

Summary
Constitutive activation of MEK-ERK signaling is often found in melanomas. Here, we identify a mechanism that links ERK with JNK signaling in human melanoma. Constitutively active ERK increases c-Jun transcription and stability, which are mediated by CREB and GSK3, respectively. Subsequently, c-Jun increases transcription of target genes, including RACK1, an adaptor protein that enables PKC to phosphorylate and enhance JNK activity, enforcing a feed-forward mechanism of the JNK-Jun pathway. Activated c-Jun is also responsible for elevated cyclin D1 expression, which is frequently overexpressed in human melanoma. Our data reveal that in human melanoma the rewired ERK signaling pathway upregulates JNK and activates the c-Jun oncogene and its downstream targets including RACK1 and cyclin D1.
Significance
Although constitutively active ERK-MAPK signaling has been found in a large fraction of human melanoma tumors, how this pathway contributes to melanoma development remains largely elusive. Here we reveal the blueprint for rewiring of key signal transduction pathways in melanoma. In this re-wiring program, constitutively active ERK affects the c-Jun oncogene, its upstream kinase JNK, and its downstream targets RACK1 and cyclin D1. Understanding how key signaling pathways are re-wired in melanoma offers new targets for therapy of this tumor type.
Introduction
Current understanding of changes commonly seen in human melanoma suggests that this tumor type may serve as a paradigm of signaling cascades that undergo a rewiring program. A large percentage of melanomas harbor super-active kinases, primarily of the mitogen-activated protein kinase (MAPK) family, as a result of activating mutations in B-RAF or N-RAS genes (Davies et al., 2004; Pollock and Meltzer, 2002; Gorden et al., 2003). Normally, kinases of the MAPK family control a diverse array of cellular functions such as gene expression, the inflammatory response, differentiation, the cell cycle, cell proliferation, and apoptosis (Weston and Davis, 2002). The extracellular-signal-regulated kinase (ERK), which is normally activated primarily by mitogens (Johnson and Lapadat, 2002), has been implicated in differentiation, senescence, and survival. Mutations in B-RAF or N-RAS genes, found in >70% of intermittently sun-exposed melanomas (Maldonado et al., 2003), cause super-activation of ERK, which has been implicated in activation of MMP1 and MITF (Molina et al., 2005; Park et al, 2004), although the precise mechanism linking ERK and the downstream targets remains in many cases elusive. In the present study we demonstrate that highly active ERK affects the degree of c-Jun NH2-terminal kinase (JNK) activity. As a result it upregulates the c-Jun oncogene and consequently the c-Jun target cyclin D1, which is frequently upregulated in human melanoma.
JNK, which is among the major subgroups of MAPK, is activated primarily by inflammatory cytokines and environmental stress (Karin, 1995; Weston and Davis, 2002). These stimuli induce JNK phosphorylation on Thr183 and Tyr185 residues by the dual specific kinases MKK4 and MKK7 (Davis, 2000). Among key JNK substrates is the transcription factor c-Jun. Phosphorylation by JNK enables c-Jun’s cooperation with members of the b-ZIP family, including c-Fos and ATF2, resulting in concomitant activation of a wide set of substrates that control the cell cycle as well as cell proliferation, differentiation and death (Vogt, 2001; Shaulian and Karin, 2002). c-Jun cooperates with both cellular and viral oncogenes (e.g., mutant Ras) to mediate transformation of cells in culture (Johnson et al., 1996) and tumor development in animal models (Eferl et al., 2003), activities consistent with its deregulation in various human tumors (Mathas et al., 2002). As a protein that plays a key role in cell growth and transformation, c-Jun is tightly regulated at the levels of expression and activity. Among key factors in c-Jun regulation is its phosphorylation, which affects its stability and activity (Morton et al., 2003; Laine and Ronai, 2005). Phosphorylation of c-Jun at Ser63, Ser73, Thr91 and Thr93, primarily mediated by JNK, is required for its transcriptional activity (Minden et al., 1994) and protects c-Jun from ubiquitination and subsequent degradation (Fuchs et al., 1996). Conversely, phosphorylation of c-Jun on Thr239 and Ser243 targets it for ubiquitination and subsequent degradation by Fbw7 (Wei et al., 2005), a modification that is important in c-Jun’s ability to elicit its oncogenic activity. Here we demonstrate that ERK utilizes two distinct mechanisms to upregulate c-Jun expression, which, in turn, further increase JNK signaling and induce high levels of cyclin D1 expression.
The degree of JNK activation in response to diverse stimuli can be augmented by its phosphorylation via protein kinase C (Lopez Bergami et al., 2005; Liu et al., 2006). JNK activation by PKC is required for its maximal induction by diverse stimuli including cytokines (e.g., TNFα) and external stress (e.g., UV-irradiation). PKC’s effect on JNK requires RACK1, a 7 WD40 repeat scaffold implicated in PKC and Src signaling (Ron et al., 1994). Changes in activity or levels of the PKC-RACK1 module affect the magnitude of JNK activation (Lopez-Bergami et al., 2005). Thus, changes in RACK1 expression are expected to affect the degree of JNK activity. Elevated RACK1 expression was observed in non-small cell lung carcinoma (Berns et al., 2000), colon carcinoma (Berns et al., 2000; Saito et al., 2002) and melanoma (Lopez-Bergami et al., 2005). The link between RACK1 and JNK is also expected to play a key role in tumorigenicity, as inhibition of RACK1 expression sensitizes melanoma cells to treatment and reduces their tumorigenicity in a xenograft tumor model (Lopez Bergami et al., 2005). Here, we demonstrate that ERK upregulates JNK activity via its effect on c-Jun and concomitant c-Jun activation of RACK1 transcription. RACK1, in turn, increases the degree of JNK activation by PKC, resulting in further activation of c-Jun and its downstream transcriptional targets including cyclin D1.
Cyclin D1 is a 34-kDa nuclear protein coded by the CCND1 gene, which is located at 11q13. Cyclin D1 is an important positive regulator of the G1-S cell cycle transition, which is achieved through its binding and activation of its kinase partners cdk4/6, contributing to phosphorylation and inactivation of pRb, blocking its growth-inhibitory activity and promoting release of bound E2F, leading to cell cycle progression (Bartek et al., 1996). Amplification of CCND1 was found in primary melanomas and metastases to varying degrees (Utikal et al., 2005), suggesting a role for the CCND1 gene in the pathogenesis of melanoma. Whereas increased copy number of the CCND1 gene is associated with overexpression of cyclin D1, over 25% of melanomas that overexpressed cyclin D1 have a normal copy number of the CCND1 gene (Sauter et al., 2002), suggesting that expression levels of cyclin D1 are modulated by mechanisms other than gene amplification. Expression of cyclin D1 has been demonstrated at constitutively high levels in melanoma cell lines (Bartek et al., 1993) as well as in melanoma metastases (Maelandsmo et al., 1996; Errico et al., 2003; Sauter et al., 2002), suggesting that it has an oncogenic role in melanoma pathogenesis. The present studies provide mechanistic understanding of upregulation of c-Jun and consequently its target genes, including cyclin D1 in melanoma, as part of a re-wiring of signaling cascades involving ERK and JNK.
Results
The activated MAPK pathway is responsible for elevated c-Jun levels in melanoma
We recently demonstrated that JNK activation can be augmented upon its phosphorylation by PKC, which requires the adaptor protein RACK1 (Lopez Bergami et al., 2005). Since the mechanism underlying upregulation of PKC or RACK1 is not known, we explored the possible effects of the ERK signaling pathway, which is notoriously upregulated in this tumor type, on RACK1 expression. Analysis of melanoma cell lines revealed that those expressing high levels of RACK1 contain higher levels of active PKCα/β, ERK and JNK, as reflected by their phosphorylation on residues required for their activity (Fig. 1A). Since active ERK is also associated with increased levels of c-Jun (Fig. 1A), we assessed the possible impact of ERK on c-Jun expression.
Figure 1. ERK signaling affects c-Jun expression.
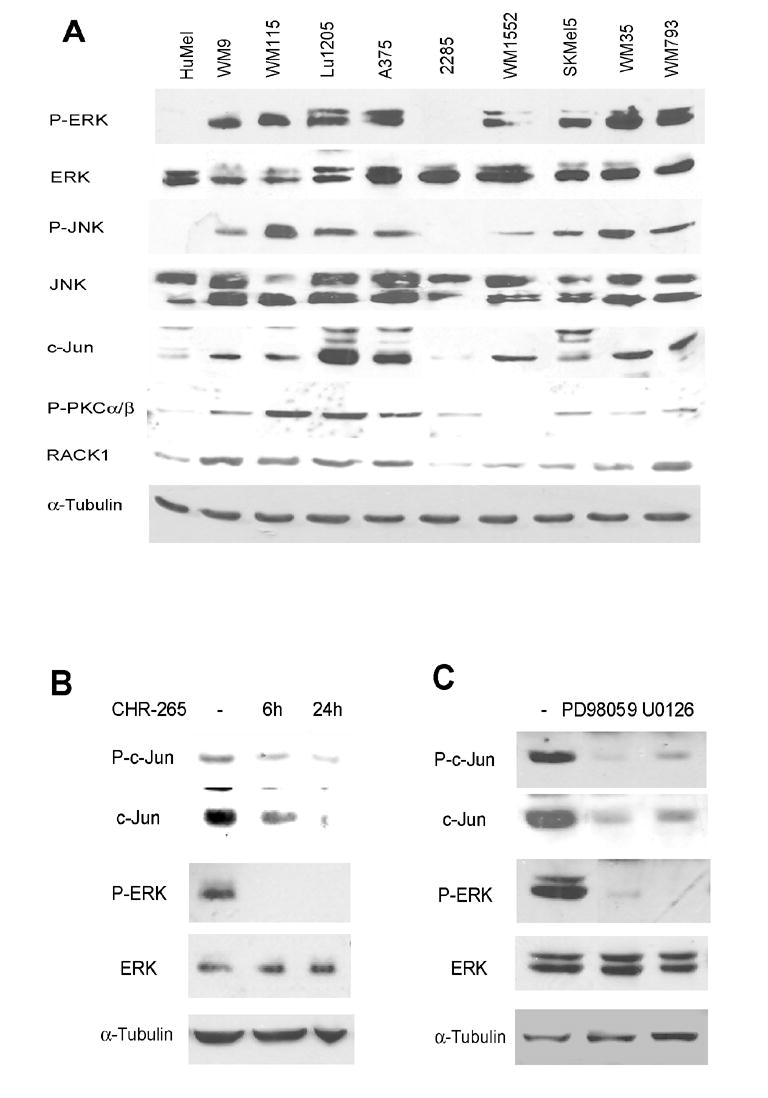
A. Levels of P-ERK, P-JNK and proteins relevant to the PKC/JNK pathway in melanoma cell lines. Protein extracts (40μg) from melanoma cell lines were analyzed by Western blots using the indicated antibodies. Human melanocytes (HuMel) were used as a control. α-Tubulin reveals equal loading.
B. Inhibition of B-Raf reduces c-Jun levels. Lu1205 cells were treated with 300nM CHR-265 for 6h and 24h. Protein samples were analyzed by Western blots using the indicated antibodies. α-Tubulin reveals equal loading.
C. Inhibition of the MEK/ERK pathway reduces c-Jun levels. A375 cells were treated with 50μM PD98059 and 20μM U0126 for 24h. Protein samples were analyzed by Western blots using the indicated antibodies.
To directly assess a causative link between ERK and c-Jun, we used 3 inhibitors known to affect B-RAF and MEK, major components in this pathway which is deregulated in melanoma. Treatment of melanoma Lu1205 cells with CHR-265, a BRAF inhibitor, reduced c-Jun levels (Fig. 1B). This effect was mediated by the MEK/ERK pathway since a similar decrease was observed following treatment with the specific MEK inhibitors U0126 and PD98059 (Fig. 1C). The effect of ERK on c-Jun expression was specific to melanoma cells, as it was not seen in HEK293T cells (Fig. S1) or in human melanocytes (Fig. S2) in which MEK/ERK are not constitutively upregulated. The possibility that changes in c-Jun expression following MEK/ERK inhibition were due to altered cell cycle distribution was ruled out (Fig. S3). These data suggest that ERK signaling plays a central role in up-regulation of c-Jun expression in human melanoma cells in which this signaling cascade is constitutively active. We therefore set out to identify the mechanism underlying ERK’s effect on c-Jun expression and activity.
ERK regulates c-Jun stability
To assess the mechanism by which ERK sustains elevated levels of c-Jun, we tested possible effects of ERK inhibition on transcription and stability of c-Jun. Addition of actinomycin D or PD98059 caused a noticeable decreased in the level of c-Jun expression. However, after a 2h treatment, the MEK/ERK inhibitor decreased c-Jun levels more efficiently than actinomycin D (Fig. 2A). These differences implied the possible involvement of both transcriptional and post-transcriptional mechanisms in ERK’s regulation of c-Jun. Since c-Jun is an unstable protein that is regulated by several ubiquitin ligases (reviewed in Laine and Ronai, 2005), we first tested the possible effect of ERK on c-Jun stability. Proteasome inhibitors attenuated the effect of the ERK inhibitors and further increased the level of c-Jun expression (Fig. S4), suggesting that c-Jun stability is important for ERK-mediated effects on c-Jun expression. To better understand the effect of ERK on stability vs. transcription of c-Jun, we monitored changes in the expression of exogenously expressed c-Jun, which is not expected to reflect changes in c-Jun transcription. As seen with the endogenous protein, inhibition of ERK activity markedly decreased the level of exogenous c-Jun expression, which was monitored using two different promoters (CMV and EF; Fig. 2B). As a control, levels of exogenous ATF2 were not affected by PD98059 (Fig. 2B). These results suggest that the MEK/ERK pathway affects c-Jun stability.
Figure 2. ERK regulates c-Jun stability.
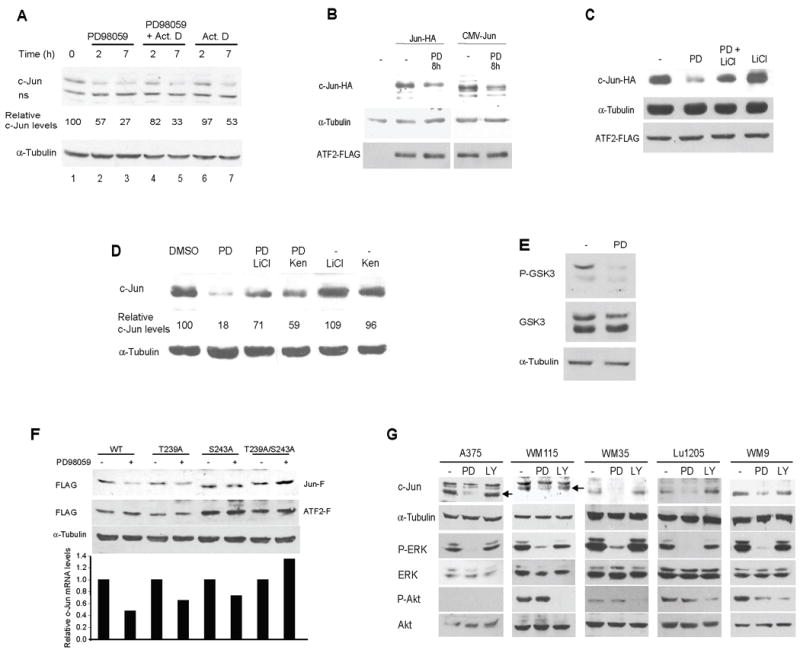
A. Regulation of c-Jun by the MEK/ERK pathway is via transcriptional and post-transcriptional mechanisms. A375 cells were treated with 50μM PD98059 (PD) at the indicated time points in the presence or absence of Actinomycin D (Act D) (see Methods). Protein samples were analyzed by Western blots using the indicated antibodies. α-Tubulin reveals equal loading. ns: non-specific band. Quantification of c-Jun level is shown. For all quantifications, c-Jun levels were normalized using α-Tubulin levels. The experiment shown is representative of the four experiments performed.
B. Inhibition of the MEK/ERK pathway affects the levels of exogenously expressed c-Jun. Lu1205 cells were transfected with either 0.5μg Jun-HA (an expression vector containing the EF [Elongation Factor] promoter or 1μg CMVHA-Jun (an expression vector using a CMV promoter). ATF2-Flag (1μg) was used to monitor transfection efficiency. Twenty-four hours after transfection, cells were treated with 50μM PD98059 (PD) for 8h. Protein samples were analyzed by Western blots using HA or Flag antibody as indicated. α-Tubulin reveals equal loading.
C. LiCl restores c-Jun levels reduced by MEK/ERK inhibitors. Lu1205 cells were transfected with c-Jun-HA and ATF2-Flag as indicated in B. Twenty four hours after transfection, cells were treated with 50μM PD98059 (PD) in the presence or absence of 10mM LiCl for 12h. Protein samples were analyzed by Western blots using HA and Flag antibodies. α-Tubulin reveals equal loading.
D. LiCl and Kenpaullone attenuate the effect of MEK/ERK inhibitors on endogenous c-Jun levels. A375 cells were treated with 50μM PD98059 (PD) for 12h in the presence or absence of 10mM LiCl or 10μM Kenpaullone (Ken). Protein samples were analyzed by Western blots using the indicated antibodies. α-Tubulin reveals equal loading. c-Jun protein levels were normalized to α-Tubulin levels. The experiment shown is representative of the four experiments performed.
E. Phosphorylation of GSK3(S9/S21) is inhibited by PD98059. A375 cells were treated with 50μM PD98059 (PD) for 12h. Protein samples were analyzed by Western blots using the indicated antibodies. α-Tubulin reveals equal loading.
F. T239 and S243 are required for regulation of c-Jun by MEK/ERK. Lu1205 cells were transfected with 0.5μg of Flag-tagged c-Jun WT, c-Jun T239A, c-Jun S243A or c-Jun T239A/S243A. ATF2-Flag (1μg) was used to monitor transfection efficiency. Twenty four hours after transfection, cells were treated with 50μM PD98059 for 12h. Protein samples were analyzed by Western blots using Flag antibody. α-Tubulin reveals equal loading. Graph shows c-Jun protein levels normalized to α-Tubulin levels. One experiment representative of three performed is shown.
G. ERK regulates c-Jun levels in melanoma. The indicated melanoma cell lines were treated with 50μM PD98059 (PD) or 10μM LY294002 (LY) for 12h in the absence of serum. Protein samples were analyzed by Western blots using the indicated antibodies. α-Tubulin reveals equal loading.
Since c-Jun degradation requires phosphorylation of T239 by GSK3 (Wei et al., 2005), we investigated whether ERK elicits its effect on c-Jun stability via inactivation of GSK3. In the event that ERK inhibition destabilizes c-Jun via GSK3 phosphorylation, LiCl (a GSK3 inhibitor) would be expected to reverse the effect of PD98059. Indeed, treatment of cells with LiCl attenuated PD98059’s effect, resulting in almost complete restoration of initial c-Jun levels (Fig. 2C). Since LiCl also inhibits other protein kinases with only slightly less potency than GSK3 (Davies et al., 2000), it has been suggested that a structurally unrelated GSK3 inhibitor such as Kenpaullone should be used in combination with LiCl to confirm a GSK3 involvement (Bain et al., 2003). As shown in Fig. 2D, both Kenpaullone and LiCl partially prevented the decrease in endogenous c-Jun levels due to PD98059, confirming that GSK3 mediates the effects of ERK on c-Jun. The antagonistic effect of GSK3 and MEK/ERK on c-Jun levels suggests that PD98059 blocks GSK3 inactivation by ERK. In fact, analysis performed with pGSK3 (S9/S21) antibody revealed that the level of pGSK3 (non-active form) in control cells was reduced upon treatment with PD98059 (Fig. 2E). Consistent with the effect of GSK3 on regulation of c-Jun stability by ERK is the finding that c-Jun mutated on the GSK3 phosphoacceptor site(s) was partially (single mutants T239A and S243A) or completely (double mutant T239A/S243A) resistant to degradation induced by the ERK pathway inhibitor (Fig. 2F).
In its role as a GSK3 upstream kinase, Akt has been proposed as a major regulator of cellular levels of c-Jun (Wei et al., 2005). Since our data indicate that c-Jun is regulated by ERK, we compared the relative contribution of Akt and ERK to c-Jun levels. To this end, melanoma cells displaying different levels of active Akt were treated with PD98059 and LY294002, a PI3K inhibitor, and the effect on c-Jun expression was determined. An evident reduction in c-Jun levels upon treatment with the ERK pathway inhibitor was observed in all five melanoma cell lines assayed (Fig. 2G). Interestingly, inhibition of the PI3K/Akt pathway by LY294002 did not affect c-Jun levels in A375, WM35 and Lu1205 cells and only slightly reduced it in WM115 and WM9 cells (Fig. 2G). Taken together, these data indicate that constitutive ERK activation in melanoma cells is the primary pathway that regulates the levels of c-Jun expression, in part, by inactivating GSK3 and consequently preventing c-Jun degradation.
ERK affects c-Jun transcription via CREB
Real-Time PCR analysis of c-Jun mRNA revealed a marked inhibition of c-Jun transcription upon treatment with the ERK pathway inhibitor (Fig. 3A), suggesting that ERK regulates c-Jun mRNA levels. Since c-Jun is responsible for its own transcription (Angel et al., 1988), changes in c-Jun activity are expected to affect c-Jun mRNA levels as well. Therefore, we first evaluated ERK-dependent c-Jun activation by measuring phosphorylation of N-terminus sites, Ser63 and Thr91/Thr93. This analysis revealed that inhibition of ERK activity for 30min does not affect S63 (Fig. S5) and Thr91/Thr93 (Fig. S6) phosphorylation. In contrast, phosphorylation of Ser63 and Thr91/Thr93 was reduced by addition of a JNK inhibitor (Fig. S5 and S6; note that within 30min, the time frame used for this analysis, c-Jun levels are not affected). These data suggest that ERK is not directly involved in c-Jun phosphorylation and activation. Consistent with this possibility, the levels of exogenous wt and S63A forms of c-Jun were affected to the same degree upon inhibition of ERK (Fig. S7). An alternative explanation for ERK-dependent changes in c-Jun transcription is that this process is mediated by other transcription factor(s) downstream of ERK. Among the possible transcription factors that could mediate ERK-dependent c-Jun transcription is ATF1 (Gupta and Prywes, 2002). Inhibition of ATF1 expression by its corresponding siRNA did not affect c-Jun expression (Fig. S8), suggesting that ATF1 is not involved in regulation of c-Jun transcription in melanoma cells. We next examined a possible role for CREB, another transcription factor activated by ERK (Wiggin et al., 2002; Johannessen et al., 2004) which could potentially affect AP1/CRE elements known to be important in c-Jun transcription. Thus, we tested the effect of the ERK inhibitor on CREB phosphorylation. CREB phosphorylation on S133, which is required for its transcriptional activities, was efficiently inhibited by PD98059 (Fig. 3B) indicating positive regulation by ERK. Moreover, inhibition of CREB expression by siRNA markedly decreased c-Jun expression at both the protein (Fig. 3C) and mRNA (Fig. 3D) levels. Additionally, a dominant negative mutant of CREB (A-CREB) also reduced c-Jun protein levels (Fig. S9). In line with these results, CREB siRNA inhibited transcriptional activity driven by the c-Jun promoter (Fig. 3E). These findings reveal that in melanoma, ERK controls c-Jun RNA expression levels via its activation of CREB.
Figure 3. ERK regulates c-Jun transcription.

A. Inhibition of the MEK/ERK pathway affects c-Jun mRNA levels. A375 cells were treated with 50μM PD98059 for the indicated time periods. c-Jun mRNA levels were analyzed with Real-Time PCR. Data were normalized using levels of β-Actin mRNA. Results are shown as the mean (bar) ± standard deviation (SD) of the respective relative concentrations. A representative experiment (of three performed) is shown.
B. Inhibition of the MEK/ERK pathway affects CREB phosphorylation. A375 cells were treated with 50μM PD98059 for 12h. Protein samples were analyzed by Western blots using the indicated antibodies. α-Tubulin reveals equal loading.
C. CREB siRNA affects c-Jun protein levels in melanoma. Protein extracts from A375SM cells stably transfected with non-targeting vector (NT) or with a vector encoding CREB siRNA were analyzed by Western blots using the indicated antibodies. α-Tubulin reveals equal loading.
D. CREB siRNA affects c-Jun mRNA levels. RNA was extracted from A375SM cells stably transfected with non-targeting vector (NT) or with a vector encoding CREB siRNA. c-Jun mRNA levels were analyzed with Real-Time PCR. Data were normalized using levels of β-Actin mRNA. Results are shown as the mean (bar) ± SD of the respective relative concentrations. A representative experiment (of three performed) is shown.
E. CREB siRNA affects transcription driven by the c-Jun promoter. A375SM cells stably transfected with non-targeting vector (NT) or with a vector encoding CREB siRNA were transiently transfected with 0.2μM of luciferase reporter containing the c-Jun WT promoter or c-Jun promoters mutated on the Jun2 site (Jun2-M), the TRE site (TRE-M) or the double mutant. Results are shown as the mean ± SD. Data were standardized using β-galactosidase activity.
c-Jun is required for RACK1 expression
Increased c-Jun expression by ERK is expected to contribute to its transcriptional activities. Yet for c-Jun to function as a potent transcription factor, phosphorylation on Ser63 and Ser73 is required, which in melanoma is primarily mediated by JNK (Fig. S5, S6). Since both PKC and JNK are constitutively active in most melanoma cell lines (Fig. 1A), one would expect that ERK contribution’s to c-Jun expression will be complemented by c-Jun phosphorylation by JNK, with a possible contribution of PKC. To test this possibility, we inhibited PKC activity with a pharmacological inhibitor. Consistent with our earlier report on the role of PKC in activation of JNK and c-Jun in melanoma cells, inhibition of PKC by Go6976 or a dominant negative form of PKCβ markedly decreased basal phosphorylation of JNK1 and JNK2 in melanoma cell lines A375 and Lu1205 (Fig. 4A; S10) and lowered c-Jun transcriptional activity, assessed using a TRE-Luc reporter assay (Fig. 4B). TPA, which induces PKC at an early time point and downregulates it later, increased RACK1 expression in a manner that coincided with JNK activity (Fig. S11). This observation prompted us to examine possible changes in RACK1 transcription, following inhibition of PKC and JNK activity. Significantly, incubation with Go6976 inhibited the luciferase activity driven by the RACK1 promoter, similar to what was seen for the TRE promoter (Fig. 4B). Inhibition of PKC did not affect transcriptional activity driven by a β-catenin-Luc reporter plasmid, used here as a control (Fig. 4B, TOP). This observation implies that RACK1 transcription may be dependent on c-Jun.
Figure 4. c-Jun regulates RACK1 transcription.
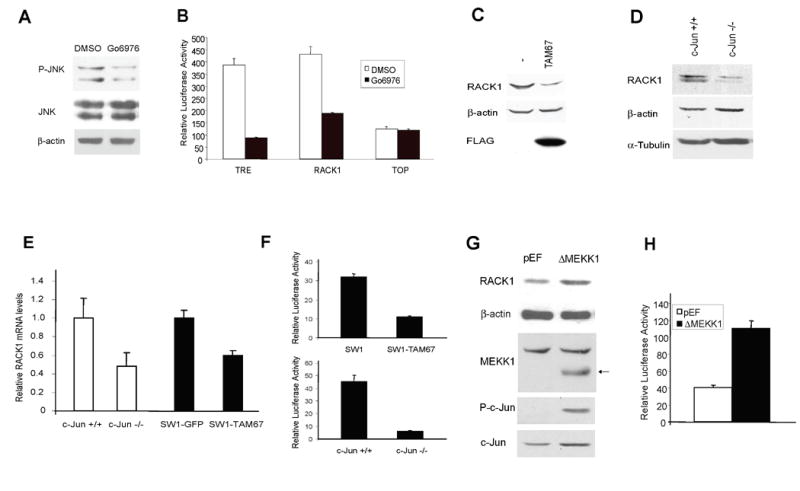
A. Go6976 inhibits JNK activation. A375 cells were treated with Go6976 for 2h before protein extracts were prepared. Levels of P-JNK and total JNK were assessed by Western blot. β-actin was used to monitor equal loading.
B. Inhibition of PKC reduces RACK1-driven luciferase activity. The luciferase activity driven by the RACK1 promoter (2Kb of the proximal RACK1 promoter cloned into a pGL2 vector) was assessed in Lu1205 cells in the presence of Go6976 (3μM for 8h). TRE and TOP vectors were used as positive and negative controls, respectively. Results are shown as the mean ± SD. Data were standardized on the basis of β-galactosidase activity.
C. c-Jun regulates RACK1 expression. Protein extracts from SW1 cells stably transfected with FLAG-TAM67 were blotted with RACK1 and FLAG antibodies. β-actin antibody was used to monitor equal protein loading.
D. RACK1 expression is reduced in c-Jun-/- fibroblasts. Protein extracts from c-Jun -/- and control fibroblasts were blotted with RACK1 antibody. β-actin and α-Tubulin antibodies were used to monitor equal protein loading.
E. Decrease in relative mRNAs levels of RACK1 in c-Jun -/- MEF and in SW1-TAM67 cells. Relative levels of RACK1 mRNA were determined by Real-Time quantitative PCR. Reactions were run in triplicate. β-actin was used as a control. Results are shown as the mean (bar) ± SD of the respective relative concentrations. A representative experiment (of three performed) is shown.
F. c-Jun regulates RACK1-driven transcription. The luciferase activity driven by the RACK1 promoter (2Kb of the proximal RACK1 promoter cloned into a pGL2 vector) was assessed in SW1 cells expressing TAM67 as well as in wt and c-Jun mutant fibroblasts. Results are shown as the mean ± SD. Data were standardized on the basis of β-gal activity.
G. Activation of the JNK/c-Jun pathway increases RACK1 expression. HEK293T cells were transfected with ΔMEKK1 or empty pEF plasmid. Protein extracts were obtained 48h post-transfection and blotted with the indicated antibodies. β-actin antibody was used to monitor loading. Arrow indicates position of ΔMEKK1.
H. ΔMEKK1 increases RACK1-driven transcription. The luciferase activity driven by the RACK1 promoter (2Kb of the proximal RACK1 promoter cloned into a pGL2 vector) was assessed in HEK293T cells transfected with ΔMEKK1. Results are shown as the mean ± SD. Data were standardized on the basis of β-gal activity.
To directly address the possible role of c-Jun in RACK1 expression, we used cells deficient in c-Jun or in which c-Jun transcriptional activities were effectively inhibited by a dominant-negative c-Jun construct (TAM67). Inhibition of c-Jun transcriptional activities or the lack of c-Jun expression sufficed to attenuate RACK1, at both protein (Fig. 4C and 4D) and mRNA levels (Fig. 4E). Consistent with the mRNA data, luciferase activity driven by the RACK1 promoter was also reduced in both TAM67-expressing cells and in fibroblasts that lack c-Jun (Fig. 4F). Moreover, cells in which activated c-Jun was reduced by using JNK siRNA also revealed lower levels of RACK1 (Fig. S12). To further substantiate the contribution of JNK/c-Jun signaling to RACK1 expression, we monitored changes in RACK1 expression in cells transfected with the constitutively active form of MEKK1 (ΔMEKK1), which is known to elicit strong activation of JNK and c-Jun. As shown in Fig. 4G, ΔMEKK1 efficiently increased activation of c-Jun and elevated the level of RACK1 expression. Similarly, ΔMEKK1 expression also increased the level of RACK1-driven luciferase activity (Fig. 4H). Together, these data indicate that JNK signaling, via c-Jun, is a positive regulator of RACK1 expression.
Mapping the AP-1 site on the RACK1 promoter
We next mapped the c-Jun response element(s) on the RACK1 promoter. Analysis of the proximal region revealed the presence of c-Jun target sequences at positions -1814, -863, -733 and -60; another possible site was found at +517 in the first intron (marked with letters A-E in Fig. 5A). Nested deletions of the RACK1 promoter containing these elements were cloned into a luciferase promoter-less vector, and the degree of luciferase activity was monitored along with the effect of PKC inhibitor Go6976. This analysis identified the AP-1 site at position -60 (TGAATCA) as sufficient to mediate PKC/JNK-dependent activation of the RACK1 promoter (Fig. 5B). Gel shift analysis demonstrated the ability of c-Jun to bind to an oligonucleotide containing the AP-1 site (Fig. 5C and S13). Consistent with this finding, mutation within this site attenuated the basal level of reporter activity (Fig. 5D) and the binding of c-Jun (Fig. 5C) confirming that c-Jun regulates RACK1 transcription via its response element at the -60 site. Further support for the role of c-Jun in regulation of RACK1 transcription comes from ChIP analysis. Sheared chromatin was immunoprecipitated with antibodies to c-Jun (or control IgG) followed by PCR amplification of RACK1 promoter sequences bearing the AP1 response element. Immunoprecipitation of c-Jun enabled amplification of RACK1 promoter sequences (Fig. 5E), demonstrating in vivo binding of c-Jun to the RACK1 promoter. Amplification of RACK1 promoter sequences was not observed in cells expressing TAM67 (Fig. 5E) or upon transfection of siRNA against c-Jun (Fig. 5F) and decreased following treatment with PD98059 (Fig. 5G). These data substantiates the finding that c-Jun mediates RACK1 transcription.
Figure 5. Mapping the c-Jun response element on the RACK1 promoter.
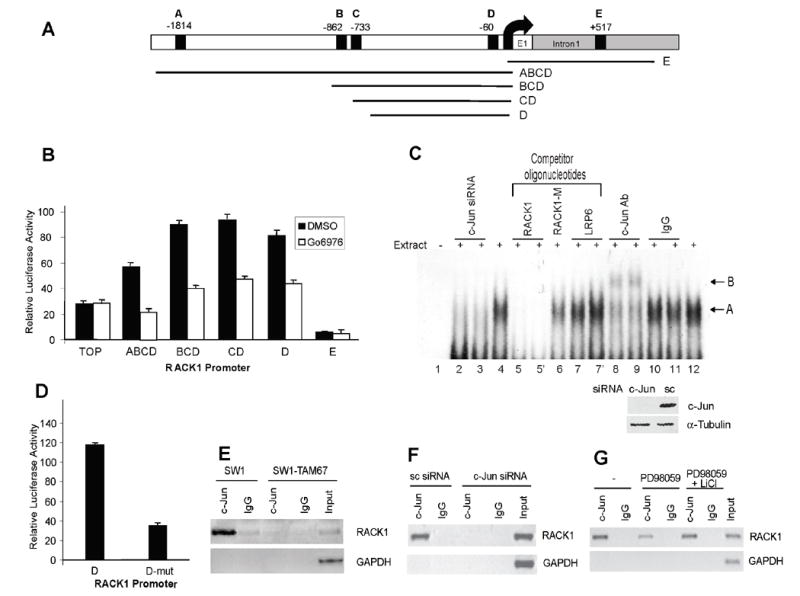
A. Structure of the RACK1 promoter. Putative c-Jun response elements and fragments of the promoter that were cloned are depicted (black boxes, A-E).
B. Mapping of the functional AP-1 site of the mouse RACK1 promoter. DNA fragments containing various response elements (shown in A) were cloned into pGL2 and their luciferase activity was assayed in Lu1205 cells in the presence of Go6976 (3μM for 8h). TOP vector was used as negative control. Results are shown as the mean ± SD.
C. c-Jun binds to the AP-1 element located at -60 on the RACK1 promoter. Ten micrograms of nuclear extracts from control cells were incubated with a doublestranded DNA fragment probe corresponding to the –67 to –54 region of the RACK1 promoter carrying a single AP-1 motif (lanes 4 to 12). Unlabeled competitors (RACK1, wt oligonucleotide (lane 5), RACK1-M, RACK1 oligonucleotide containing two bp changes in the AP-1 element (lane 6) and LRP6, non-related competitor (lane 7)) were added at a 10-fold excess as indicated (lanes 5 and 7 were run in duplicates). For supershift assays, 0.4 (lanes 8 and 10) or 2 μg (lanes 9 and 11) of c-Jun Ab or control IgG (Santa Cruz Biotechnology, Santa Cruz, CA) were included in the reaction. Lanes 4 and 12 show the shift in the absence of antibodies or competitors. The lower arrow (A) indicates a specific retarded band corresponding to c-Jun in complex with the probe. The upper arrow (B) indicates the supershifted complex. Reactions using nuclear extracts from c-Jun siRNA treated cells were included as a control (lane 2 and 3, 20 and 10μg respectively). The lower panel shows changes in c-Jun protein levels in cells transfected with control (sc) and c-Jun siRNA. A picture of the complete gel is shown in Fig. S13.
D. Mutation of c-Jun element at -60 inhibits reporter activity. The AP-1 response element D was mutated and the relative luciferase activity of the WT and mutant construct was assessed in Lu1205 cells. Results are shown as the mean ± SD. Data were standardized on the basis of β-galactosidase activity.
E. Chromatin immunoprecipitation assay. Sheared chromatin from SW1 cells or SW1 stably transfected with TAM67 was immunoprecipitated with appropriate antibody (anti-c-Jun or control IgG). Immunoprecipitated DNA was used as the template in PCR using primers corresponding to the proximal region of the RACK1 promoter. GAPDH primers were used as control.
F. c-Jun is not present at the RACK1 promoter in cells treated with c-Jun siRNA. Sheared chromatin from SW1 cells or SW1 transfected with c-Jun siRNA was immunoprecipitated with appropriate antibody (anti-c-Jun or control IgG). Samples were processed as in Fig. 5E.
G. MEK inhibitor affects c-Jun binding to RACK1 promoter. Sheared chromatin from SW1 cells or SW1 treated with 50μM PD98059 (PD) in the presence or absence of 10mM LiCl was immunoprecipitated with appropriate antibody (anti-c-Jun or control IgG). Samples were processed as in Fig. 5E.
Earlier studies demonstrated that RACK1 is required for PKC signaling (Schechman and Mochly-Rosen, 2001) as well as for PKC-dependent activation of JNK (Lopez Bergami et al., 2005; Liu et al., 2006). In accordance with these data, inhibition of RACK1 expression by a specific siRNA attenuated JNK activation (Lopez Bergami et al., 2005) and c-Jun phosphorylation (Fig. S14). Taken together, our data suggest that c-Jun is part of a feed-forward loop mechanism that maintains high levels of RACK1 expression to support PKC-dependent JNK activation of c-Jun. Constitutive activation of c-Jun is expected to contribute to its oncogenic potential.
ERK increases JNK activity via the RACK1/PKC/JNK/c-Jun feedback loop
The data above showed that c-Jun levels in melanoma are controlled by constitutive ERK activity. In turn, c-Jun induces expression of RACK1, which is required for JNK activation by PKC, pointing to a c-Jun/RACK1/PKC/JNK feedback loop. Accordingly, interference with the MEK/ERK pathway is expected to attenuate the effect of ERK on JNK activity. To test this possibility, ERK and JNK inhibitors were compared with regard to their effect c-Jun expression levels. Whereas short exposure to an ERK inhibitor did not affect c-Jun expression in A375 melanoma cells, longer treatment blocked c-Jun expression (Fig. 6A) and reduced RACK1 expression and JNK activity (Fig. 6A). Interestingly, treatment with a JNK inhibitor also decreased c-Jun levels although with slower kinetics compared with PD98059. Similarly, analysis of four melanoma cell lines treated with PD98059 for 16h showed a decrease in c-Jun levels with a concomitant decrease in P-JNK levels (Fig. 6B). Consistent with these findings, immunokinase reactions using JNK immunopurified from melanoma cells subjected to treatment with the ERK inhibitor revealed a 4-fold decrease in JNK activity (Fig. S15). Further, constitutive expression of the dominant negative c-Jun construct, TAM67, in SW1 melanoma cells attenuated the degree of JNK activity, substantiating the role of c-Jun in JNK activity (Fig. 6C). All in all, our results establish that regulation of c-Jun by ERK ultimately affects JNK in melanoma.
Figure 6. ERK induces JNK activation as part of cross-talk and feed-forward mechanisms.

A. Inhibition of the MEK/ERK pathway affects RACK1 and P-JNK levels. A375 cells were treated with 50μM PD98059 (PD) or 10μM of JNK inhibitor for the indicated times. Protein samples were analyzed by Western blots using the indicated antibodies. α-Tubulin reveals equal loading.
B. Inhibition of the MEK/ERK pathway affects c-Jun, P-JNK and cyclin D1 levels. The indicated melanoma cell lines were treated with 50μM PD98059 for 16h. Protein samples were analyzed by Western blots using the indicated antibodies. α-Tubulin reveals equal loading.
C. Inhibition of c-Jun attenuates JNK activity in SW1 mouse melanoma cells. SW1 cells were transfected to establish stable clones expressing the dominant negative form of c-Jun, TAM67. Cells were used to monitor JNK phosphorylation on aa 183/5, which reflect JNK activity.
D. c-Jun regulates cyclin D1 expression. Protein extracts from Lu1205 and SW1 cells stably transfected with FLAG-TAM67 were blotted with the indicated antibodies. α-Tubulin antibody was used to monitor equal protein loading.
E. LiCl restores c-Jun and cyclin D1 levels that are reduced by MEK/ERK inhibitors. Lu1205 cells were treated with 50μM PD98059 (PD) in the presence or absence of 10mM LiCl for 12h. Protein samples were analyzed by Western blots using the indicated antibodies. α-Tubulin reveals equal loading.
F. CREB siRNA affects both c-Jun and cyclin D1 protein levels in melanoma. Protein extracts from A375SM cells stably transfected with non-targeting vector (NT) or with a vector encoding CREB siRNA were analyzed by Western blots using the indicated antibodies. α-Tubulin reveals equal loading.
c-Jun activation by ERK and JNK induces cyclin D1 transcription
c-Jun expression and phosphorylation by ERK and JNK are expected to result in a constitutively transcriptionally active c-Jun. To test this possibility, we assessed the roles of ERK and JNK in relation to the c-Jun transcriptional target, cyclin D1 (Sabbah et al., 1999), which is frequently overexpressed in melanoma (Bartek et al., 1993; Maelandsmo et al., 1996; Pardo et al., 2004; Coupland et al., 1998, Sauter et al., 2002). Inhibition of c-Jun activity by TAM67 efficiently attenuated expression of cyclin D1 in human and mouse melanoma cells (Fig. 6D), confirming its primary role in regulation of cyclin D1 expression. Similarly, inhibition of ERK activity in 4 human melanoma cell lines attenuated the level of cyclin D1 expression (Fig. 6B). In line with our model indicating that ERK regulates c-Jun via GSK3 and CREB, inhibition of GSK3 attenuated the effect of ERK inhibitor on c-Jun and cyclin D1 expression (Fig. 6E), and siRNA of CREB reduced c-Jun and cyclin D1 expression (Fig. 6F).
ERK activity associates with JNK, Jun and RACK1 levels and activity in human tumor samples
To substantiate a causative link between the ERK and JNK signaling pathways, we analyzed levels of active ERK and JNK and expression levels of c-Jun, cyclin D1 and RACK1 in a set of 24 melanoma tumors (Fig. 7A). Bcl-2 expression was assessed as outlier in statistical analysis. Of the 24 melanoma samples 16 were found to exhibit a robust P-ERK staining (66%). Of these, 13 (81%) presented high levels of c-Jun (Fig. 7A and S16). Of the remaining 8 samples, which showed negative or very weak P-ERK staining, none displayed high c-Jun levels (Fig. 7A, S16, S17). This finding provides important support for the link between ERK and c-Jun, as revealed in the present studies. Furthermore, as our data also point to a possible cooperation between ERK and JNK that would result in high levels of transcriptionally active c-Jun, we assessed the possible relationship between ERK and JNK activities and c-Jun and RACK1 expression levels in the melanoma tumor set. Twelve of the 24 samples (50%) were found to exhibit positive staining for both P-ERK and P-JNK together with high levels of c-Jun. Of those, 11 (92%) also exhibited high levels of RACK1 (Fig. 7A, S16), substantiating the link among P-ERK, P-JNK, c-Jun and RACK1, pointed out in the present study. Interestingly, of the remaining 12 samples (samples that showed either negative P-ERK or negative P-JNK), only 3 exhibited high levels of RACK1, suggesting that possibly another mechanism upregulates RACK1 expression in a small fraction of samples of this tumor type.
Figure 7. Analysis of RACK1, c-Jun, cyclin D, P-ERK and P-JNK levels in melanoma tumor samples.
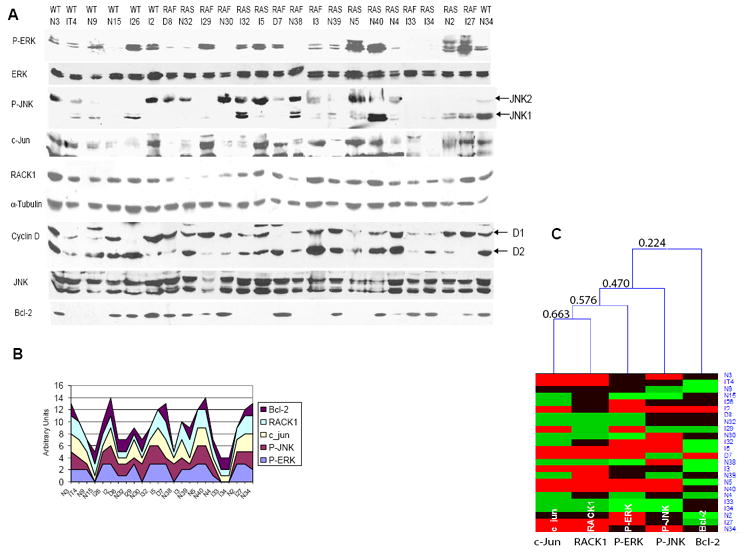
A.ERK activity is associated with JNK, c-Jun and RACK1 levels and activity in human tumor samples. Protein samples (80μg) obtained from melanoma tumors were analyzed by Western blot using the indicated antibodies. For the RACK1, Bcl-2 and α-Tubulin blots, 20μg of protein was used. The status of B-RAF and N-RAS genes is shown above the tumor ID. RAS indicates a Q61K or Q61R mutation. RAF indicates a V599E mutation. WT indicates the absence of Q61 and V599 mutation in N-RAS or B-RAF.
B. Stack graph represents relative levels of P-ERK, c-Jun, RACK1 and P-JNK that were quantified (Fig. S16) and converted into numbers (0, 1, 2, 3) to reflect protein expression level qualitatively.
C. Hierarchical clustering of the samples shown in A was performed with the aid of HCE program (Seo et al., 2002), using average linkage and Euclidean distance measure between expression levels. Different expression levels are shown in different colors: 0- light green, 1- green, 2- black and 3- red. The order of linking corresponds to the proximity of expression levels in different cell types. Minimal similarity values (computed in HCE program using Euclidean distance and normalized to a scale from 0 (no similarity) to 1 (identical profile)) are shown on the tree to reflect qualitatively similarities in expression patterns. The analysis revealed significant relations between P-ERK and c-Jun (0.672), c-Jun and RACK1 (0.617) and between P-ERK and RACK1 (0.680) levels. These values are significant with 99% confidence (the critical value for 1% significance level for 22 degrees of freedom (24-2) is 0.515, using Student’s t-statistics). Bcl2 levels, which are not directly associated with the other proteins in this analysis, served as an outlier for the statistical analysis.
Relative levels of P-ERK, c-Jun, RACK1 and P-JNK are represented in a stacked graph, which reveals the trend of the contribution of each of the above five variables over different samples (Fig. 7B). These data clearly reveal positive correlation between the 4 variables, substantiating the relationship between PER-K-c-Jun-RACK1 and P-JNK (Fig. 7C). This analysis provides strong support for re-wiring between the ERK and JNK signaling pathways identified in human melanoma.
Of note, some tumor samples that harbor mutations on either B-RAF or N-RAS showed negative or very weak staining of P-ERK that correlated with negative or weak P-MEK staining (Fig. 7A and data not shown).
As shown for the melanoma cell lines tested, the link between ERK and c-Jun expression and activity affects RACK1 as well as Cyclin D1 expression. Consistent with the notion that in a certain percentage of melanoma tumors cyclin D1 is upregulated as a result of gene amplification, melanoma samples exhibited elevated expression of cyclin D1, or cyclin D2, regardless of the status of P-ERK or c-Jun. The latter finding reveals that both transcriptional upregulation and gene amplification increase cyclin D1 in melanoma tumors.
Discussion
Understanding mechanisms underlying cross-talk among signal transduction pathways is key to unveiling the dynamics of multidimensional regulatory signaling networks. Although such networks fine-tune cellular function under normal growth as well as following stress and DNA damage, it has been a challenge to understand the nature of changes in this complex mechanism that occur in pathological cases, including human cancer. Melanoma, an aggressive form of skin cancer that often harbors mutant BRAF or N-RAS, and consequently increased ERK-MAPK activity, serves as a paradigm of re-wiring signaling pathways.
Here we provide a blueprint for a re-wired signaling pathway in melanoma. Our data reveal that ERK increases the level of c-Jun expression by affecting its transcription and stability. We further demonstrate that c-Jun increases the transcription of RACK1, an adaptor protein required for activation of JNK by PKC, which constitutes a feed-forward mechanism that increases c-Jun transcriptional activity. Lastly, we demonstrate that the ERK-Jun signaling cascade is required for c-Jun-mediated transcription of cyclin D1, which is often found to be overexpressed in human melanomas. Analysis of melanoma tumors confirms the changes in these signaling pathways, for which our studies provide the underlying mechanisms.
Our study identifies the mechanisms underlying upregulation of c-Jun expression by ERK, as we demonstrate the effect of ERK on GSK3 inactivation, resulting in c-Jun protein stabilization, and on CREB activation, which increases c-Jun transcription (model proposed in Fig. 8). Activation of ERK results in phosphorylation of CREB at Ser133, either directly by ERK or by ERK-stimulated MSK (Wiggin et al., 2002). The finding that ERK contributes to stabilization of c-Jun through inactivation of GSK3 is consistent with those reported by Wei et al. (2005), who demonstrated that phosphorylation of c-Jun on T239 by GSK3β is critical in c-Jun degradation. Yet whereas Akt was shown to affect GSK3β phosphorylation in HeLa cells (Wei et al., 2005), in melanoma cells, ERK signaling was found to mediate primarily GSK3 phosphorylation, resulting in c-Jun stabilization. ERK-dependent GSK3 phosphorylation at S9/S21 is likely to be mediated by an intermediate kinase, although we have ruled out RSK (siRNA of RSK did not impact c-Jun stability in these cells; data not shown). Alternatively, association of ERK with GSK3 and subsequent phosphorylation of GSK3 at T43 was shown to enhance the rate of phosphorylation on Ser9 by a third kinase (Ding et. al, 2005).
Figure 8. Model of ERK cross-talk with JNK signaling.

Super-active ERK as seen in melanomas that carry mutant B-RAF or N-RAS genes increases stability of c-Jun via its phosphorylation-dependent inactivation of GSK3, and induces c-Jun transcription via its activation of CREB. In turn, c-Jun induces transcription of RACK1, which, in concert with active PKC and MKK4/7, augment the degree of JNK activity to further increase (and maintain) c-Jun stability and activity, constituting a feed-forward loop mechanism induced by ERK. Our data support the existence of this model in melanoma, but not in cells in which ERK activity is not sufficiently high.
Our data identify RACK1 as a c-Jun transcriptional target. It is plausible that other c-Jun target genes may contribute to the feed-forward loop mechanism. Among those are PDK1, the PKC upstream kinase, whose transcription also appears to be c-Jun-dependent (Lopez-Bergami et al., unpublished data) and c-Jun-dependent changes in expression of protein phosphatases that affect JNK activity (Sprowles et al., 2005). Several requirements must be satisfied for the feed-forward loop to exist. First is that c-Jun is also subjected to phosphorylation on residues S63, S73 (and possibly T91 and T93), thus acquiring its transcriptional capabilities. Such phosphorylation is primarily attributed to JNK, although it has been also ascribed to ERK in JNK-deficient cells (Morton et al., 2003); our data rule out ERK involvement in this process in melanoma (Fig. S5; S6), further substantiating the need for the ERK-Jun link with subsequent activation of the RACK1-JNK-PKC module. The second requirement is that JNK must be activated by the canonical MKK4/7 pathway. This requirement is crucial since the contribution of PKC to overall JNK activity depends on JNK’s own phosphorylation on amino acids 183/5 by MKK4/7. Since active JNK is commonly seen in melanoma (Fig. 1A; Fig. 7A; Lopez Bergami et al., 2005b), this requirement is satisfied, although the reason for the upregulation of JNK in melanoma is not completely clear. The finding that some tumor samples display positive P-JNK staining in the absence of ERK activity suggests that JNK activation proceeds through ERKindependent mechanisms. In any case, in agreement with our model, JNK activity by itself is not sufficient to maintain high levels of c-Jun. The third requirement relates to PKC’s own activity. To cooperate with RACK1 in augmenting JNK activity, PKC must be active. Earlier studies pointed out that expression of several PKC isoforms is elevated in melanoma (Oka and Kikkawa, 2005; Selzer et al., 2002). Here we demonstrate that PKCα/β are among the isoforms that appear to be predominantly active in melanoma (Fig. 1A and Fig. S17). That PKC is important for growth and metastasis of melanoma was shown in studies where pharmacological inhibitors of PKC effectively inhibited growth of melanoma in mouse xenograft models (Dumont et al., 1992; Mapelli et al., 1994). That PKC and JNK are active in this tumor type satisfies the above-stated requirements and supports the existence of the feed-forward loop mechanism triggered by ERK’s effect on c-Jun expression levels (Fig. 8).
The cell lines we used were obtained, for the most part, from vertical growth phase tumors (A375, WM115, Lu1205, WM9), which are associated with more metastatic phenotypes. Consistent with our findings, CREB activity was implicated in melanoma growth and metastasis (reviewed in Nyormoi and Bar-Eli, 2003). Similarly, via its heterodimerization with ATF2, c-Jun was also shown to play an important role in melanoma growth (Bhoumik et al., 2004). Importantly, regulation of c-Jun by the ERK pathway was not seen in cells that lack constitutively high levels of ERK activity (e.g., melanocytes or HEK293 cells), suggesting that this regulation reflects selective rewiring of melanoma tumors in which upstream MAPK are constitutively active. It is likely that our finding extends beyond the cases of mutant B-RAF and N-RAS since activation of the MEK/ERK pathway and upregulation of cyclin D1 are also seen in tumors where such mutations do not exist. Thus, one would expect that the newly established link between ERK and JNK, with its implications regarding cyclin D1 expression, may allow identifying additional deregulated signaling components along the JNK or ERK signaling pathways. One would also expect that the link between ERK and JNK would also exist in other tumors where ERK is upregulated, a common occurrence in human cancer (Davies et al., 2002; Downward, 2003).
The effect of ERK on both transcriptional activation and stabilization of c-Jun via CREB activation and GSK3 inactivation, respectively points to the importance of securing high levels of c-Jun expression, as illustrated in melanoma-derived cells. The fact that two pathways cooperate in increasing c-Jun expression levels reflects independent mechanisms whose impact on c-Jun expression might involve different kinetics and possibly magnitudes (Murphy and Blenis, 2006). This consideration may be important in fine-tuning the degree of inhibition of ERK activity in melanomas where ERK is among the primary targets of therapy.
In summary, the study presented here provides an undisclosed insight into the mechanism underlying the link between the ERK and JNK pathways that is mediated by ERK’s effect on c-Jun expression levels. This re-wiring has been demonstrated in human melanoma, where mutant N-RAS or B-RAF affect downstream ERK signaling. In providing an initial blueprint for re-wiring key signal transduction pathways in melanoma, our findings point to additional targets for therapy of this tumor type.
Experimental Procedures
Melanoma samples and cell lines
Melanoma samples were obtained in the operating room and were snap frozen in liquid nitrogen within 5min. of harvest. The tumor samples were stored in liquid nitrogen until processed. All human tumor samples were collected by The Tissue Retrieval Service, a shared resource of The Cancer Institute of New Jersey, following UMDNJ-RWJ Medical School IRB and HIPPA guidelines. All patients signed informed consent for the harvesting, storage, and subsequent use of their tissue samples in research related projects. All specimens were encoded so that no patient identifying data is available as per IRB and HIPPA regulations. Samples were obtained from regional dermal metastases, nodal metastases or distant sites of metastases. The status of BRAF (V599E) and N-RAS genes was assessed as described (Alsina et al., 2003; Goydos et al., 2005). Activation of the MEK/ERK pathway was confirmed by Western blot using P-ERK and P-MEK antibodies. Tumors that showed no correlation of P-ERK and P-MEK staining were not considered in our analysis. Melanoma cell lines were kindly provided by Dr. M. Herlyn and maintained as indicated (Satyamoorthy et al., 1997). Melanoma cell lines A375, Lu1205, and WM9 present the V599E mutation on the B-RAF gene (Satyamoorthy et al., 2003). The WM115 cell line presents a V599D mutation on B-RAF (Tanami et al., 2004). The WM35 cell line is wt for both Ras and B-RAF (exons 11 and 15) but presents positive P-ERK staining due to an autocrine loop (Li et al., 2003; Satyamoorthy et al., 2003). c-Jun mutant fibroblasts were kindly provided by R. Wisdom. Cells were transfected with calcium phosphate or by using LipofectAMINE PLUS Reagent (Invitrogene) following the manufacturer’s protocol.
Constructs
Constructs encoding GST-Jun1–89, MEKK and Flag-TAM67 were previously described (Bhoumik et al., 2004 and Lopez Bergami et al., 2005). The plasmids encoding c-Jun-Flag and c-Jun mutants were kindly provided by W. Kaelin. The A-CREB construct (Ahn et al., 1998) was kindly provided by C. Vinson. Fragments from the mouse RACK1 promoter were cloned from the BAC clone RP23-14F5 by PCR into the KpnI-XhoI sites of the pGL2-basic luciferase reporter (Promega). DNA sequences were confirmed by DNA sequencing. Mutation on the RACK1 promoter was introduced using the Quick Change Site-Directed Mutagenesis Kit (Stratagene) and confirmed by DNA sequencing.
In vitro kinase assay
JNK immunokinase assays were performed as described (Lopez Bergami et al., 2005). Briefly, proteins were extracted, immunoprecipitated with anti-JNK1 Ab and subjected to an in vitro kinase assay using GST-Jun1–89 as a substrate. Reaction mixtures were separated on SDS–PAGE and transferred to a nitrocellulose membrane and the phosphorylation state of GST-Jun1–89 detected and quantified using a phosphorimager. The same membranes were used for immunoblotting with anti-JNK Ab and for Ponceau S staining of GST-Jun1–89.
RNA interference
Control, c-Jun-, RACK1- and ATF1-specific siRNA oligonucleotides were obtained from Ambion. CREB siRNA sequence GAGAGAGGTCCGTCTAAGTT was cloned into pRSUPER and used to generate melanoma cells that stably express this siRNA.
Cell cycle analysis
Cell cycle distribution was assessed by propidium iodide staining as described (Ivanov et al., 2001).
Antibodies and reagents
Immunoblotting and immunohistochemistry, Luciferase assays, Real Time PCR, Chromatin immunoprecipitation and Electrophoretic mobility shift assays are detailed in Supplemental data.
Supplementary Material
Acknowledgments
We thank Dr. W. Kaelin for providing expression vectors for c-Jun mutant proteins, Ron Wisdom for the Jun-deleted cells, C. Vinson for the A-CREB construct and Michael Bittner for the TAM67 construct. We thank Sapna Vijayakumar for assistance with the Real-Time PCR determinations and Kelly Blehm for the melanoma cells expressing CREB siRNA. We also thank members of the Ronai lab for active discussions. Support by NCI grants (CA51995 to ZR; and CA76098 to MBE) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol. 1998;18:967–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsina J, Gorsk DH, Germino FJ, Shih W, Lu SE, Zhang ZG, Yang JM, Hait WN, Goydos JS. Detection of mutations in the mitogen-activated protein kinase pathway in human melanoma. Clin Cancer Res. 2003;9:6419–6425. [PubMed] [Google Scholar]
- Angel P, Hattori K, Smeal T, Karin M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Staskova Z, Draetta G, Lukas J. Molecular pathology of the cell cycle in human cancer cells. Stem Cell. 1993;11:51–58. doi: 10.1002/stem.5530110611. [DOI] [PubMed] [Google Scholar]
- Bartek J, Bartkova J, Lukas J. The retinoblastoma protein pathway and the restriction point. Curr Opin Cell Biol. 1996;8:805–814. doi: 10.1016/s0955-0674(96)80081-0. [DOI] [PubMed] [Google Scholar]
- Berns H, Humar R, Hengerer B, Kiefer FN, Battegay EJ. RACK1 is up-regulated in angiogenesis and human carcinomas. FASEB J. 2000;14:2549–2558. doi: 10.1096/fj.99-1038com. [DOI] [PubMed] [Google Scholar]
- Bhoumik A, Jones N, Ronai Z. Transcriptional switch by activating transcription factor 2-derived peptide sensitizes melanoma cells to apoptosis and inhibits their tumorigenicity. Proc Natl Acad Sci USA. 2004;101:4222–4227. doi: 10.1073/pnas.0400195101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;13:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Davies H, et al. Mutations in the B-RAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Ding Q, Xia W, et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol Cell. 2005;19:159–170. doi: 10.1016/j.molcel.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signaling pathways in cancer therapy. Nature Rev Cancer. 2003;3:11–12. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Dumont JA, Jones WD, Jr, Bitonti AJ. Inhibition of experimental metastasis and cell adhesion of B16F1 melanoma cells by inhibitors of protein kinase C. Cancer Res. 1992;52:1195–2000. [PubMed] [Google Scholar]
- Eferl R, Ricci R, Kenner L, Zenz R, David JP, Rath M, Wagner EF. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell. 2003;112:181–192. doi: 10.1016/s0092-8674(03)00042-4. [DOI] [PubMed] [Google Scholar]
- Errico ME, Staibano S, Tranfa F, et al. Expression of cyclin-D1 in uveal malignant melanoma. Anticancer Res. 2003;23:2701–2706. [PubMed] [Google Scholar]
- Fuchs SY, Dolan L, Davis RJ, Ronai Z. Phosphorylation-dependent targeting of c-Jun ubiquitination by Jun N-kinase. Oncogene. 1996;13:1531–1535. [PubMed] [Google Scholar]
- Gorden A, Osman I, Gai W, He D, Huang W, Davidson A, Houghton AN, Busam K, Polsky D. Analysis of BRAF and N-RAS mutations in metastatic melanoma tissues. Cancer Res. 2003;63:3955–3957. [PubMed] [Google Scholar]
- Goydos JS, Mann B, Kim HJ, Gabriel EM, Alsina J, Germino FJ, Shih W, Gorski DH. Detection of B-RAF and N-RAS mutations in human melanoma. J Am Coll Surg. 2005;200:362–370. doi: 10.1016/j.jamcollsurg.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Gupta P, Prywes R. ATF1 phosphorylation by the ERK MAPK pathway is required for epidermal growth factor-induced c-jun expression. J Biol Chem. 2002;277:50550–50556. doi: 10.1074/jbc.M209799200. [DOI] [PubMed] [Google Scholar]
- Ivanov VN, Bhoumik A, Krasilnikov M, Raz R, Owen-Schaub LB, Levy D, Horvath CM, Ronai Z. Cooperation between STAT3 and c-jun suppresses Fas transcription. Mol Cell. 2001;7:517–528. doi: 10.1016/s1097-2765(01)00199-x. [DOI] [PubMed] [Google Scholar]
- Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cell Signal. 2004;16:1211–1227. doi: 10.1016/j.cellsig.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Johnson R, Spiegelman B, Hanahan D, Wisdom R. Cellular transformation and malignancy induced by ras require c-jun. Mol Cell Biol. 1996;16:4504–4511. doi: 10.1128/mcb.16.8.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Laine A, Ronai Z. Ubiquitin chains in the ladder of MAPK signaling. Sci STKE. 2005;281:re5. doi: 10.1126/stke.2812005re5. [DOI] [PubMed] [Google Scholar]
- Li G, Kalabis J, Xu X, Meier F, Oka M, Bogenrieder T, Herlyn M. Reciprocal regulation of MelCAM and AKT in human melanoma. Oncogene. 2003;22:6891–6899. doi: 10.1038/sj.onc.1206819. [DOI] [PubMed] [Google Scholar]
- Liu J, Yang D, Minemoto Y, Leitges M, Rosner MR, Lin A. NFkappaB is required for UV-induced JNK activation via induction of PKCdelta. Mol Cell. 2006;21:467–480. doi: 10.1016/j.molcel.2005.12.020. [DOI] [PubMed] [Google Scholar]
- Lopez-Bergami P, Habelhah H, Bhoumik A, Zhang W, Wang LH, Ronai Z. RACK1 mediates activation of JNK by protein kinase. C Mol Cell. 2005;19:309–320. doi: 10.1016/j.molcel.2005.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bergami P, Bhoumik A, Ronai Z. Altered Signal Transduction in Melanoma. In: Hearing VJ, Leong SPL, editors. In From Melanocytes to Malignant Melanoma. Humana Press; Totowa, New Jersey, USA: 2005 b. pp. 119–147. [Google Scholar]
- Maelandsmo GM, Florenes VA, Hovig E, et al. Involvement of the pRb/p16/cdk4/cyclin D1 pathway in the tumorigenesis of sporadic malignant melanomas. Br J Cancer. 1996;73:909–916. doi: 10.1038/bjc.1996.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T, Ono T, Albertson DG, Pinkel D, Bastian BC. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. 2003;95:1878–1890. doi: 10.1093/jnci/djg123. [DOI] [PubMed] [Google Scholar]
- Mathas S, Hinz M, et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J. 2002;21:4104–4113. doi: 10.1093/emboj/cdf389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli E, Banfi P, Sala E, Sensi M, Supino R, Zunino F, Gambetta RA. Effect of protein kinase C inhibitors on invasiveness of human melanoma clones expressing different levels of protein kinase C isoenzymes. Int J Cancer. 1994;57:281–286. doi: 10.1002/ijc.2910570225. [DOI] [PubMed] [Google Scholar]
- Minden A, Lin A, Smeal B, Derijard M, Cobb R, Davis R, Karin M. c-Jun N-terminal phosphorylation correlates with activation of the JNK subgroup but not the ERK subgroup of mitogen-activated protein kinase. Mol Cell Biol. 1994;14:6683–6688. doi: 10.1128/mcb.14.10.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina DM, Grewal S, Bardwell L. Characterization of an ERK-binding domain in microphthalmia-associated transcription factor and differential inhibition of ERK2-mediated substrate phosphorylation. J Biol Chem. 2005;280:42051–42060. doi: 10.1074/jbc.M510590200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton S, Davis RJ, McLaren A, Cohen P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 2003;22:3876–3886. doi: 10.1093/emboj/cdg388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Nyormoi O, Bar-Eli M. Transcriptional regulation of metastasis-related genes in human melanoma. Clin Exp Metastasis. 2003;20:251–263. doi: 10.1023/a:1022991302172. [DOI] [PubMed] [Google Scholar]
- Oka M, Kikkawa U. Protein kinase C in melanoma. Cancer Metastasis Rev. 2005;24:287–300. doi: 10.1007/s10555-005-1578-8. [DOI] [PubMed] [Google Scholar]
- Park CH, Lee MJ, Ahn J, Kim S, Kim HH, Kim KH, Eunm HC, Chung JH. Heat shock-induced matrix metalloproteinase (MMP)-1 and MMP-3 are mediated through ERK and JNK activation and via an autocrine interleukin-6 loop. J Invest Dermatol. 2004;123:1012–1019. doi: 10.1111/j.0022-202X.2004.23487.x. [DOI] [PubMed] [Google Scholar]
- Pollock PM, Meltzer PS. A genome-based strategy uncovers frequent BRAF mutations in melanoma. Cancer Cell. 2002;2:5–7. doi: 10.1016/s1535-6108(02)00089-2. [DOI] [PubMed] [Google Scholar]
- Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D. Cloning of an intracellular receptor for protein kinase C, a homolog of the beta subunit of G proteins. Proc Natl Acad Sci USA. 1994;91:839–843. doi: 10.1073/pnas.91.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah M, Courilleau D, Mester J, Redeuilh G. Estrogen induction of the cyclin D1 promoter : involvement of a cAMP response-like element. Proc Natl Acad Sci USA. 1999;96:11217–11222. doi: 10.1073/pnas.96.20.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A, Fujii G, Sato Y, Gotoh M, Sakamoto M, Toda G, Hirohashi S. Detection of genes expressed in primary colon cancers by in situ hybridisation, overexpression of RACK 1. Mol Pathol. 2002;55:34–39. doi: 10.1136/mp.55.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter ER, Yeo UC, von Stemm A, et al. Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res. 2002;62:3200–3206. [PubMed] [Google Scholar]
- Schechtman D, Mochly-Rosen D. Adaptor proteins in protein kinase C-mediated signal transduction. Oncogene. 2001;20:6339–6347. doi: 10.1038/sj.onc.1204778. [DOI] [PubMed] [Google Scholar]
- Selzer E, Okamoto I, Lucas T, Kodym R, Pehamberger H, Jansen B. Protein kinase C isoforms in normal and transformed cells of the melanocytic lineage. Melanoma Res. 2002;12:201–209. doi: 10.1097/00008390-200206000-00003. [DOI] [PubMed] [Google Scholar]
- Seo J, Shneiderman B. Interactively Exploring Hierarchical Clustering Results. IEEE Computer. 2002;35:80–86. [Google Scholar]
- Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:131–136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- Sprowles A, Robinson D, Wu YM, Kung HJ, Wisdom R. c-Jun controls the efficiency of MAP kinase signaling by transcriptional repression of MAP kinase phosphatases. Exp Cell Res. 2005;308:459–468. doi: 10.1016/j.yexcr.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Tanami H, Imoto I, Hirasawa A, Yuki Y, Sonoda I, Inoue J, Yasui K, Misawa-Furihata A, Kawakami Y, Inazawa J. Involvement of overexpressed wild-type BRAF in the growth of malignant melanoma cell lines. Oncogene. 2004;23:8796–8804. doi: 10.1038/sj.onc.1208152. [DOI] [PubMed] [Google Scholar]
- Utikal J, Udart M, Leiter U, Peter RU, Krahn G. Additional Cyclin D(1) gene copies associated with chromosome 11 aberrations in cutaneous malignant melanoma. Int J Oncol. 2005;26:597–605. [PubMed] [Google Scholar]
- Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol. 2003;13:680–685. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- Vogt PK. Jun, the oncoprotein. Oncogene. 2001;20:2365–2377. doi: 10.1038/sj.onc.1204443. [DOI] [PubMed] [Google Scholar]
- Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12:14–21. doi: 10.1016/s0959-437x(01)00258-1. [DOI] [PubMed] [Google Scholar]
- Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol. 2002;22:2871–2881. doi: 10.1128/MCB.22.8.2871-2881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.