained as the slope of the straight line curve obtained by plotting the number of observed chromosomal aberrations against the dose of x-rays as indicated in Fig. 2 (2, 8).
Figure 2.

Demonstration of the linearity of the plot of chromosomal aberration yield vs. γ-radiation dose for immortalized human lymphocytes in the presence of 1.0 mg/ml of caffeine. Construction of the same curve in the absence of caffeine was not possible because of the mitotic delay that suppresses the number of mitotic cells available for cytogenetic analysis. This curve remains linear down to 5 cGy of irradiation. Results from two separate experiments have been plotted to demonstrate the reproducibility. It appears that even lower doses could be monitored if larger numbers of mitotic cells were analyzed.
For a dose, D, appreciably less than DR0, the survival is close to 100%. For doses sufficiently great so that the limiting slope is achieved, the linear logarithmic region of the survival curve is given by the following equation:
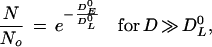 |
1 |
where DE0, the effective dose, is defined by:
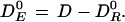 |
2 |
(Similarly, if mutagen-neutralizing substances are present in the medium,
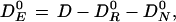 |
3 |
where DN0 represents the amount of dose neutralized by the antimutagenic agents.)
These equations represent cell survival only in the region of the survival curve where the limiting slope has been achieved. For doses lower than this, survival may be calculated by taking into account the probabilistic nature of the process. The exact survival curve for any dose can be represented by the appropriate equations to be presented in the next paper in this series.
Cells or agents that for various reasons do not exhibit the typical survival curve like that in Fig. 1 have to be analyzed individually. Obviously, agents that produce high mutagenesis and low cell killing are the most dangerous from the standpoint of disease causation.
Measurement of the Mitotic Index.
The next step is to prepare slides for cytogenetic analysis and to determine the mitotic index on such slides. A positive indication of mutagenesis activity at any dose is given if, and only if, both the following conditions are satisfied: (i) demonstration of a clear drop in the mitotic index caused by the test agent in the absence of caffeine, and (ii) reversal of this drop in mitotic index by the action of caffeine (Table 1). For example, estradiol causes a G2 lag and consequent drop in mitotic index in this procedure, but the effect is not reversed by caffeine. Tests with estradiol showed no mutagenic activity at these doses. Thus, the mitotic index test can be used to rule out agents that are not mutagenic in the dose employed. A rapid scanning for chromosome aberrations in such cases can confirm the absence of mutations and eliminate the need for painstaking counting of aberrations. Treatments exhibiting patterns of mitotic index effects different from those shown in Table 1 should be scored carefully for mutagenesis. While a variety of chemical agents including 1-methyl-3-nitro-1-nitrosoguanidine, hydrogen peroxide, and 4-nitroquinoline have been successfully tested in this fashion, further testing is necessary to determine the universality of the use of mitotic index as a preliminary mutation indicator. It should be emphasized that it is necessary to adhere strictly to the procedure given here to maximize the mitotic index effect.
The Standard Mutagenesis Curve.
Quantitative delineation of mutagenesis is achieved by scoring clearly defined chromosomal aberrations in the treated cells, both in the presence and absence of caffeine (11). Theoretically, one should obtain from these experiments not only a value for DM0, but also a confirmation of the value for DR0. Practically, however, the latter value is difficult to obtain from this procedure because the G2 delay in the absence of caffeine causes so large a drop in the mitotic index even for small radiation doses that it is difficult to obtain enough mitotic figures to provide an accurate value of the aberrations index (Table 1 and Fig. 2). Because the points form a straight line through the origin, each aberration determination permits a value of DM0. In a series of four experiments with radiation doses varying from 25 to 200 cGy, DM0 calculated from 10 separate determinations in the presence of caffeine yielded a value of DM(caff)0 = 12.3 ± 1.5 cGy. In the absence of caffeine, DM0 was determinable only for 25 and 50 cGy of irradiation, and yielded a value of 41.8 ± 12 cGy.
From the curve in Fig. 2, one can calculate that DM0, the average dose needed to produce one recognizable mutation per G2 cell, is ≈12.3 cGy. Similar but different values for the various parameters are obtained if a different test cell like CHO is employed. Activated or immortalized human lymphocytes have been chosen by us because of their great convenience in cytogenetic analysis. It may be noted that chromosome aberration yields for different doses of x-irradiation observed by us almost 40 years ago using normal human fibroblasts agrees well with that shown in Fig. 2 in the absence of caffeine (6). Use of lymphocytes has the added advantage of making possible a rapid test for agent mutagenesis and cellular repair capacity for any person from whom a blood sample is available (18–22).
Measurement of Effects on the Repair Reaction.
The fourth procedure is to determine the effect of the agent to be tested on the repair of mutations produced by other agents. For example, a 2-hr exposure to 1 mg/ml of caffeine has no effect on cell survival, but has a very large effect on the repair of mutations produced by sublethal amounts of x-rays. A mutation yield curve is constructed for γ-irradiated cells in the presence and absence of the agent to be studied, which is tested in various concentrations in the procedure exactly like that used for caffeine in determining its effect on the mutagenesis of γ-rays. Thus, agents like caffeine, which inhibit repair, and the critical concentrations needed for activity, can be identified. This procedure will also identify agents and treatments that enhance repair.
Conclusions
From the data of Table 1, the following points are evident. (i) Both the chromosomal aberration count and the drop in mitotic index, which is reversed by caffeine, clearly reveal that doses of 10 cGy or more are highly mutagenic. (ii) The DM0 value is 12.3 cGy for the G2 cells of the human lymphocyte. (iii) The DR0 values varying between 35 cGy for a representative human lymphocyte and 120 cGy or more for the HeLa cell indicate the variability in repair potential of different human cells. (iv) The aberration yield for lymphocytes in the presence of caffeine is highly linear even up to a dose of 500 cGy of irradiation. The mutagenic effects of even 2 cGy are detectable by this procedure, as others have also noted (16), and undoubtedly smaller doses could be detected if the number of mitoses analyzed were to be increased sufficiently. (v) At doses between 5 and 50 cGy, well below the DR0 value, appreciable numbers of mutations are obtained even in the absence of caffeine, indicating that even in this comparatively low dose region, appreciable numbers of mutations can escape repair.
From these data, one can calculate that the mean lethal dose (DL0) for these cells of about 80–100 cGy represents about seven aberrations per cell needed for lethality. (If cells in G1 and S are less susceptible to killing in proportion to their decreased DNA content and therefore smaller target size, then the average number of aberrations needed to kill a G2 cell will be somewhat less and for G1-S cells somewhat more than this figure.)
Discussion
It is necessary to consider what types of mutations are actually scored in this system. It has been conventional to regard chromosomal aberrations of the type considered here as gross changes in chromosomal structure usually involving breaks in the DNA chain. The implication of such considerations is that smaller so-called “point” mutations would be missed by this procedure.
It should be pointed out that the procedure described here is extremely sensitive, easily able to detect doses as little as 2–5 cGy of x-rays, in a fairly rapid fashion. In part the increased sensitivity is due to use of the entire genome as a target instead of just a single marker gene. The reason for the ability to score single mutational events can be understood from analysis of the underlying metabolic processes. Scoring of mutations is carried out in mitotic cells in which each chromosome has been condensed 20,000 times and its thickness correspondingly increased so that the chromosomes at this part of the life cycle become visible in the light microscope. This condensation is achieved by coiling, supercoiling, and super-supercoiling of the DNA. Throughout this process, proteins attach to particular sites in a highly specific fashion. This entire sequence of molecular events is carried out in an exceedingly orderly way, as evidenced by the uniform diameter achieved throughout the chromosomal length despite the enormous condensation that has occurred, and by the highly reproducible fashion in which the various banded regions on every chromosome are displayed.
If a change in structure at any point of any chromosome is produced by a mutagenic agent such that the specific condensation reaction at that point is inhibited, DNA around that point will remain uncondensed or poorly condensed. Because uncondensed DNA is invisible under the light microscope, the result will be an apparent discontinuity in the chromosome or some other departure from normal chromosomal structure like chromosome unravelling. Of course some mutations may indeed produce an actual chromosomal discontinuity but these are not the only lesions monitored by the procedure described. By far the most frequent visible effect of mutagenesis is an apparent mitotic chromosomal discontinuity, and these are so easily recognized that they have constituted the principal basis for our measuring system so far.
The methodology described here can also be used for chemical mutagens (2), and detailed data will be presented elsewhere. Automation appears feasible and should make the scoring procedure even more rapid and sensitive. It becomes necessary to consider effects of changing dose rate, particularly with respect to chemicals whose entry into the cell, or whose modification inside the cell, may lend further complications to the process.
Results similar to those reported here have been obtained by us on lymphocytes from freshly drawn blood, which have been induced to initiate reproduction by treatment with phytohemagglutinin, as has also been previously reported by Parshad, Sanford, and coworkers (18–22).
The approach described here appears useful for the following applications. (i) Detection and measurement of the effects of different doses of mutagenic agents. (ii) Detection and measurement of the effects of different doses of antimutagens. (iii) Detection and measurement of the effects of mixtures of agents like those including tobacco, asbestos, alpha-emitters, and other substances that appear able to synergize mutational effects. (iv) Discovery and characterization of agents that inhibit or enhance repair. (v) Analysis of mutation and repair in various pathologic conditions like aging and diseases in which repair is defective (18–22). (vi) Analysis of mutational risk factors in selected human populations and in family members of persons with demonstrated genetic disease (18–22).
The question arises as to why DM0 is definitely smaller than DL0. If, as we propose, the mutational lesion is indeed what triggers apoptosis/necrosis, it may be that only about one-seventh of the lesions we examine contribute to cell lethality. Several possible factors may contribute to this difference. (i) We visualize that mitotic cells entering a new reproductive cycle undergo DNA monitoring for mutations. Presumably monitoring molecules traverse the exposed DNA regions. On encountering a mutation, the monitoring process halts and either the lesion is repaired or apoptosis/necrosis is initiated. Repaired mutations presumably do not lead to death. Thus, one unrepaired mutation may be sufficient to trigger the lethal process. (ii) Cells in G1 have only a fraction of their DNA exposed to react with the molecules involved in the monitoring function (33). Thus mutations in areas that become sequestered in G1 may fail to contribute to cell death. (iii) The value of 12.3 cGy for DM0 was obtained for cells that were in the G2 phase of the cell cycle, whereas the survival curve is determined for cells distributed over the entire life cycle. The amount of DNA in G2 cells is twice that of G1 cells, while in S the DNA content varies from that of the G1 cell at the beginning of S to that of the G2 cells at its end. Thus cells in G1 and early S will present a smaller target than those in G2. (iv Finally other factors including repair may operate differently for cells irradiated in G2 as compared with other parts of the cycle. We prefer to use the G2 cell for mutation monitoring because of its convenience and the fact that its greater DNA content should provide maximum target size and therefore sensitivity.
For most cells the survival curve drops in simple exponential fashion after a dose in the neighborhood of several DL0’s has been given. Presumably then, a single unrepaired but exposed mutation is enough to cause cell killing by apoptosis/necrosis. Cells in different parts of the life cycle may vary in the effectiveness of repair so that some may be able to repair more mutations than others. When the repair capacity has been saturated, however, the presence of a single further (and therefore unrepaired) mutation would appear to be sufficient to initiate the lethal processes after the cell moves into its next reproductive cycle.
It should be pointed out that the term “repair” does not imply that the cell genome has been returned to its original state. All that this analysis indicates is that the mutational changes that have occurred in the structure of the cellular genome that prevent abnormal appearance of chromosome condensation in mitosis and can contribute to cell reproductive death, have been altered metabolically so as to permit the cell to resume unlimited reproduction instead of undergoing apoptosis/necrosis.
Because caffeine under the conditions prescribed by Waldren and Rasko (28) produces a linear logarithmic survival curve with approximately the same slope as the limiting slope achieved without caffeine, we conclude that caffeine prevents repair of all (or almost all) lesions contributing to cell death under these specific experimental conditions. There may be other types of DNA lesions produced by the mutagenic action that are not repaired or do not contribute to cell death which are not affected by caffeine.
Radiation doses that cause mutation in G2 cells also cause a delay in reaching mitosis, during which time the repair is effected. This delay, which is at least roughly proportional to the dose, can be detected for doses of approximately 9 rads or less (34, 35). Caffeine causes bypass of the repair process so that both DNA repair and its accompanying G2 lag are eliminated.
The condensation process in the mitotic chromosomes thus furnishes visual amplification of the results of mutation and so permits the very high sensitivity that is required for detecting nonlethal mutagenesis responsible for disease. Earlier views regarded discontinuities in mitotic chromosomes as an indication of an actual break in the DNA chain. The fact that such an apparent break does not necessarily indicate a DNA discontinuity is borne out by recent studies in fragile X syndrome in which the apparent discontinuity in the X chromosome has been shown to represent a continuous chromosome in which amplification of a repetitive sequence is responsible for the aberrant structure of the mitotic X chromosome (36, 37).
The procedure used here might miss certain classes of mutation. If such are discovered, they will have to be identified and corrected for in monitoring situations important in human disease. The three quantities, DL0, DM0, and DR0, would appear to define the cell’s reaction to a mutagenic agent applied in a very short time. The concept of the effective dose, DE0, permits calculation of the effects of cellular repair and the presence of added antimutagens.
Effects of chemical agents will be considered in detail later. Some complexity arises because such a dose usually cannot be administered in as short a time as with radiation. However, the approach outlined here remains applicable. Survival curves for chemical agents are well known. Representative mutagenic data for 1-methyl-3-nitro-1-nitrosoguanidine (MNNG) have been presented earlier (2). It can be stated that mitotic index effects for this agent have been found to resemble those illustrated here for γ-rays.
The most dangerous dose range in the production of human disease would appear to be in the lower dose ranges where large numbers of viable cells remain, but in which the repair mechanism has been unable to neutralize all of the mutations produced. It is obvious, then, that agents like caffeine, which inhibit repair, may be contributing as much as mutagenic agents themselves in causing disease. In higher concentrations caffeine itself also causes mutation. The molecular nature of caffeine action at different concentrations and exposure times requires more detailed study.
Finally it should be noted that mutagenic doses of γ-irradiation well below the maximum repair dose as interpreted here are not necessarily harmless. While doses of 50 cGy or less produce considerably less mutation in the absence of caffeine than in its presence, they still produce significant numbers of mutations even when normal repair operates. When one considers the huge number of cells in the body, and that presumably a single cell mutated appropriately can lead to cancer or genetic disease, the magnitude of the problem can be appreciated. Persons exposed to mutagens like radiation or cigarette smoke would be well advised to avoid simultaneous intake of caffeine.
Acknowledgments
We thank P. Webb and M. H. Puck for editorial assistance. This work was aided by U.S. Department of Energy Subcontract 6267 P0015–3C with Los Alamos National Laboratory Life Sciences Division, the Disease Prevention Fund, and by a National Institute of Environmental Health Sciences contract.
References
- 1.Beardsley T. Sci Am. 1994;270:130–138. doi: 10.1038/scientificamerican0194-130. [DOI] [PubMed] [Google Scholar]
- 2.Puck T T, Harvey W F. Mutat Res. 1995;329:173–181. doi: 10.1016/0027-5107(95)00028-h. [DOI] [PubMed] [Google Scholar]
- 3.Puck T T. Lett Math Phys. 1985;10:225–230. [Google Scholar]
- 4.Thilly W G. Environ Mutagen. 1985;7:255–258. doi: 10.1002/em.2860070212. [DOI] [PubMed] [Google Scholar]
- 5.Bender M S. Science. 1957;12:974–975. doi: 10.1126/science.126.3280.974. [DOI] [PubMed] [Google Scholar]
- 6.Puck T T. Proc Natl Acad Sci USA. 1958;44:772–780. doi: 10.1073/pnas.44.8.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson L H, Hoy C A. In: Chemical Mutagens: Principles and Methods for Their Detection. de Serres F J, editor. New York: Plenum; 1986. pp. 285–325. [Google Scholar]
- 8.Natarajan A T, Obe G, Dulout F N. Hum Genet. 1980;54:183–189. doi: 10.1007/BF00278969. [DOI] [PubMed] [Google Scholar]
- 9.Pincheira J, Lopez-Saez J F. Mutat Res. 1991;251:71–77. doi: 10.1016/0027-5107(91)90216-b. [DOI] [PubMed] [Google Scholar]
- 10.Kihlman B A, Andersson H D. Mutat Res. 1985;150:313–325. doi: 10.1016/0027-5107(85)90128-9. [DOI] [PubMed] [Google Scholar]
- 11.Puck T T, Morse H, Johnson R, Waldren C A. Somatic Cell Mol Genet. 1993;19:423–429. doi: 10.1007/BF01233247. [DOI] [PubMed] [Google Scholar]
- 12.Dewey W D, Miller H H, Leeper D B. Proc Natl Acad Sci USA. 1971;68:667–671. doi: 10.1073/pnas.68.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanson R, Palitti F, Kihlman A, Karlsson M B. Heriditae. 1982;97:51–58. doi: 10.1111/j.1601-5223.1982.tb00711.x. [DOI] [PubMed] [Google Scholar]
- 14.Tolmach L J, Busse P M. Radiat Res. 1980;82:374–392. [PubMed] [Google Scholar]
- 15.Walters R A, Gurley L R, Tobey R A. Biophys J. 1974;14:99–118. doi: 10.1016/S0006-3495(74)70002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andersson H C, Kihlman B A. Mutat Res. 1984;141:45–48. doi: 10.1016/0165-7992(84)90036-8. [DOI] [PubMed] [Google Scholar]
- 17.McGuinness S M, Shibuya M L, Ueno A M, Vannais D B, Waldren C A. Radiat Res. 1995;142:247–255. [PubMed] [Google Scholar]
- 18.Parshad R, Sanford K K, Jones G M, Tarone R E. Cancer Genet Cytogenet. 1985;14:163–165. doi: 10.1016/0165-4608(85)90227-4. [DOI] [PubMed] [Google Scholar]
- 19.Takai S, Price F M, Sanford K K, Tarone R E, Parshad R. Carcinogenesis. 1990;11:1425–1428. doi: 10.1093/carcin/11.8.1425. [DOI] [PubMed] [Google Scholar]
- 20.Sanford K K, Parshad R, Gantt R R, Tarone R E. Crit Rev Oncog. 1989;1:323–341. [PubMed] [Google Scholar]
- 21.Parshad R, Price F M, Bohr V A, Cowans K H, Zujewski J A, Sanford K K. Br J Cancer. 1996;74:1–5. doi: 10.1038/bjc.1996.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helzlsouer K J, Harris E L, Parshad R, Fogel S, Bigbee W L, Sanford K K. Int J Cancer. 1995;64:14–17. doi: 10.1002/ijc.2910640105. [DOI] [PubMed] [Google Scholar]
- 23.Mossman T. J Immunobiol Methods. 1983;65:55–63. [Google Scholar]
- 24.Gustafson D L, Beall H D, Bolton E M, Ross D, Waldren C A. Mol Pharmacol. 1996;50:728–735. [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmend S A, Gogal R, Jr, Wash J. J Immunol Methods. 1994;170:211–224. doi: 10.1016/0022-1759(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 26.Page B, Page M, Noël C. Int J Oncol. 1993;3:473–476. [PubMed] [Google Scholar]
- 27.Puck T T, Marcus P I. J Exp Med. 1956;103:653–666. doi: 10.1084/jem.103.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waldren C A, Rasko I. Radiat Res. 1978;73:95–110. [PubMed] [Google Scholar]
- 29.Puck T T, Morkovin D, Marcus P I, Cieciura S J. J Exp Med. 1957;106:845–500. doi: 10.1084/jem.106.4.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kihlman B A. Caffeine and Chromosomes. Amsterdam: Elsevier; 1977. [Google Scholar]
- 31.Kihlman B A, Andersson H C. In: Genetic Toxicology of Environmental Chemicals. Ramel C, Lambert B, Magnussen, editors. New York: Liss; 1986. Part A, pp. 395–402. [Google Scholar]
- 32.Natarajan A T, van Zeeland A A, Palitti F, Meijers M, Verdegaal-Immerzed E A M. Hum Genet. 1980;54:183–189. [Google Scholar]
- 33.Puck T T, Krystosek A. Int Rev Cytol. 1992;132:75–108. doi: 10.1016/s0074-7696(08)62454-7. [DOI] [PubMed] [Google Scholar]
- 34.Yamada M-A, Puck T T. Proc Natl Acad Sci USA. 1961;47:1181–1191. doi: 10.1073/pnas.47.8.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puck T T, Steffen J. Biophys J. 1963;3:379–397. doi: 10.1016/s0006-3495(63)86828-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pearson C E, Sinden R R. Biochemistry. 1996;35:5041–5053. doi: 10.1021/bi9601013. [DOI] [PubMed] [Google Scholar]
- 37.Kramer R R, Pearson C E, Sinden R R. Hum Genet. 1996;98:151–157. doi: 10.1007/s004390050179. [DOI] [PubMed] [Google Scholar]