

Summary
PilT is a hexameric ATPase required for bacterial Type IV pilus retraction and surface motility. Crystal structures of ADP and ATP-bound Aquifex aeolicus PilT at 2.8 and 3.2 Å resolution show N-terminal PAS-like and C-terminal RecA-like ATPase domains followed by a set of short C-terminal helices. The hexamer is formed by extensive polar subunit interactions between the ATPase core of one monomer and the N-terminal domain of the next. An additional structure captures a non-symmetric PilT hexamer in which approach of invariant arginines from two subunits to the bound nucleotide forms an enzymatically competent active site. A panel of pilT mutations highlights the importance of the arginines, the PAS-like domain, the polar subunit interface, and the C-terminal helices for retraction. We present a model for ATP binding leading to dramatic PilT domain motions, engagement of the arginine wire, and subunit communication in this hexameric motor. Our conclusions apply to the entire Type II/IV secretion ATPase family.
Introduction
Type IV pili are grappling lines that extend from bacterial cells to solid substrates to be subsequently reeled in by membrane-associated motors, thereby promoting motility on surfaces (Merz et al., 2000; Skerker and Berg, 2001). The ~100 pN forces exerted by individual pilus filaments (Maier et al., 2002) are enormous given their tiny (~60 Å) diameter and alone can initiate signaling events in eukaryotic target cells (Howie et al., 2005). Presumably retraction proceeds as pilin subunit monomers are disassembled from the base of an extended type IV pilus polymer and dispersed into the membrane for later reuse in pilus elongation (Morand et al., 2004; Skerker and Berg, 2001). Type IV pilus-mediated motility is vital to the lifestyle of many bacteria, including the free-living Myxococcus xanthus (Li et al., 2003; Wu et al., 1997) and plant and animal pathogens Ralstonia solanacearum, Xylella fastidiosa, Pseudomonas syringae, Pseudomonas aeruginosa, Neisseria meningitidis, and Neisseria gonorrhoeae (Comolli et al., 1999; Kang et al., 2002; Liu et al., 2001; Meng et al., 2005; Merz and So, 2000; Pujol et al., 1999). Therefore, a study of the properties of this machinery is important for basic understanding of bacterial physiology and as a potential discovery tool for preventing the spread of bacterial infections. Although the structures of several Type IV pilin monomers have been solved and the polymers formed by them accurately modeled (reviewed in Craig et al., 2004; Hansen and Forest, 2006), the structure of the assembly and retraction machinery is largely unknown. We have thus undertaken a biochemical and structural characterization of the pilus retraction motor, PilT.
PilT proteins form a subgroup of the bacterial Type II secretion ATPases. These in turn are members of a larger family of Type II/Type IV hexameric secretion ATPases, required for transport of a diverse set of soluble proteins across the bacterial envelope of a wide variety of pathogenic and environmental bacteria (Cianciotto, 2005; Planet et al., 2001). Based on homology with type II secretion ATPases, each PilT monomer is expected to be composed of two major structural domains. The N-terminal domain (NTD), containing ~100–115 amino acids, is required for membrane association and for polar localization of PilT (Chiang et al., 2005). The C-terminal domain (CTD), ~240 amino acids in length, contains the sequences commonly associated with NTPase activity in hexameric ATPases of the RecA superfamily (Figure 1), including the Walker A phosphate binding (P-) loop and a loosely defined Walker B Box for this ASCE (Additional Strand Catalytic Glutamate (Iyer et al., 2004)) ATPase. The CTD also contains Asp and His Boxes defined for Type II and Type IV secretion ATPases (Iyer et al., 2004; Planet et al., 2001; Rivas et al., 1997). Accordingly, PilT is a hexameric ring with low NTPase activity in vitro (Forest et al., 2004; Herdendorf et al., 2002; Okamoto and Ohmori, 2002). The ATPase activity is likely important for its in vivo function, as mutation of the P-loop lysine prevents twitching motility (Aukema et al., 2005). Beyond the ~170 amino acids that make up the ATPase core, the C-terminal ~70 amino acids also contain a well-conserved PilT-specific AIRNLIRE motif which is required for retraction but not ATPase activity (Aukema et al., 2005).
Figure 1.

The sequence and secondary structure of A. aeolicus PilT (AaPilT) aligned with P. aeruginosa PilT (PaPilT), N. gonorrhoeae PilT (NgPilT), V. cholerae EpsE (EpsE) and H. pylori HP0525 (HP0525). Alignments are based on 3-D superpositions, sequence homology, and manual optimization. Observed A. aeolicus PilT α-helices (h) and β-strands (>) are denoted above the sequence, and indicated by blue and red letters, respectively, for the three known structures. Type II/IV secretion ATPase motifs are indicated by shaded boxes: Walker A, Asp Box, Walker B and His Box, in that order. Other symbols mark the CTDn:NTDn+1 interface (diamond), recognition of adenine (plus sign), arginine wire (quotation), other residues presumed to participate in ATP recognition and catalysis (pound sign), and positions of changes in other non-functional PilT variants from this study (dagger). Dots indicate every 10th amino acid in the A. aeolicus PilT sequence. The EpsE zinc-binding tetracysteine motif (CM subdomain) is marked by a carat; the alignment continues after this 40 amino acid insertion. Residues not resolved in the refined structures are typed in grey. EpsE and HP0525 sequences are not shown upstream of the region of structural homology.
Structures of three traffic ATPases are known (Hare et al., 2006; Robien et al., 2003; Savvides et al., 2003; Yeo et al., 2000). HP0525 is the hexameric Type IV secretion ATPase responsible for gating the flow of CagA out of the Gram-negative bacterium Helicobacter pylori. The structure of this VirB11 homologue has been solved in apo, ADP- and ATPγS-bound forms, which taken together suggest that pivoting of the CTD relative to the NTD upon nucleotide binding drives assembly of the secretion apparatus and/or secretion of CagA. The Bordetella suis VirB11 homologue has the same domain folds and hexamer assembly despite a dramatic domain swap and a novel subunit interface, due partly to a 30 residue domain linker which wraps around the adjacent CTD (Hare et al., 2006). EpsE is a Type II secretion ATPase from Vibrio cholera, required for secretion of cholera toxin and chitinase (Sandkvist, 2001). The EpsE apo and AMPPNP-bound structures revealed three-dimensional similarity to HP0525 within the NTD and the ATPase core (subdomain C1), with the addition of a zinc-binding structural motif (subdomain CM) and four α-helices at the far C-terminus (subdomain C2) (Robien et al., 2003). Although PilT and EpsE amino acid sequences are 30% identical overall, PilT does not contain the residues that define the CM domain of EpsE, and its amino acid sequence is only 13% identical to EpsE over the residues that encompass the C2 domain of EpsE. Thus, while the HP0525 and EpsE structures provide clues to the architecture of PilT, and in particular may allow relatively accurate prediction of the structure of the ATPase core, they are not suitable for detailed modeling of the Type IV pilus system.
Here we present four independent X-ray crystal structures of PilT from the hyperthermophilic bacterium Aquifex aeolicus. This PilT was chosen for its stable biochemical properties and =50% sequence identity over its entire length with PilT from P. aeruginosa, N. gonorrhoeae, N. meningitidis and M. xanthus, three human pathogens and a free-living bacterium in which much work on Type IV pilus assembly and retraction has been carried out. A comparison of the structures of independent monomers within the PilT hexamer yields results about the dramatic conformational changes that this motor may undergo in vivo.
Results
Domain Folds of NTD and CTD
The 2.8 Å resolution structure of full-length nucleotide-bound A. aeolicus PilT (Figure 2A) was determined in space group P6, with one molecule per asymmetric unit. The fitting was greatly aided by the results of the structure determination at 1.87 Å resolution of the CTD alone. The PilT NTD forms a 6-stranded all-antiparallel β-sheet flanked on one side by three short α-helices (Figures 2A,B). Not unexpectedly, the closest structural matches in the Protein Data Bank to the NTD of PilT are EpsE (2.3 Å r.m.s.d. over 88 amino acids) and HP0525 (2.9 Å r.m.s.d. over 91 amino acids). Additionally, the fold of the NTD resembles the well-known Per/Arndt/Sim (PAS) domain, with the addition of one edge strand (β3) to the canonical 5-stranded sheet (Figure 3A). The PilT PAS similarity is more evident than that of EpsE or HP0525, as only PilT contains αC preceding β4 to complete the PAS-like fold.
Figure 2.
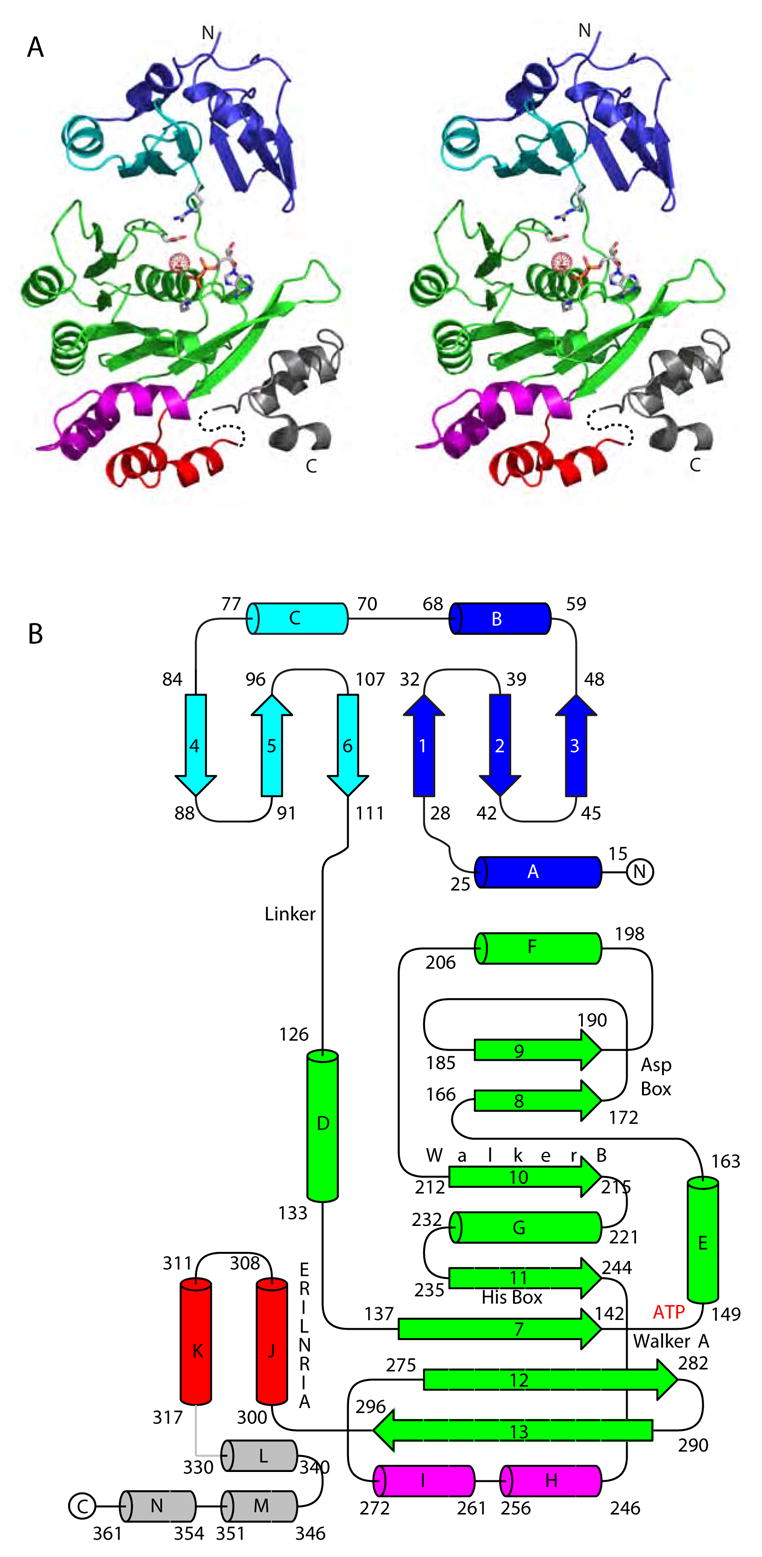
Structure of PilT monomer. (A) Stereo view of the refined structure of the PilT:ADP monomer. The N-terminal PAS-like domain is blue and cyan to indicate quasi internal 2-fold symmetry. The CTD secondary structure elements form the RecA fold (green), helices αH and αI which form the hexamer constriction and are an insertion in the RecA fold (magenta), helices αJ and αK (red), and the far C-terminal helices αL, αM, and αN (grey). Conserved residues Arg110, Lys149, Ser 150 and Glu176, as well as the bound water and ADP, signpost the nucleotide-binding pocket. (B) Schematic of the PilT secondary structure elements with conserved ATP binding motifs indicated. The coloring scheme is as in (A).
Figure 3.
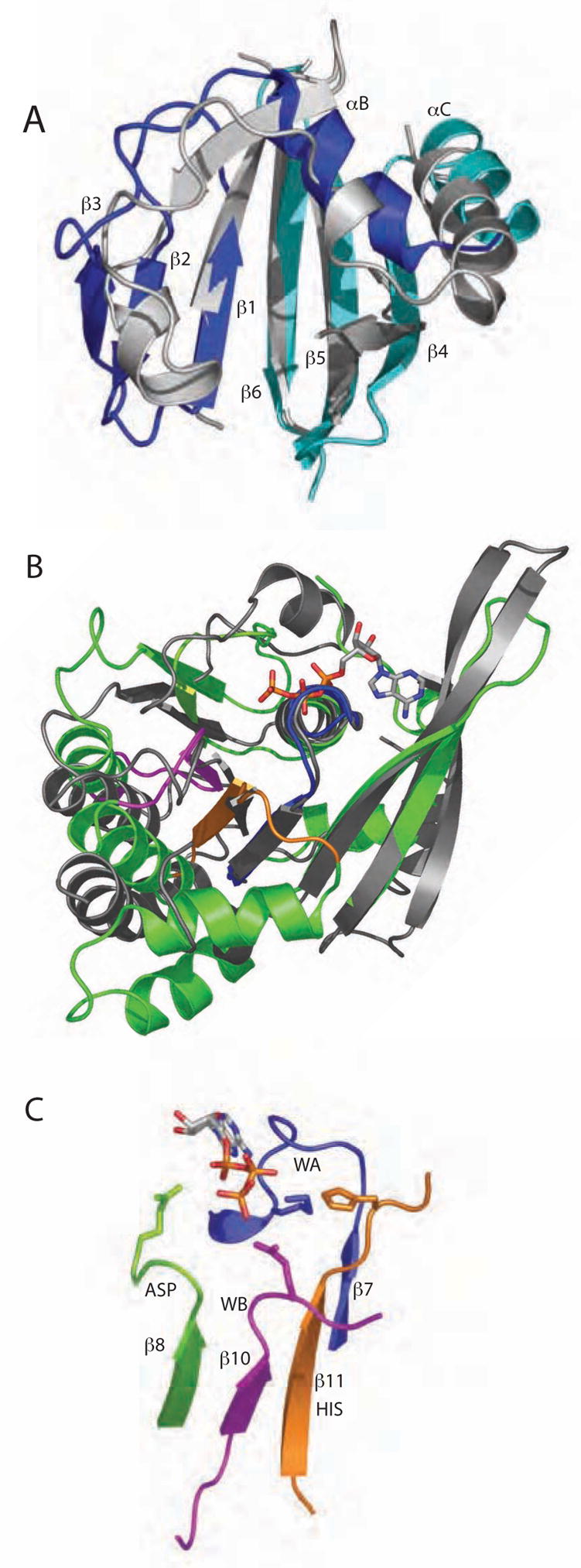
Conserved elements of PilT fold. (A) The NTD of PilT (colored as in Fig. 2A) resembles the well-known PAS domain (grey, represented by the circadian clock protein Period (Yildiz et al., 2005)). Non-canonical PAS elements (PilT αA and loops within Period) are removed for clarity. (B) The core ATPase subdomain of PilT (green) is readily superimposable upon RecA (Story and Steitz, 1992) (grey). In this view the least squares calculation is over P-loop residues only. Type II/IV secretion ATPase family motifs Walker A (blue), Asp Box (lime green), Walker B (magenta) and His Box (orange) neighbor the bound nucleotide. (C) Isolated, magnified view of the four sequence motifs described in (B), with ADP and signature invariant residues Lys149, Glu176, Glu217 and His242 depicted (blue, green, magenta, and orange, respectively).
This is an appropriate place to note that the PilT NTD does not adopt the fold of the “PilT N-terminus” or PIN domain, nor is there clear sequence homology among these proteins and PilT. Possible roles for PIN domains as RNAses involved in RNAi in eukaryotes and as part of toxin/antitoxin pairs in prokaryotes have been proposed (Arcus et al., 2004; Clissold and Ponting, 2000). The structures of several PIN domain proteins from extremeophiles have been recently reported as part of structural genomics efforts (Arcus et al., 2004; Jeyakanthan et al., 2005; Levin et al., 2004). While the authentic PilT NTD and the reported PIN domains are both α,β proteins, the similarity ends there, as the PIN domain is a 5-stranded parallel β-sheet with β3-β2-β1-β4-β5 topology, helical hairpins between β1 and β2 and between β2 and β3, and single helices between β3 and β4 and between β4 and β5 (compare Figure 3A with Figure 1 of (Levin et al., 2004)). Thus, it is not entirely clear how the PIN domain got its name, and one should use caution when assuming that the growing wealth of structural genomics information has been exhaustively annotated. We propose to reassign the PIN acronym to Putative Interaction with Nucleic acid, thus ensuring that research and literature on the PIN domain will remain accessible and searchable but that the letters have an appropriate meaning.
The PilT ATPase core is formed by a six-stranded parallel β-sheet with a seventh antiparallel strand, sandwiched between α-helices (Figures 2A,B). This fold can be superimposed upon RecA (Story and Steitz, 1992) with an r.m.s.d. of 3.5 Å over 152 core residues (Figure 3B). Since PilT and EpsE have similar sequences and structural elements in the CTDs, a 1.3 Å r.m.s.d was calculated over 203 structurally similar Cα atoms including PilT helices αJ, αK and αN (Figure 3B). For the PilT/HP0525 CTD superposition, lower sequence homology and lack of a C2 subdomain led to a 1.5 Å r.m.s.d. using 121 Cα atoms. Within the shared fold are the four well-recognized secretion ATPase sequence motifs, all of which neighbor the bound ATP (Figures 2B, 3B,C).
Seven short α-helices decorate the base of the CTD. The AIRNLIRE region, a PilT signature required for pilus retraction (Aukema et al., 2005), forms αJ (Figure 2).
Nucleotide Binding at NTD-CTD junction
Both ATP- and ADP-bound structures of PilT have been solved (Table 1), with the intent to understand how substrate binding and/or hydrolysis affect the structure. In both, the ligand binding pocket is indeed occupied by either ATP (Figure 4A) or ADP (Figure 4B) and water, allowing us to examine protein:nucleotide interactions (Figure 4A–C). Within the highly conserved P-loop, Gly146-Ser-Gly-Lys-Ser-Thr151, backbone nitrogen atoms of each amino acid form hydrogen bonds with phosphate oxygens. Sequence-specific side chain interactions are formed by the amino group of Lys149 (Nζ salt bridge to O2β) and the hydroxyls of Ser150 (Oγ hydrogen bonds to O3β and γ phosphate oxygens) and Thr151 (Oγ hydrogen bond to O1α phosphate oxygen) (Figure 4A–C). These interactions are not expected to be relevant during catalysis, as the lysine in canonical P-loops bridges β and γ phosphates while the serine coordinates the β phosphate and Mg2+. ATP hydrolysis almost certainly proceeds via carboxylate side chain-mediated activation of a nearby catalytic water, as seen in many ATPase sites including RecA and F1 ATPase (Abrahams et al., 1994; Story and Steitz, 1992). In the P6 PilT case the Asp Box Glu176 is 4.5 Å from Oγ and the Walker B Glu217 is 5.3 Å distant (Figure 4C). Either or both of these carboxylate side chains are also candidates to stabilize Mg2+ binding in the active form of the enzyme. These two closely approaching side chains have invariant counterparts in HP0525 (Glu209, Glu248) and EpsE (Glu296, Glu334) (Figure 1).
Table 1.
X-ray data collection and refinement statistics.
| DataCollectiona | ||||||
| Data Setb | CTD:High | CTD:Peak | CTD:Inflection | CTD:Low | CTD:Crystal 2 | Hexamer |
| Wavelength (Å) | 0.95650 | 0.97934 | 0.97957 | 1.0031 | 0.9792 | 0.97896 |
| Resolution (Å) | 40-2.1 (2.18–2.10) | 40-2.1 (2.18–2.10) | 40-2.1 (2.18–2.10) | 40-2.1 (2.18–2.10) | 20-1.87 (1.92–1.87) | 50-4.2 (4.42–4.25) |
| Nunique | 31929 (3136) | 31924 (3153) | 31958 (3145) | 35057 (3155) | 42952 (3473) | 32266 (3591) |
| Complete (%) | 99.7 (98.5) | 99.7 (99.1) | 99.7 (98.9) | 99.7 (98.7) | 93.8 (82.8) | 95.7 (97.2) |
| Redundancy | 6.2 (5.5) | 6.1 (5.4) | 6.2 (5.7) | 6.2 (5.6) | 9.6 (7.4) | 5.7 (5.5) |
| Rsym (%)c | 5.7 (22.5) | 6.4 (22.3) | 5.5 (21.3) | 5.1 (21.4) | 8.0 (19.7) | 7.8 (22.2) |
| <I/σ(I)> | 35.2 (5.6) | 35.6 (5.7) | 37.1 (6.3) | 37.9 (6.2) | 37.3 (9.1) | 14.8 (5.2) |
| Structure Determination | ||||||
| Structure | PilT-ATP | PilT-ADP | CTD | PilT-Hexamer | ||
| Refinement | REFMAC5 | CNS/twin | SHELX/twin | REFMAC5 | ||
| Resolution (Å) | 20-3.2 (3.28–3.20) | 20-2.8 (2.90–2.80) | 20-1.87 (1.97–1.87) | 25-4.2 (4.30–4.20) | ||
| Rwork/Rfree (%)r.m.s.d. | 24.2/31.0 (30.1/43.5) | 18.5/26.7 (37.4/38.3) | 17.1/24.5 (20.8/na) | 34.4/41.0 (39.9/41.5) | ||
| bonds (Å) | 0.007 | 0.008 | 0.007 | 0.019 | ||
| angles (°) | 1.5 | 1.5 | 2.3 | 1.74 | ||
| avg. B-value (Å2) | 70.7 | 76.8 | 34.3 | 120.9 | ||
| twin factor | n/a | 0.29 | 0.535 | n/a | ||
| twin symmetry | n/a | h,-h-k,-l | h,-k,-l | n/a | ||
Numbers in parentheses are for the highest resolution shell.
All data sets were processed without merging anomalous pairs for MAD phasing, these statistics are reported. Structure refinements were against merged datasets.
Rsym= Σ | I − <I> | / ΣI, where I is the observed intensity, <I> is the average intensity for symmetry-related reflections, and reflections that were measured only once are excluded from the calculations.
Figure 4.
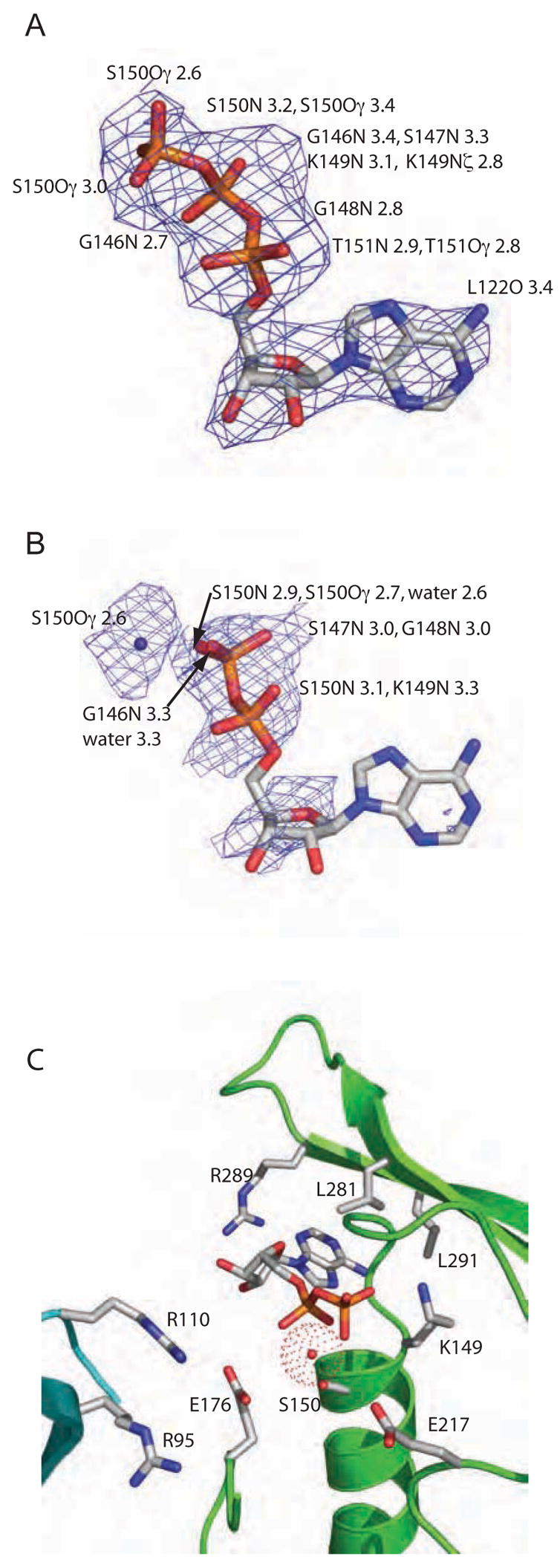
Nucleotide binding in PilT. Composite Fo-Fc omit electron density maps show (A) ATP (countoured at 2.0σ) and (B) ADP (contoured at 1.0σ) in the nucleotide-binding site of the respective PilT P6 structures. Protein atoms within 3.4 Å of ATP are enumerated in (A). In (B), the water is also indicated and protein atoms within 3.4 Å of the water, phosphates, or ribose are enumerated. (C) Close-up view of side chains and water adjacent to the nucleotide binding site in PilT:ADP, with backbone ribbon colored as in Figure 1A.
In order to complete the ATPase reaction, one or more basic residues are required to help orient the γ phosphate and stabilize the negative charges of the Pi leaving group. In PilT, candidates for this role are β5 Arg95 and/or β6 Arg110. Both residues are in the NTD and oriented toward the ligand, although too distant in the PilT:ATP structure to participate in catalysis (Figure 4C). Rather than providing a stabilizing charge for a leaving group, in these PilT structures the guanidinium side chain of β6 Arg110 forms an electrostatic interaction with Asp Box Glu176 (Figure 4C). Because there are no significant conformational differences between our ADP and ATP-bound structures we conclude they represent the same step in the retraction cycle, in which the ATPase active site is in a nonfunctional conformation.
PilT is promiscuous with regard to base for the nucleoside triphosphate substrate, with a slight preference for pyrimidines over purines (Herdendorf et al., 2002). This lack of specificity is explained by the few specific interactions and solvent-exposed nature of the base (Figure 4C). In the more well-defined ATP-containing ligand pocket, the only hydrogen bond to the purine ring is from the Leu122 backbone O to AdeN6. Relatively non-specific van der Waals interactions are formed between the adenine ring and side chains of Leu122, Leu281, Leu291, and the aliphatic portion of Arg289. The Arg289 guanidinium group is 3.8 Å from the sugar ring O3.
CTDn:NTDn+1 interface establishes hexamer
Within the P6 PilT crystal form, the six-fold rotation axis generates a symmetric hexamer from the 41 kDa monomer (Fig. 5A). The outer diameter is ~118 Å and the narrowest inner diameter ~32 Å, in agreement with our earlier results (Forest et al., 2004). The ATP binding site is easily accessible from the outside of the complex, while the pore of the PilT ring is constrained by αH-loop-αI.
Figure 5.
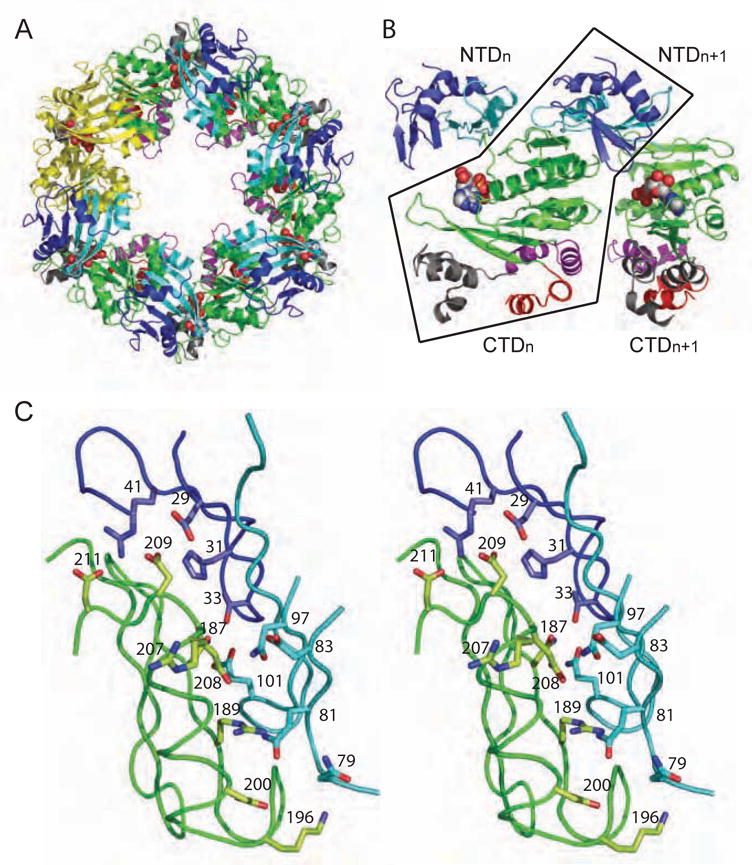
Hexamer formation. (A) The P6 space group constrains the hexamer to be symmetric, as shown in this view of the assembly in which one subunit is highlighted in yellow. (B) In both ADP and ATP-bound P6 crystal forms, the main intermolecular interface is CTDn:NTDn+1 as seen in these two adjacent subunits, viewed perpendicular to the 6-fold symmetry axis. Black outlines the non-covalent CTDn:NTDn+1 pair. (C) Zoomed-in stereo view of the intermolecular CTDn:NTDn+1 interface, rotated to highlight a subset of the amino acids in the polar interface. The coloring in all panels is as in Figure 2.
Within this crystallographic oligomer, each two-domain PilT monomer contacts a neighboring subunit across a CTDn:NTDn+1 interface, burying a total solvent-accessible surface area of 1782 Å2 (Fig. 5B). 1157 Å2 of the interface are provided by polar and charged interactions involving backbone atoms and side chains of nine NTD residues (Asp29, His31, Thr33, Arg41, Asn79, Gln81, Asp83, Gln97 and Gln101) and nine CTD residues (Ser165, Asn187, Arg189, Lys196, Asp200, Arg207, Glu208, Asp209, Asp211) (Figure 5C). The remaining ten NTD and nine CTD residues involved in the interaction are either hydrophobic (e.g. Phe48 and Phe99) or charged residues that form van der Waals interactions. Outside this surface, only 153 Å2 are buried in the minimal NTDn:NTDn+1 interface, and there are no CTDn:CTDn+1 interactions involving either the ATPase core or the short C-terminal helices. Thus, the PilT P6 hexameric ring is formed exclusively by zig-zagging covalent NTDn-CTDn linkers and CTDn:NTDn+1 intermolecular interactions. Although EpsE and HP0525 oligomers do contain additional substantial CTDn:CTDn+1 and NTDn:NTDn+1 intersubunit contacts, the overall electrostatic nature, large buried surface area and some individual residues in this CTDn:NTDn+1 interface are conserved among PilT, EpsE and HP0525 assemblies (Figure 1) (Robien et al., 2003; Yeo et al., 2000).
Dramatic Domain Motions in Hexamer Structure
We obtained an additional crystal form of full-length PilT, with six monomers in each C2 asymmetric unit (Table 1). Despite the low resolution of this structure several striking conclusions can be drawn about the asymmetry, domain orientations, ATP binding sites, and subunit interactions within this PilT hexamer.
The six PilT subunits form an ellipsoid (Fig. 6A). Four monomers (A, C, D, and F) have equivalent domain orientations in which the NTD-CTD cleft is closed by 16° compared to the P6 monomer. This juxtaposition of the NTD and CTD causes the close approach of αC on the edge of the NTD to the loop between β9 and αH of the CTD (Fig. 6B). The remaining two opposing subunits (E and B) have undergone a remarkable 65–69° domain rotation about the covalent linker in comparison to the four closed ones (Fig. 6B). These wide-open subunits are displaced from the center of the hexamer, lending quasi two-fold symmetry to the hexamer. This symmetry is observed in the residual ligand electron density as well. Subunits A and D are empty while subunits B and E, despite being open, contain density that may be sulfate from the crystallization buffer. Subunits C and F most closely approach the lumen, but although both are clamped shut subunit F has stronger density and contains ADP in the binding pocket with some evidence for an additional sulfate molecule which has prompted us to model released Pi (Fig. 6C). At 4.2 Å resolution we have not drawn detailed conclusions about the significance of the occupancy of ligand in each subunit.
Figure 6.
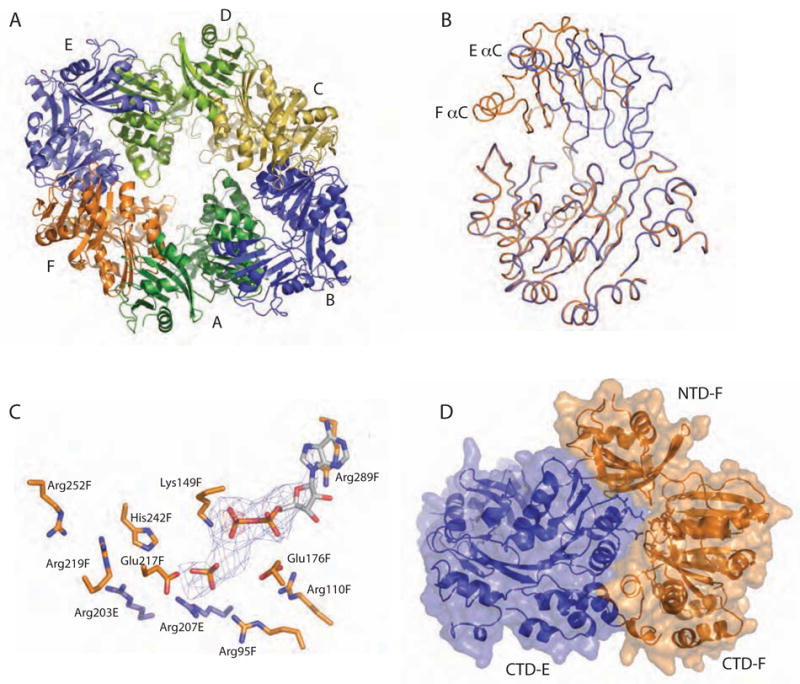
Dramatic domain orientation differences in the PilT C2 hexamer. (A) 6-fold symmetry is broken. Subunits E and B (light and dark blue, respectively) are displaced from the center of the hexamer. (B) Adjacent subunits E (blue) and F (orange) were superimposed over CTD residues 116–361. The NTDs of these monomers are then related by a 69° rotation about an axis through the domain linker. Helices C are labeled to highlight the exent of the movement. (C) Close-up view of the nucleotide-binding site at the three-way interface of NTDF (orange, below) and CTDF (orange, above) from the closed, liganded subunit and the CTDE (blue) from the adjacent open subunit, with refined ADP and modeled Pi shown in Fo-Fc omit electron density calculated without nucleotide (contoured at 2.5σ (blue) and 5σ (red)). Note the arginine wire. Given the low resolution of the data used for this refinement, it is appropriate to consider this figure as one model that is consistent with our data. (D) At the CTDE:CTDF interface, arginine finger residues are engaged and 950 Å2 are buried.
Within subunit F, the nucleotide-binding site is complete. β5 Arg95 and β6 Arg110, both discussed above in the context of the P6 structure, now reach across from the NTD to form one side of the nucleotide binding pocket, coming closest to the β and α phosphates of ADP, respectively (Fig. 6C). Both side chains are also less than 5 Å from Glu176 (Fig. 6C). Other expected interactions between P-loop residues and ADP are seen, with Lys149 pointing directly at the presumed position of the γ phosphate. Glu217 is oriented toward the modeled γ phosphate, appropriately positioned to activate water. His242 also approaches the modeled γ phosphate (Fig. 6C).
In additon to the domain orientation differences and completed active site, a novel feature of the asymmetric PilT hexamer is the complementary buried surface between adjacent CTDs, which encompasses ~750 to ~1,350 Å2 (Fig. 6D). At the four smaller intersubunit interfaces, the asymmetric hexamer causes the juxtaposition of the ATP binding site of one molecule with αF Arg203 and Arg207 of the adjacent molecule (Fig. 6C, D). Arg207 in particular is an invariant residue within the Walker B motif that positions its charged Nε groups within 8 Å of β PO4 of ADP at the CTDE:CTDF interface. The noteworthy concentration of Arg sidechains, including Arg95 and Arg110 from the NTD as well as Arg203 and Arg207 from the adjacent CTD, is undoubtedly relevant to ATP binding and hydrolysis, and suggests that an arginine wire may lead the released Pi product out of the active site (as modeled in Figure 6C), as well as ultimately control large scale movements that drive pilus retraction.
Additional interactions are observed between αM and αN of the binding site subunits and αI and αJ of the adjacent subunits, thus suggesting a role in protein:protein interactions for the far C-terminal αM and αN, which are the least well-conserved and also least poorly ordered part of all the PilT structures.
The two largest CTDn:CTDn+1 interfaces are created by the packing of the backside of the central resting subunits A or D into the domain cleft of the peripheral open subunits B or E, respectively, and these interfaces do not constitute active ATPase geometries. These CTDA:CTDB and CTDD:CTDE interfaces involve both polar and hydrophobic interactions of the loops preceding the Walker B and His Box motifs in subunit A or D with residues within or immediately following all four of the conserved sequence motifs in subunits B or E, as well as the packing of αI from A or D against the Walker B motif in subunits B or E. There are also packing interactions of short C-terminal helices αI and αJ from subunits A or D against αH and αI from subunits B or E (e.g. Asn304 sidechain to Leu259 backbone).
The lumen of the asymmetric hexamer is considerably smaller than that of the symmetric P6 hexamer, although the constriction is likewise formed by the αH-αI loops. The lining of the lumen remains overwhelmingly negatively charged (data not shown). The smaller opening on the base formed by the short C-terminal helices is ~17 Å, whereas the wider entrance on the NTD side of the hexamer is ~30 Å. The C-terminal helices and the αH-αI constriction of subunits A, C, D, and F occupy this central location.
Structural Implications Extended by Random and Site-Directed Mutagenesis
In order to link in vitro structural results to the in vivo function of PilT in pilus retraction, we generated a random library of variant P. aeruginosa pilT genes and screened for nonfunctional PilT proteins in P. aeruginosa by colony morphology and twitching motility. Although 42 single loss-of-function point mutants were isolated, only those three for which in vivo protein levels were comparable to the wild type protein, and which are identical amino acid residues in A. aeolicus and in P. aeruginosa, are described here (Table 2 and Figure 7).
Table 2.
Amino acid substitutions in P. aeruginosa PilT.
| Residue changed | A.a. # | Morph | Twitching | Location |
|---|---|---|---|---|
| WT | n/a | F | yes | n/a |
| D17G | 29 | S | no | NTDn:CTDn+1 |
| H222R | 235 | S | no | His Box |
| K297E | 310 | S | no | αJ-αK loop |
| R82Q | 95 | S | no | β5 Arg |
| R82S | 95 | S | no | β5 Arg |
| R97Q | 110 | S | no | β5 Arg |
| R97S | 110 | S | no | β5 Arg |
| R194Q | 207 | S | no | Arg Finger |
| R194S | 207 | S | no | Arg Finger |
Each of the P. aeruginosa PilT variants listed (with corresponding A. aeolicus amino acid numbers) was isolated in a random screen or created by site-directed mutagenesis (R82/R97/R194 variants). Colony morphology of P. aeruginosa expressing PilT variants is tabulated as F (fingered, like wild-type strain PAK), or S (smooth like pilT). Twitching motility at an agar-plastic surface was not observed for any mutant.
Figure 7.
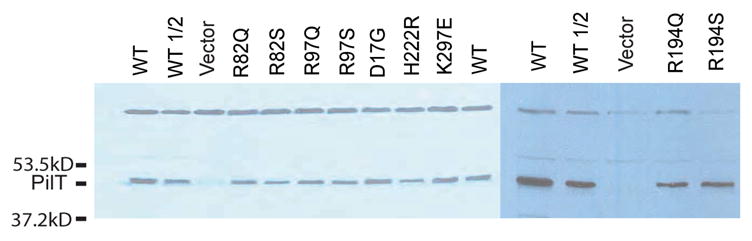
Protein levels of variant PilTs in vivo. Western blot analysis, using a polyclonal anti-P. aeruginosa PilT antibody, was used to assess relative levels of plasmid-encoded PilT in a PAK pilT background. The binding of the antibody to an unidentified constant band at higher molecular weight serves as a loading control. The wild type protein is shown with full and half-strength sample concentrations (WT and WT 1/2). Samples come from P. aeruginosa cells expressing the indicated PilT protein.
The P. aeruginosa mutations fall onto each of the three structural regions of PilT; the NTD, ATPase core and far C-terminal short α-helices. Asp17Gly in the NTD (A. aeolicus Asp29) highlights the requirement for hexamer formation in vivo. A. aeolicus β1 Asp29 falls within the CTDn:NTDn+1 interface, and makes a 3.3 Å approach to the carboxylate side chain of Asp209 within the extended Walker B sequence (Figure 5C). This seemingly unfavorable interaction is offset by the highly distributed charge network and/or by the fact that the side chain appropriately fills the available volume in the interface; the nonfunctional variant has Gly at this position. The His222Arg (His235 in A. aeolicus) change affects the first of two namesake residues in the His Box at the N-terminal end of β11 (Figure 3C), which approaches the neighboring subunit in the CTDA:CTDB and CTDD:CTDE interfaces. We postulate that this residue is involved in signaling between subunits, and in this context it is notable that the complementary side of the interface includes the second A. aeolicus His Box histidine, His242 at the C-terminal end of β11. The third variant is Lys297Glu (A. aeolicus Lys310) in the solvent-exposed αJ-αK loop. These short helices swing dramatically closer to the center of the assembly in subunits A, C, D and F than in the peripheral monomers B and E. A. aeolicus Lys310 itself is 35 Å closer to the hexamer center. Together with a previously reported point mutation in αI (Wu et al., 1997), substitutions in αJ (Aukema et al., 2005), and a three-residue deletion in αL (Wolfgang et al., 1998) which prevent pilus-mediated motility in vivo, the P. aeruginosa Lys297Glu PilT insinuates the C-terminal helices in a central role in pilus retraction.
To substantiate the hypothesis that invariant β5 and β6 arginines in PilT play a critical role in PilT function we created site-directed P. aeruginosa PilT variants Arg82Ser, Arg82Gln, Arg97Ser and Arg97Gln (equivalent to A. aeolicus positions Arg95 and Arg110). In all four cases we found that protein was made in P. aeruginosa pilT cells harboring the mutant genes on plasmids (Figure 7). Yet, in no case was PilT function normal when assayed by colony morphology or twitching motility (Table 2).
The most far-reaching implication of the asymmetric hexamer structure for the overall motor mechanism of PilT and Type II/IV secretion ATPases may be the involvement of Arg203 and/or Arg 207 in the ATP binding site of the adjacent subunit. By analogy to Ras, F1F0ATPase and PcrA (Dittrich and Schulten, 2006) one intriguing speculation is that these residues could play the role of the arginine finger in this motor, communicating the presence of ATP between subunits. Site directed mutagenesis of the Arg207 equivalent (P. aeruginosa Arg194) to either a Gln or Ser codon abolished twitching motility but not protein accumulation in vivo (Table 2, Figure 7). The same substitutions at the Arg203 equivalent (P. aeruginosa Arg190) had only a more minor affect on the phenotypes, suggesting either that this non-conserved residue is unimportant for activity or that other positively charged side chains in the arginine wire provide redundancy (data not shown).
Discussion
We have been able to halt the pilus retraction motor PilT for X-ray crystallographic analysis in several packing contexts. Our results reveal a dynamic machine in which large-scale motions between an N-terminal PAS-like domain and C-terminal Rec-A like ATPase core drive motility. The well-ordered P6 crystalline conformation of A. aeolicus PilT lacks an active three-dimensional ATPase site, although ligand-binding has provided symmetry and led to 2.8 Å diffraction (Forest et al., 2004). These structures allow us to accurately describe the fold of the PilT molecule (Figure 2) and to examine the extensive polar CTDn:NTDn+1 interface (Figure 5). Our low-resolution asymmetric engaged hexamer structure (Fig. 6), on the other hand, provides striking insight into the PilT retraction mechanism and potentially into the overall mode of operation of this entire class of ATPase (Figure 8).
Figure 8.
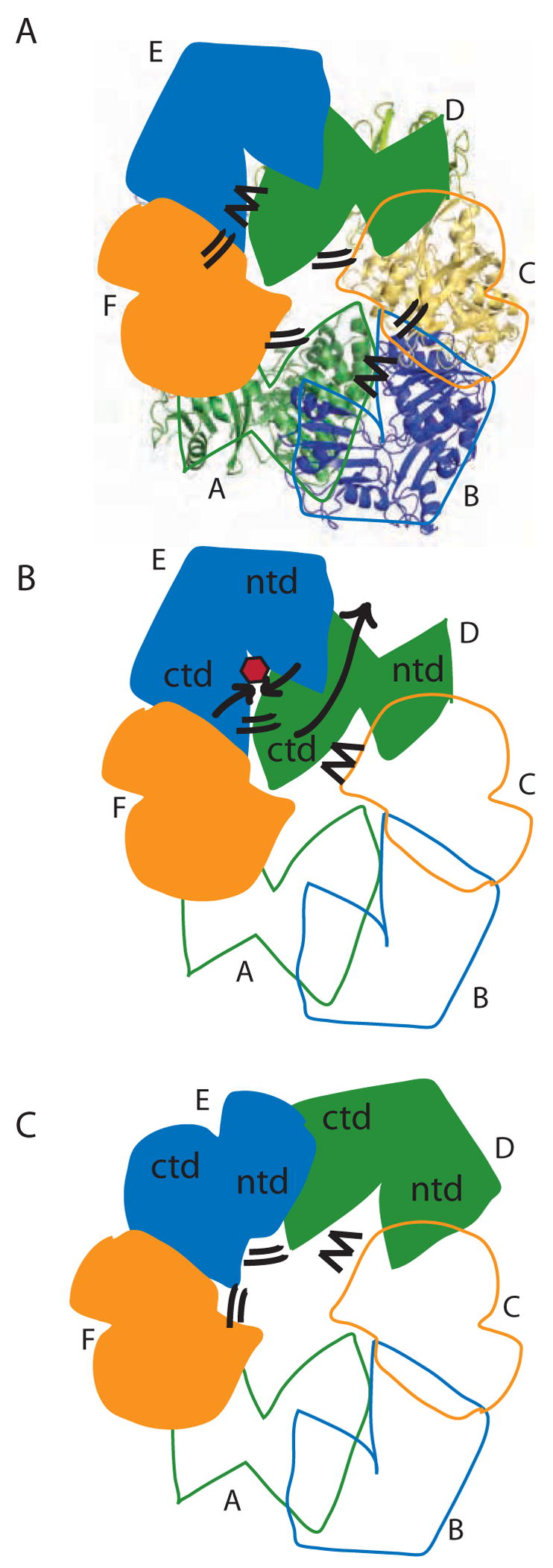
Model for concerted PilT motions. (A) The quasi 2-fold symmetric C2 crystal structure has two peripheral wide-open subunits (B, E; blue), two central “active” subunits (C, F; orange) and two central “resting” subunits (A, D; green). Four CTD:CTD interfaces have engaged arginine fingers (double lines). The remaining two have disengaged fingers (zig-zag). Subunit F is clamped around bound nucleotide. (B) When ATP (red) binds in the E cleft, the two domains close around the ligand (short black arrows), causing the β5/β6 arginines to approach the ATP. Because of the extensive CTDD:NTDE interface, the motion of NTDE forces the swiveling of CTDD (in particular the C-terminal helices) toward the periphery of the hexamer (long grey arrow). Consequently, the D arginine fingers engage in the E active site (double lines). On the other side of CTDD, the CTDC:CTDD interface likewise rearranges, disengaging the C arginine fingers from the D active site (zig-zag). (C) Subunit D is now poised as the most peripheral, wide open subunit and ready to bind nucleotide; E is clamped around nucleotide and contributing to an engaged CTD:CTD interface on either side.
The critical nature of several invariant arginine residues is an important part of the mechanism of action for PilT. Six arginine residues from two subunits together form an arginine wire around the ATP binding site (Fig. 6C). These positive charges probably play several roles in the functional cycle of this family of proteins in vivo. Within a single subunit, the “arginine switch” (Arg95 and 110 from the NTD) drives conformational change by interacting with nucleotide triphosphate in the CTD binding site and may fulfill the ATPase activity requirement for positive charge to stabilize the Pi leaving group. Both side chains are required for twitching motility in vivo (Table 2). As the equivalent β5 Arg113 and β6 Arg133 in HP0525 were previously shown to be required for ATPase activity in vitro and secretion function in vivo (Savvides et al., 2003) we can conclude that invariant β5/β6 arginine switch residues in the NTD of the Type II/Type IV secretion ATPase family are a common critical requirement for biological function.
Secondly, αG and αF with its arginine residues from the adjacent subunit approach the nucleotide binding site in the closed subunit conformation seen in monomers A, C, D, and F of our C2 structure. Arg207, in particular, is an invariant residue in the Walker B motif of the Type II/Type IV secretion ATPase family. We have shown that variant proteins carrying Ser or Gln at position 207 prevent Type IV pilus-dependent surface motility (Table 2). We speculate that this residue may function in PilT like the arginine finger of GTPase-activating proteins and AAA+ ATPases (Scheffzek et al., 1998; Zhang et al., 2002), ensuring communication between adjacent subunits. Potentially it also drives one-way retraction. Although caution must be exercised in interpreting our low resolution structure, when Pi is modeled into residual electron density in our ADP-bound subunit F, it is surrounded by several charged Arg side chains, including those of Arg203 and Arg207 (Fig. 6C). A cluster of acidic residues is also found near bound nucleotide at the subunit interface in structures of the VirB11 ATPase HP0525 in which it was proposed that ATP prevents the electrostatic clash among the arginines thus allowing domain closure (Yeo et al., 2000). Interestingly, only Arg207 is invariant; the specific position of neighboring arginines is different in PilT vs. the VirB11 ATPases (Fig. 1). To our knowledge, this is the first time the arginine finger role has been suggested in PilT or any other member of the Type II/Type IV secretion ATPase family. Although our results do not permit us to draw detailed conclusions about the role of Arg207 in subunit interactions, they do provide testable hypotheses regarding the roles of the arginine wire in pilus retraction.
The functional PilT retraction cycle must be a series of transient steps in vivo (Figure 8). Unliganded PilT is a heterogeneous population of hexamers as evidenced by poor crystalline order (Forest et al., 2004). Active PilT apparently has quasi 2-fold symmetry, with two subunits open and displaced from the center of the ellipsoidal hexamer. This is reminiscent of a similar behavior for apo-HPO525, the Type IV secretion ATPase which serves as the founding member of the small set of Type II/Type IV secretion ATPases whose structures have been investigated, and for which monomers also obey an approximate 2-fold symmetry with respect to overall structure deviations and buried surface areas (Savvides, et al., 2003). As each new retraction step begins, binding of ATP to one of these subunits would promote dramatic domain closure (Figure 8), potentially by neutralizing otherwise unfavorable electrostatic interactions (Yeo et al., 2000), leading to a closed, ATP-bound form of this subunit. An open question is whether all six subunits work in concert or whether a subset can be active and still promote motor function, as appears to be the case for the ClpX-mediated unfolding of ClpP substrates (Martin et al., 2005).
A defining feature of the retraction motion is the role of the CTDn:NTDn+1 interface in linking motions of two adjacent subunits: the pivoting motion of the NTDn+1 toward the center of the hexamer upon ATP binding will flip the CTDn toward the hexamer periphery (Fig. 8). Thus in PilT the set of C-terminal helices will swing outward, with some residues moving tens of Ångstroms. Residues at either end of αJ (Ala301 and Lys310) are required for motility in vivo (Aukema et al., 2005 and Table 2). As PilT undergoes its domain motion it could drag a protein bound to this exposed surface 7–25 Å (the range of differences in the center-to-Ala301 distance for the three non-equivalent subunits in our structure) or possibly further depending on the geometry of the interaction. This large-scale motion recalls the myosin linear motor, which undergoes a similar 45–70° domain rotation with a step size of ~50 Å as it moves an actin filament during its power stroke (Cooke, 1997; Xiao et al., 1998). We cannot say to what extent the wide-open conformation of subunits B and E is representative of PilT’s active conformation in solution; the domain orientation differences could be exaggerated by crystal packing in our C2 crystals. In the non-symmetric apo structure of the Type IV secretion ATPase HP0525, observed domain orientation differences were likewise observed but were significantly smaller, 2–15° (Savvides et al., 2003). Nonetheless, the strong conclusion is that domain motions of PilT lead to substantial movements of C-terminal helices that could translate bound proteins during pilus retraction. If all six PilT monomers within the hexamer are capable of such large-scale motions, even a modest 10 Å power stroke per ATP binding event would lead to very significant motions of a presumed pilus substrate. Further investigation into interaction partners, including structural studies of protein complexes, will be required to answer the fundamental question how pilin monomers themselves - either directly or indirectly - interact with this machinery to couple PilT cycling to pilus retraction.
Experimental Procedures
Purification and Crystallization
An expression plasmid for PilT CTD was created from the expression vector for full-length PilT (pTJH1000 (Herdendorf et al., 2002)) by introduction of an NdeI restriction site via full circle mutagenesis (Stratagene) at the coding region for the domain junction to match one already present in the 5’ multi cloning site, followed by digestion and ligation to delete the 330 base pair coding region for the NTD (Caruthers et al., 2000). The cloning of pET-23a(+)/CTD-6xHis A. aeolicus pilT (pGAW1004) was verified through automated DNA sequencing, and it was discovered that a mutation encoding a Glu294Gly substitution had been inadvertently introduced.
For CTD expression, pGAW1004 was transformed into chemically-competent BL21(DE3) cells (Novagen) for native protein expression or into B834(DE3)pLysS (Novagen), a methionine auxotroph (Leahy, et al., 1992), for seleno-L-methionine (selenomet) incorporation. In the native case, growth and expression were in LB-ampicillin. For selenomet labeled protein, modified M9 minimal media plus antibiotics and a nutrient mix containing amino acids and key vitamins (SelenoMet Nutrient Mix, Molecular Dimensions Inc.) was used for growth and 40 mg of selenomet (Molecular Dimensions Inc) were added prior to induction (Ramakrishnan, et al., 1993). In both cases, expression was induced with 1 mM IPTG for 20 hrs at 20°C.
CTD protein was purified by standard Ni2+ agarose affinity chromatography followed by anion exchange chromatography at pH 8.5. Peak fractions were dialyzed against 50 mM HEPES pH 8.0, 150 mM KCl, and concentrated to 10 mg/ml. Crystal Screens I and II (Hampton Research) with 0.1 to 0.2 M ammonium sulfate as precipitating agent produced diamond shaped crystals. Final crystallization solution was 0.1 M MES, pH 6.5, 10% dioxane and 1.6 M ammonium sulfate, with 2.5 M sodium malonate in mother liquor for cryoprotection. The space group of these CTD crystals is P41 with two molecules per asymmetric unit and cell constants a=b=89.2 Å, c=70.6 Å, α=β=γ=90°.
Full length A. aeolicus PilT was overexpressed, purified and crystallized with nucleotide in space group P6 as described (Forest et al., 2004). Unexpectedly, within our ATP-soaked structure, ATP does not appear to be hydrolyzed, potentially due to a non-active enzyme at room temperature (Herdendorf et al., 2002) protecting ATP from non-catalytic hydrolysis (Figure 4A). When ATPγS was present during crystal growth, our electron density indicated that ADP occupied the nucleotide-binding site (Figure 4B). These crystals were obtained after heating the protein:ATPγS solution. Mass spectrometry experiments showed unequivocally that ATPγS is hydrolyzed to ADP under these conditions (data not shown). There is no residual density for γ phosphate or sulfate in the electron density maps. Lacking strong evidence from coordination geometry for a Mg2+ ion, we have fit a water molecule into the difference electron density (Figure 4B).
PilT crystals in space group C2 (a=206.1 b=105.2 c=123.0 Å, β=111.1°) were obtained from 7.5 mg/ml selenomet-labeled PilT in 16 mM Tris, pH 7.6, 98 mM KCl, 130 mM imidazole, 6.5% glycerol, 9.7% PEG1000, 13.0 mM TCEP, 13.0 mM LiCl, 0.3 M ammonium sulfate and 2.6 mM ATPγS. The protein solution was heated to 70° C for 30 min. Hanging droplets of 2–3 μl were placed over unbuffered reservoirs of 0.2 M ammonium sulfate and 40% PEG1000. Rod-shaped crystals appeared in three days and were flash-cooled directly.
Data Collection and Processing
CTD data used for phasing were collected on two cryocooled crystals at the Advanced Photon Source (APS), Beamline 14 ID-B, on a MarCCD detector. A 1.87 Å resolution native data set was collected on a single crystal at APS COMCAT beamline 32 ID-B, also on a MarCCD detector, in three resolution shells, with an attenuated beam for the low resolution sweep. Data were integrated and scaled with HKL2000 (Otwinowski and Minor, 1997) (Table 1).
Data collection for PilT:ATP and PilT:ATPγS crystals has been reported (Forest et al., 2004). An additional low resolution data set from a smaller crystal was scaled to the original SeMet PilT:ATP data set to improve the accuracy of the low resolution data (data not shown).
Data for the C2 crystal structure were also collected at APS beamline 14ID-B. Data were collected at the Se absorption edge, however the anomalous signal was not useful.
Structure Determination and Refinement
To assist with the structure determination of the full-length PilT molecule, the structure of a CTD construct covering PilT residues 113–366 was determined by multiwavelength anomalous dispersion (MAD) and solvent flattening using SOLVE (Terwilliger and Berendzen, 1999) and DM (Cowtan, 1994). The initial figure of merit for 14 refined selenium positions was 0.46; following solvent flattening and fragment fitting this rose to 0.61. 75% of the structure in both molecules in the asymmetric unit was traced automatically with RESOLVE, followed by manual interpretation using Xfit (McRee, 1992). As testing for twinning (www.doe-mbi.ucla.edu/Services/Twinning) revealed nearly perfect merohedral twinning, refinement at 1.87 Å resolution was carried out with the twin refinement options in CNS (Brunger et al., 1998) and SHELXL (Herbst-Irmer and Sheldrick, 1998) (Table 1). The final CTD model covers residues 117–363 for both chains, with 91.2% of residues in the favored region and 98.2% in the allowed region of the Ramachandran plot.
Two structures of full-length PilT in space group P6 were solved. For PilT:ATP, initial phases used to determine the ultrastructure of the hexamer (Forest et al., 2004) proved insufficient for structure determination. Additional low resolution data from a second crystal were critical for improved phasing and density modification; a clear heavy atom solution with eight selenium positions and a figure of merit of 0.51 was identified using SHARP (Global Phasing LTD.) and subjected to density modification using DM (Cowtan, 1994). Manual model building into the experimental electron density map using Xfit was aided by a PHASER (Storoni et al., 2004) molecular replacement solution based on the EpsE C1 and C2 subdomains (Robien et al., 2003). The final α-helices were difficult to fit and thus the CTD structure was vital for establishing an initial model of corresponding residues in the full-length PilT structure. Before the PilT:ATP structure was completed, it was used as the starting model for the higher resolution PilT:ADP refinement (see below), after which the final PilT:ADP structure was used as the starting model for the refinement of the PilT:ATP structure at 3.2 Å resolution with REFMAC5 (Murshudov et al., 1997) alternated with further manual fitting in Xfit (McRee, 1992). It contains ATP and PilT N-terminal residues 13–111 connected by a 6 residue linker to C-terminal residues 118–360 (Table 1, Figure 2A). Residues 320–326 are a disordered loop and are not included in the final model.
For PilT:ADP data, twinning was also evident. An early version of the PilT:ATP structure, stripped of nucleotide, was oriented in the PilT:ADP data using rigid body refinement and refined at 2.8 Å resolution with the CNS twinning package (Brunger et al., 1998) alternated with manual fitting. The PilT:ADP model includes ADP and residues 12–361 minus the disordered loop (Table 1, Figure 2A). In the ADP-bound structure, 81.1% of the residues are in the favored and 97.1% of the residues within the allowed regions of the Ramachandran plot. The relatively poor Φ/Ψ statistics for the CTD and PilT:ADP structures may be due in part to refinement against highly twinned data. Refinement statistics for all structures are presented in Table 1.
The C2 PilT structure was solved by molecular replacement using the non-covalent CTDn:NTDn+1 domain pair from the refined PilT:ADP structure as the search model in PHASER (Storoni et al.). Six copies were located in an unambiguous solution with an overall log likelihood gain of 1260 and no packing errors. Difference electron density for the linker residues 111–115 that were not included in the domain models was evident, as was density within four of the six ligand binding sites. While these atoms were added in Xfit, no attempt was made to manually optimize side chain rotamers anywhere else in the structure due to the very low resolution of the data. The structure was refined in REFMAC5 (Murshudov et al., 1997) using 6-fold noncrystallographic symmetry restraints (tight for main chain atoms and medium for side chain atoms) within each domain (12–110 and 116–361). Final R/Rfree values are 34.4/41.0%.
All structure figures were calculated with PYMOL (DeLano Scientific, LLC), surface areas with AREAMOL (Collaborative Computational Project, 1994), structural superpositions using TOP (Lu, 2000) and domain rotations using DynDom (Hayward, 1999).
Mutagenesis of pilT
The pilT-containing plasmid pKA22 (Aukema et al., 2005) was adapted to facilitate random mutagenesis of pilT by error-prone PCR. Two NcoI sites were removed from the pilT open reading frame and a BamHI site was introduced nine bases after the stop codon so that the entire protein coding region could be digested from and ligated into a new vector, pEK3. pilT was amplified from pEK3 under conditions chosen to lower the fidelity of the DNA polymerase (Shim et al., 2004), with final [MnCl2] 0.05 – 0.3 mM. The PCR products and pEK3 were digested with NcoI and BamHI, and ligated together. The pool of plasmids containing variant pilT genes was transformed into P. aeruginosa PA103 pilT as described (Aukema et al., 2005). Carbenicillin-resistant colonies were screened for the smooth morphology indicative of non-twitching mutants. For candidate colonies, plasmid was isolated in a 96-well format (Quiagen). In order to achieve larger quantities to serve as templates for sequencing reactions, these plasmids were transformed into E. coli and re-isolated. The pilT genes were sequenced twice using automated DNA sequencing protocols.
Site-directed mutagenesis of the pilT gene was carried out as described (Aukema et al., 2005).
PilT function in vivo was assessed by colony morphology and twitching motility (Aukema et al., 2005). Steady state protein levels were assessed by semi-quantitative Western blot of cell lysates of P. aeruginosa expressing appropriate PilT variants from a plasmid. A 1:5000 dilution of a polyclonal rabbit anti-PilT antiserum was used. This was raised against P. aeruginosa PilT expressed in E. coli; PilT for the first injection was purified by DEAE-ion exchange followed by ATP-affinity chromatography whereas PilT for the booster was purified by DEAE-ion exchange followed by hydrophobic interaction chromatography (data not shown).
Acknowledgments
We are grateful to Dr. James Keck for data collection on C2 crystals. We acknowledge the excellent technical assistance of APS staff at BioCARS (14 ID-B) and LS-CAT (32 ID-B) beamlines. This project was funded by the NIH (GM59721) and the W. M. Keck Foundation. Coordinates and structure factors have been deposited at the Protein Data Bank with ID codes 2EYU for CTD, 2EWV for PilT:ADP, 2EWW for PilT:ATP, and 2GSZ for the asymmetric PilT hexamer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahams JP, Leslie AGW, Lutter R, Walker JE. Structure at 2.8 Å resolution of the F1-ATPase from bovine heart mitochondria. Nature. 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- Arcus VL, Bäckbro|| K, Roos|| A, Daniel EL, Baker EN. Distant Structural Homology Leads to the Functional Characterization of an Archaeal PIN Domain as an Exonuclease. J Biol Chem. 2004:16471–16478. doi: 10.1074/jbc.M313833200. [DOI] [PubMed] [Google Scholar]
- Aukema KG, Kron EM, Herdendorf TJ, Forest KT. Functional dissection of a conserved motif within the pilus retraction protein PilT. J Bact. 2005;187:611–618. doi: 10.1128/JB.187.2.611-618.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A, Adams P, Clore M, Gros P, Nilges M, Read R. Crystallography & NMR System. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Caruthers JM, Johnson ER, McKay DB. Crystal structure of yeast initiation factor 4A, a DEAD-box RNA helicase. Proc Natl Acad Sci USA. 2000;97:13080–13085. doi: 10.1073/pnas.97.24.13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang P, Habash M, Burrows LL. Disparate subcellular localization patterns of Pseudomonas aeruginosa Type IV pilus ATPases involved in twitching motility. J Bact. 2005;187:829–839. doi: 10.1128/JB.187.3.829-839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianciotto NP. Type II secretion: a protein secretion system for all seasons. Trends Microbiol. 2005;13:581–588. doi: 10.1016/j.tim.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Clissold PM, Ponting CP. PIN domains in nonsense-mediated mRNA decay and RNAi. Curr Biol. 2000;10:R888–R8890. doi: 10.1016/s0960-9822(00)00858-7. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. 1994. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Comolli JC, Hauser AR, Waite L, Whitchurch C, Mattick JS, Engel JN. Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infection & Immunity. 1999;67:3625–3630. doi: 10.1128/iai.67.7.3625-3630.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke R. Actomyosin interaction in striated muscle. Physiol Rev. 1997;77:671–697. doi: 10.1152/physrev.1997.77.3.671. [DOI] [PubMed] [Google Scholar]
- Cowtan K. Joint CCP4 and ESF-EACBM Newsletter on Protein. Crystallography. 1994;31:34–38. [Google Scholar]
- Craig L, Pique ME, Tainer JA. Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol. 2004;2:363–378. doi: 10.1038/nrmicro885. [DOI] [PubMed] [Google Scholar]
- Dittrich M, Schulten K. PcrA helicase, a prototype ATP-driven molecular motor. Structure. 2006;14:1345–1353. doi: 10.1016/j.str.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Forest KT, Satyshur KA, Worzalla GA, Hansen JK, Herdendorf TJ. The pilus-retraction protein PilT: ultrastructure of the biological assembly. Acta Cryst. 2004;D60:978–982. doi: 10.1107/S0907444904006055. [DOI] [PubMed] [Google Scholar]
- Hansen JK, Forest KT. Type IV pilin structures: insights on shared architecture, fiber assembly, receptor binding and type II secretion. J Mol Microbiol Biotechnol. 2006;11:192–207. doi: 10.1159/000094054. [DOI] [PubMed] [Google Scholar]
- Hare S, Bayliss R, Baron C, Waksman G. A large domain swap in the VirB11 ATPase of Brucella suis leaves the hexameric assembly intact. J Mol Biol. 2006;360:56–66. doi: 10.1016/j.jmb.2006.04.060. [DOI] [PubMed] [Google Scholar]
- Hayward S. Structural principles governing domain motions in proteins. Proteins. 1999;36:425–435. [PubMed] [Google Scholar]
- Herbst-Irmer R, Sheldrick GM. Refinement of twinned structures with SHELXL97. Acta Cryst. 1998;B54:443–449. [Google Scholar]
- Herdendorf TJ, McCaslin D, Forest KT. A. aeolicus PilT, homologue of a twitching motility protein, is an oligomeric ATPase. J Bact. 2002;184:6465–6471. doi: 10.1128/JB.184.23.6465-6471.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie HL, Glogauer M, So M. The N. gonorrhoeae type IV pilus stimulates mechanosensitive pathways and cytoprotection through a pilT-dependent mechanism. PLoS Biol, PMID. 2005:15769184. doi: 10.1371/journal.pbio.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Leipe DD, Koonin EV, Aravind L. Evolutionary history and higher order classification of AAA+ ATPases. J Struct Biol. 2004;146:11–31. doi: 10.1016/j.jsb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Jeyakanthan J, Inagaki E, Kuroishi C, Tahirov TH. Structure of Pin-Domain Protein Ph0500 from Pyrococcus horikoshii. Acta Cryst. 2005;F61:463–468. doi: 10.1107/S1744309105012406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Liu H, Genin S, Schell MA, Denny TP. Ralstonia solanacearum requires type 4 pili to adhere to multiple surfaces and for natural transformation and virulence. Mol Micro. 2002;46:427–437. doi: 10.1046/j.1365-2958.2002.03187.x. [DOI] [PubMed] [Google Scholar]
- Levin I, Schwarzenbacher R, Page R, Abdubek P, Ambing E, Biorac T, Brinen LS, Campbell J, Canaves JM, Chiu HJ, et al. Crystal structure of a PIN (PilT N-terminus) domain (AF0591) from Archaeoglobus fulgidus at 1.90 A resolution. Proteins. 2004;56:404–408. doi: 10.1002/prot.20090. [DOI] [PubMed] [Google Scholar]
- Li Y, Sun H, Ma X, Lu A, Lux R, Zusman D, Shi W. Extracellular polysaccharides mediate pilus retraction during social motility of Myxococcus xanthus. Proc Natl Acad Sci USA. 2003;100:5443–5448. doi: 10.1073/pnas.0836639100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Kang Y, Genin S, Schell MA, Denny TP. Twitching motility of Ralstonia solanacearum requires a type IV pilus system. Microbiology. 2001;147:3215–3229. doi: 10.1099/00221287-147-12-3215. [DOI] [PubMed] [Google Scholar]
- Lu G. TOP: A new method for Protein Structure Comparisons and Similarity Searches. J Appl Cryst. 2000;33:176–183. [Google Scholar]
- Maier B, Potter L, So M, Seifert HS, Sheetz MP. Single pilus motor forces exceed 100 pN. Proc Natl Acad Sci USA. 2002;99:16012–16017. doi: 10.1073/pnas.242523299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT. Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines. Nature. 2005;437:1115–1120. doi: 10.1038/nature04031. [DOI] [PubMed] [Google Scholar]
- McRee DE. XtalView: A visual protein crystallographic software system for X11/Xview. J Mol Graphics. 1992;10:44–47. [Google Scholar]
- Meng Y, Li Y, Galvani CD, Hao G, Turner JN, Burr TJ, Hoch HC. Upstream migration of Xylella fastidiosa via pilus-driven twitching motility. J Bact. 2005;187:5560–5567. doi: 10.1128/JB.187.16.5560-5567.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz AJ, So M. Interactions of pathogenic Neisseriae with epithelial cell membranes. Ann Rev of Cell & Devel Biol. 2000;16:423–457. doi: 10.1146/annurev.cellbio.16.1.423. [DOI] [PubMed] [Google Scholar]
- Merz AJ, So M, Sheetz MP. Pilus retraction powers bacterial twitching motility. Nature. 2000;407:98–102. doi: 10.1038/35024105. [DOI] [PubMed] [Google Scholar]
- Morand PC, Bille E, Morelle S, Eugene E, Beretti JL, Wolfgang M, Meyer TF, Koomey M, Nassif X. Type IV pilus retraction in pathogenic Neisseria is regulated by the PilC proteins. EMBO. 2004;23:2009–2017. doi: 10.1038/sj.emboj.7600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Okamoto S, Ohmori M. The cyanobacterial PilT protein responsible for cell motility and transformation hydrolyzes ATP. Plant & Cell Physiol. 2002;43:1127–1136. doi: 10.1093/pcp/pcf128. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Macromolecular Crystallography, part A. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Planet PJ, Kachlany SC, DeSalle R, Figurski DH. Phylogeny of genes encoded for secretion NTPases: identification of the widespread tadA subfamily and development of a diagnostic key for gene classification. Proc Natl Acad Sci USA. 2001;98:2503–2508. doi: 10.1073/pnas.051436598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol C, Eugene E, Marceau M, Nassif X. The meningococcal PilT protein is required for induction of intimate attachment to epithelial cells following pilus-mediated adhesion. Proc Natl Acad Sci USA. 1999;96:4017–4022. doi: 10.1073/pnas.96.7.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas S, Bolland S, Cabezón E, Goñi FM, Cruz Fdl. TrwD, a protein encoded by the IncW plasmid R388, displays an ATP hydrolase activity essential for bacterial conjugation. J Biol Chem. 1997;272:25583–25590. doi: 10.1074/jbc.272.41.25583. [DOI] [PubMed] [Google Scholar]
- Robien MA, Krumm BE, Sandkvist M, Hol WGJ. Crystal structure of the extracellular protein secretion NTPase EpsE of Vibrio cholerae. J Mol Biol. 2003;333:657–674. doi: 10.1016/j.jmb.2003.07.015. [DOI] [PubMed] [Google Scholar]
- Sandkvist M. Biology of type II secretion. Mol Microbiol. 2001;40:271–283. doi: 10.1046/j.1365-2958.2001.02403.x. [DOI] [PubMed] [Google Scholar]
- Savvides SN, Yeo HJ, Beck MR, Blaesing F, Lurz R, Lanka E, Buhrdorf R, Fischer W, Haas R, Waksman G. VirB11 ATPases are dynamic hexameric assemblies: new insights into bacterial type IV secretion. EMBO. 2003;22:1969–1980. doi: 10.1093/emboj/cdg223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffzek K, Ahmadian MR, Wittinghofer A. GTPase-activating proteins: helping hands to complement an active site. Trends Biochem Sci. 1998;23:257–262. doi: 10.1016/s0968-0004(98)01224-9. [DOI] [PubMed] [Google Scholar]
- Shim JH, Kim YW, Kim TJ, Chae HY, Park JH, Cha H, Kim JW, Kim YR, Schaefer T, Spendler T, et al. Improvement of cyclodextrin glucanotransferase as an antistalling enzyme by error-prone PCR. Protein Eng Des Sel. 2004;17:205–211. doi: 10.1093/protein/gzh035. [DOI] [PubMed] [Google Scholar]
- Skerker JM, Berg HC. Direct observation of extension and retraction of type IV pili. Proc Natl Acad Sci USA. 2001;98:6901–6904. doi: 10.1073/pnas.121171698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Cryst. 2004;60D:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- Story RM, Steitz TA. Structure of the RecA protein-ADP complex. Nature. 1992;355:374–376. doi: 10.1038/355374a0. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Cryst. 1999;D55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang M, Lauer P, Park HS, Brossay L, Hebert J, Koomey M. PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Mol Micro. 1998;29:321–330. doi: 10.1046/j.1365-2958.1998.00935.x. [DOI] [PubMed] [Google Scholar]
- Wu SS, Wu J, Kaiser D. The Myxococcus xanthus pilT locus is required for social gliding motility although pili are still produced. Mol Microbiol. 1997;23:109–121. doi: 10.1046/j.1365-2958.1997.1791550.x. [DOI] [PubMed] [Google Scholar]
- Xiao M, Li H, Snyder GE, Cooke R, Yount RG, Selvin PR. Conformational changes between the active-site and regulatory light chain of myosin as determined by luminescence resonance energy transfer: the effect of nucleotides and actin. Proc Natl Acad Sci USA. 1998;95:15309–15314. doi: 10.1073/pnas.95.26.15309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo HJ, Savvides SN, Herr AB, Lanka E, Waksman G. Crystal structure of the hexameric traffic ATPase of the Helicobacter pylori type IV secretion system. Mol Cell. 2000;6:1461–1472. doi: 10.1016/s1097-2765(00)00142-8. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chaney M, Wigneshweraraj SR, Schumacher J, Bordes P, Cannon W, Buck M. Mechanochemical ATPases and transcriptional activation. Mol Microbiol. 2002;45:895–903. doi: 10.1046/j.1365-2958.2002.03065.x. [DOI] [PubMed] [Google Scholar]