

Abstract
This study was performed to determine whether adult male patients with Fabry disease who demonstrate a continuing decline in renal function despite 2 to 4 yr of conventionally dosed agalsidase alfa therapy (0.2 mg/kg every other week [EOW]) show an improved slope of decline with weekly administration using the same dosage. Eleven (27%) of 41 adult male patients with Fabry disease who participated in long-term agalsidase alfa clinical trials and who had demonstrated a slope of decline in estimated GFR (eGFR) of ≥5 ml/min per 1.73 m2/yr while receiving long-term treatment with agalsidase alfa at the currently recommended dosage of 0.2 mg/kg, infused EOW, were enrolled in this open-label, prospective study. Patients were switched from EOW to weekly infusions and followed for an additional 24 mo. Before switching to weekly dosing, eGFR was 53.7 ± 6.3 ml/min per 1.73 m2 (mean ± SEM), and mean rate of change in eGFR was −8.0 ± 0.8 ml/min per 1.73 m2/yr. During the 24-mo follow-up period after switching to weekly dosing, the mean rate of change in eGFR was observed to slow to −3.3 ± 1.4 ml/min/1.73 m2/yr (P = 0.01 versus EOW). After switching to weekly dosing, three patients demonstrated an improvement in eGFR and six patients demonstrated a slowing in the rate of eGFR decline; only two patients failed to improve their eGFR slope. A multiple regression model confirmed that the weekly infusion regimen was the strongest explanatory variable for the change in eGFR (P = 0.0008), with a weaker contribution from the concomitant use of angiotensin converting enzyme inhibitors/angiotensin receptor blockers (P = 0.02). These results suggest that weekly infusions of agalsidase alfa at a dosage of 0.2 mg/kg may be beneficial in the subgroup of patients who have Fabry disease and whose kidney function continues to decline after 2 to 4 yr or more of standard EOW dosing.
Fabry disease is an X-linked glycosphingolipid disorder that is caused by an insufficient activity of the lysosomal enzyme α-galactosidase A (GALA) (1). This deficiency results in a systemic accumulation of α-D-galactosyl conjugates, particularly globotriaosylceramide (Gb3), in vascular endothelial cells, pericytes, and smooth muscle cells of the vascular system; renal epithelial cells; myocardial cells; and dorsal root ganglion neuronal cells (2). The incidence of Fabry disease in male individuals has been estimated to be 1:117,000 births (3).
Hemizygous male patients with Fabry disease have a subtle but characteristic facial appearance (4). Clinical onset of the disease in both male hemizygotes and female heterozygotes typically occurs during childhood or adolescence with recurrent episodes of severe, often debilitating neuropathic pain in the extremities (5). However, the age of symptom onset and the age of diagnosis tend to be approximately 10 yr later in female compared with male individuals (6). Hypohidrosis and neuropathic pain contribute to poor tolerance to exercise and to heat (7,8). Proteinuria and progressive renal deterioration develop in nearly all male patients with Fabry disease (9,10) and are the result of intraglomerular deposition of Gb3, which is associated with histopathologic findings of mesangial widening and ultimately segmental and global glomerulosclerosis (10–12). Hypertrophic cardiomyopathy and coronary and cerebrovascular disease also contribute to early death in men at a median age of death of 50 to 55 yr (9).
In recent years, enzyme replacement therapy (ERT) has become part of the standard medical care for Fabry disease (13,14). Although ERT reduces neuropathic pain and improves thermal sensing threshold (15) and gastrointestinal symptoms (16), its long-term effects on the progressive kidney dysfunction, the incidence of stroke, and progressive cardiac disease have not been established (13,15,17,18). Patients in controlled clinical trials showed significant reductions in certain biomarkers: Approximately 50% lowering of Gb3 levels in plasma as well as of Gb3 in urinary sediment and in renal interstitial capillary endothelium (13,19). Reduction of Gb3 storage in renal vascular endothelial cells was associated with an overall improvement in glomerular morphology, as assessed by a significant decrease in the percentage of glomeruli with mesangial widening and a significant increase in the percentage of normal glomeruli (13). Despite this histologic evidence of a renal benefit during ERT, conclusive evidence that ERT slows the progressive decline in renal function in Fabry disease has not been reported. In the long-term, open-label extensions of pivotal clinical trials, renal function remained stable in the majority of patients with Fabry disease who were treated with agalsidase alfa or agalsidase beta for up to 4 yr (20,21). Most of the patients in these studies had relatively normal kidney function at baseline and might not be expected to experience a decline in kidney function during the studies. However, there are patients who demonstrated a continued progressive decline in renal function despite administration of either form of ERT (agalsidase alfa or beta) administered every other week (EOW) (20,21). Nevertheless, the rate of decline in the subgroup of patients with more severe renal dysfunction at pretreatment baseline did seem to be slower with agalsidase alfa treatment compared with the rate of decline in a comparable cohort of patients with untreated Fabry disease (10,20). In this study, we report the results of a clinical trial that was designed to test the hypothesis that increasing the frequency of ERT with 0.2 mg/kg agalsidase alfa from EOW to weekly could improve the slope of decline of estimated GFR (eGFR) in patients who have Fabry disease and whose eGFR had continued to decline at a relatively high rate despite treatment with agalsidase alfa 0.2 mg/kg EOW for 2 to 4 yr.
Materials and Methods
Study Design
This study was an open-label, prospective clinical trial. The screened population consisted of 41 adult hemizygous male patients who had Fabry disease and had received agalsidase alfa (Replagal; Shire Human Genetic Therapies, Cambridge, MA) at 0.2 mg/kg EOW in long-term (2 to 4 yr) clinical trials. Patients who demonstrated a rate of decline of eGFR of 5 ml/min per 1.73 m2 or more per year while receiving this conventional EOW dosing were eligible for inclusion in this study. The choice of this threshold for eGFR slope was based on its being <50% of the published mean rate of decline in eGFR of −12.2 ml/min per 1.73 m2/yr after the onset of chronic renal failure (defined by reaching a serum creatinine of >1.5 mg/dl) in untreated adult male patients with Fabry disease [10]). Patients who were included in this study could also have been receiving medications for hypertension and/or angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARB) for renoprotection. However, these treatment regimens must have remained stable for at least the 6 mo before beginning weekly agalsidase alfa therapy. ACE inhibitors or ARB were not to be initiated during the study period unless rapid deterioration of kidney function mandated their use. All patients gave written informed consent to the protocol, which was approved by the institutional review board of the National Institute of Neurologic Disorders and Stroke.
Intervention
Patients received weekly doses of agalsidase alfa at 0.2 mg/kg body wt administered over 40 min. The initial evaluation and the first weekly infusion were conducted at the National Institutes of Health. Thereafter, patients received their weekly infusions of agalsidase alfa at their local clinic or hospital under physician supervision.
Efficacy Assessments
Blood samples were obtained at baseline and after 1, 2, 4, 8, 12, 24, 36, 48, 62, 76, 90, and 104 wk of treatment for estimation of GFR, determination of plasma Gb3 levels, and measurement of the presence of anti–agalsidase alfa antibodies. Twenty-four-hour urine collections were obtained at baseline and every 6 mo thereafter for measurement of urine sediment Gb3 levels and urine protein levels. Plasma and urine sediment Gb3 levels were analyzed at Shire Human Genetic Therapies as described previously (22,23). Anti–agalsidase alfa antibodies were determined by ELISA at Shire Human Genetic Therapies, as described previously (13).
The primary outcome measure was mean rate of change in eGFR during the 2-yr weekly treatment period compared with the rate of change of eGFR during the preceding 2 to 4 yr of EOW infusions in the same patients. eGFR was calculated using the four-variable Modification of Diet in Renal Disease (MDRD) formula (24). Sweat function was evaluated at baseline and periodically during the study by the quantitative sudomotor axon reflex test as described previously (15).
Safety
All patients underwent a baseline evaluation that included a physical and neurologic examination, clinical laboratory tests (including complete blood count and platelet count), and urinalysis. Throughout the study, vital signs, weight, and use of concomitant medications were assessed at the weekly visits, as were any treatment-emergent adverse events (AE). The baseline evaluations were repeated periodically throughout the study. All laboratory parameters with exception of plasma Gb3 and anti–agalsidase alfa antibody determinations were done at the clinical-pathologic laboratory of the Clinical Center of the National Institutes of Health.
Statistical Analyses
Methods of descriptive statistics were applied. The slope of change of eGFR during EOW and weekly treatment in each patient was determined by linear regression using all available data points. The slope of change before and after switching from EOW to weekly dosing was compared with a paired t test. Testing for normal distribution of data was performed using the method of Kolmogorov and Smirnov. To account for possible effects of anti–agalsidase alfa antibodies or concomitant ACE inhibitors or ARB, we fitted a multivariate model, using eGFR as the outcome measure and ERT frequency (weekly/biweekly), antibody status (negative, transiently positive, permanently positive), ACE inhibitor/ARB status, and mean arterial pressure (MAP) as explanatory variables. MAP was calculated as diastolic BP + (systolic BP − diastolic BP)/3. The model was then tested for multicollinearity. Other statistical tests are noted in the text. All analyses were two-tailed using a significance level of 0.05. All values are expressed as means ± SD unless otherwise noted.
Results
Patients and Demographics
Twelve (29%) of 41 adult male patients with Fabry disease who were screened for inclusion had demonstrated a rate of decrease in eGFR in excess of 5 ml/min per 1.73 m2/yr during standard EOW treatment with agalsidase alfa and were enrolled in this study. All patients had the classic form of Fabry disease with no residual GALA activity. All patients were white: 10 of non-Hispanic and two of Hispanic ethnicity. Eleven of 12 patients completed the study. The 12th patient was excluded from the efficacy analysis because he reached ESRD shortly after starting weekly dosing. This patient’s creatinine clearance had already declined to 15 ml/min at the time that he switched to weekly dosing, and he started on peritoneal dialysis after only 3 mo of weekly dosing. This patient died of cardiac causes shortly after the initiation of dialysis. Overall compliance with weekly dosing was high, with only 4% of infusions being missed. The median age of patients was 44 yr (range 24 to 53 yr).
Effect on eGFR
The individual eGFR and the slopes of change of eGFR are presented in Tables 1 and 2 and Figure 1. Before beginning any therapy with agalsidase alfa, mean eGFR was 77.8 ± 30.4 ml/min per 1.73 m2, and at the time of switching from EOW to weekly dosing, mean eGFR had declined to 53.7 ± 21.0 ml/min per 1.73 m2. During EOW dosing, the mean rate of change of eGFR was −8.0 ± 2.8 ml/min per 1.73 m2/yr (−0.67 ± 0.23 ml/min per 1.73 m2/mo), and during 2 yr of weekly dosing, the mean rate of change of eGFR was observed to slow to −3.3 ± 4.7 ml/min per 1.73 m2/yr (−0.27 ± 0.40 ml/min per 1.73 m2/mo; P = 0.01, paired t test).
Table 1.
eGFR before and after 2 yr of weekly treatment with agalsidase alfaa
| Patient | Age (yr) | eGFR (ml/min per 1.73 m2) Pre-EOW | Duration of EOW (yr) | eGFR (ml/min per 1.73 m2)
|
|
|---|---|---|---|---|---|
| Weekly Study Entry | Weekly Study 2 yr | ||||
| 1 | 43.7 | 68.7 | 4.0 | 28.7 | 17.8 |
| 2 | 49.9 | 48.2 | 4.5 | 24.6 | 15.9 |
| 3 | 48.5 | 87.1 | 4.0 | 62.5 | 83.9 |
| 6 | 24.1 | 142.6 | 4.5 | 79.0 | 60.1 |
| 7 | 36.8 | 82.5 | 4.5 | 66.1 | 79.3 |
| 8 | 44.2 | 112.5 | 2.5 | 77.2 | 76.5 |
| 9 | 52.9 | 51.1 | 4.5 | 42.2 | 41.9 |
| 10 | 45.1 | 70.0 | 2.0 | 63.4 | 49.5 |
| 11 | 41.1 | 59.7 | 2.0 | 47.4 | 38.9 |
| 12 | 38.0 | 40.3 | 2.0 | 25.0 | 18.8 |
| 14 | 31.5 | 93.6 | 2.5 | 74.8 | 73.9 |
| Mean | 41.4 | 77.8 | 3.4 | 53.7 | 50.6 |
| SD | 8.4 | 30.4 | 1.1 | 21.0 | 26.0 |
eGFR, estimated GFR; pre-EOW, baseline before beginning every other week dosing.
Table 2.
Treatment results in 11 hemizygous patients who had Fabry disease and received EOW (2 to 4 yr) versus weekly (2 yr) ERT with 0.2 mg/kg agalsidase alfaa
| Patient | ERT Frequency | eGFR Slope (ml/min per 1.73 m2/mo) | Agalsidase alfa IgG Antibody Statusb | MAP (mmHg) | ACEi/ARB Statusc | Protein Excretion (mg/d) |
|---|---|---|---|---|---|---|
| 1 | EOW | −0.85 | 2 | 94 | 0 | 1785 |
| Weekly | −0.22 | 1 | 93 | 1 | 3302 | |
| 2 | EOW | −0.56 | 0 | 64 | 0 | 1025 |
| Weekly | −0.57 | 0 | 71 | 1 | 715 | |
| 3 | EOW | −0.41 | 1 | 94 | 0 | 123d |
| Weekly | 0.29 | 0 | 87 | 0 | 215 | |
| 6 | EOW | −0.54 | 2 | 84 | 0 | 3123 |
| Weekly | −1.14 | 2 | 80 | 1 | 1344 | |
| 7 | EOW | −0.43 | 2 | 104 | 0 | 264 |
| Weekly | 0.14 | 2 | 104 | 1 | 504 | |
| 8 | EOW | −0.78 | 0 | 76 | 0 | 199d |
| Weekly | −0.12 | 0 | 74 | 0 | 99 | |
| 9 | EOW | −0.59 | 0 | 96 | 1 | 225 |
| Weekly | 0.07 | 0 | 94 | 1 | 220 | |
| 10 | EOW | −0.51 | 0 | 73 | 0 | 1023 |
| Weekly | −0.30 | 0 | 89 | 1 | 3172 | |
| 11 | EOW | −0.97 | 0 | 95 | 1 | 2577 |
| Weekly | −0.58 | 0 | 89 | 1 | 999 | |
| 12 | EOW | −1.12 | 2 | 86 | 0 | 3774 |
| Weekly | −0.26 | 2 | 72 | 1 | 1476 | |
| 14 | EOW | −0.56 | 0 | 78 | 0 | 2220 |
| Weekly | −0.29 | 0 | 93 | 1 | 1938 |
ACEi/ARB, angiotensin-converting enzyme inhibitors/angiotensin receptor blockers; ERT, enzyme replacement therapy; MAP, mean arterial pressure.
0, none; 1, transiently positive; 2, permanently positive.
0, no; 1, yes.
Previous observation carried forward.
Figure 1.
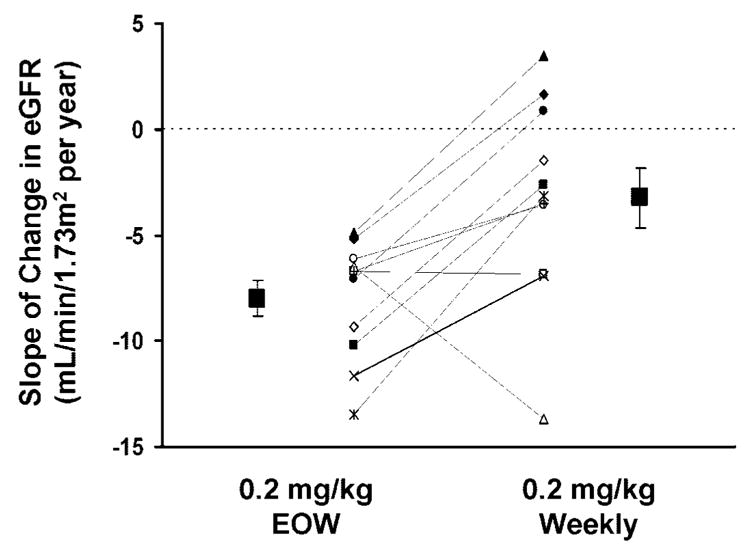
The effect of changing the dosing frequency of agalsidase alfa from every other week (EOW) to weekly in patients whose estimated GFR (eGFR) declines at >5 ml/min per 1.73 m2/yr during long-term EOW therapy. Each symbol represent an individual patient, and the squares represent the mean ± SEM during EOW and weekly dosing.
Multivariate analysis revealed that the observed slope of change in eGFR could be explained by the following equation: eGFR slope = −2.353 + 0.6486 × ERT frequency − 0.1184 × antibody status − 0.4337 × ACE inhibitor or ARB status + 0.01417 × MAP (P = 0.006, R2 = 0.55, n = 11).
The variables ERT frequency (weekly versus EOW; P = 0.0008), ACE-I/ARB medication (P = 0.02), and MAP (P = 0.03) all contributed significantly to this model. Antibody status had no significant effect.
Proteinuria and Metabolic Effects
Before beginning EOW dosing with agalsidase alfa, six patients demonstrated proteinuria in excess of 300 mg/24 h. For the entire study population, mean baseline urinary protein excretion was 1217 ± 1246 mg/24 h (range 122 to 3909 mg/24 h; median 1027 mg/24 h). Individual urinary protein excretion measurements are shown in Table 2 and Figure 2. Mean urinary protein excretion was 1485 ± 1295 mg/24 h after long-term EOW dosing and was not significantly decreased after 2 yr of weekly dosing (1271 ± 1131 mg/24 h; P = 0.41, Wilcoxon sign-rank test).
Figure 2.

Effect of EOW and weekly agalsidase alfa treatment on urinary protein excretion. The individual symbols represent the same patients depicted in Figure 1.
Baseline urine sediment Gb3 levels were elevated in all patients. The response of urine sediment Gb3 to EOW and weekly dosing of agalsidase alfa is shown in Figure 3. During EOW dosing, average urine sediment Gb3 had decreased from 2839 ± 1862 (median 2490) to 1318 ± 901 (median 1515) nmol/g creatinine. After 2 yr of weekly dosing, mean urine sediment Gb3 had decreased to 913 ± 340 (median 395) nmol/g creatinine. The urine sediment Gb3 levels measured during EOW or weekly dosing were significantly less than baseline levels (P < 0.001) but were not significantly different from each other (P = 0.12, Wilcoxon sign-rank test).
Figure 3.
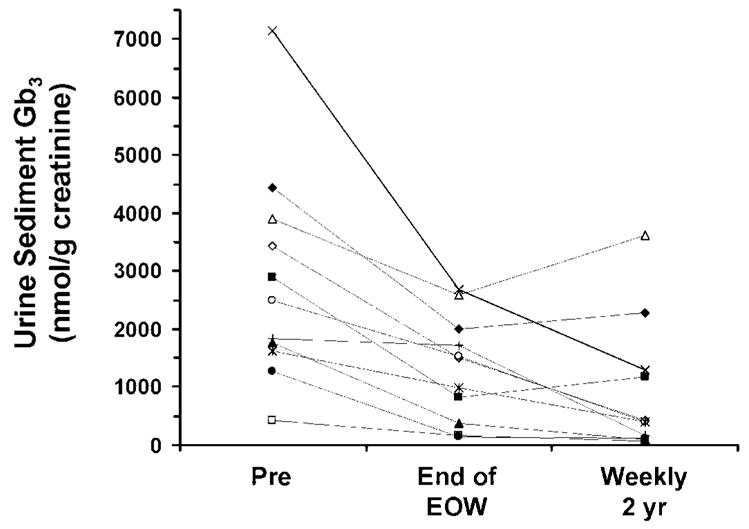
The effect of EOW and weekly agalsidase alfa on urine sediment globotriaosylceramide (Gb3) in male patients with Fabry disease. Pre indicates value before the start of EOW dosing. The individual symbols represent the same patients depicted in Figure 1.
Before beginning EOW, agalsidase alfa therapy plasma Gb3 was elevated in all patients, and mean plasma Gb3 was 11.8 ± 3.6 nmol/ml. After 2 to 4 yr of EOW dosing, plasma Gb3 had declined to 4.2 ± 1.6 nmol/ml. The small further decline to a mean value of 3.7 ± 1.3 nmol/ml during 2 yr of weekly dosing was not statistically different from that seen at the end of EOW dosing. The plasma Gb3 levels measured during EOW or weekly dosing were significantly less than baseline values (P < 0.05) but were not significantly different from each other (ANOVA followed by Newman-Keuls test).
Sweat Function
At the time of switching to weekly dosing, the average sweat excretion before receiving the infusion of agalsidase alfa (7 d after the previous dose) was 0.29 ± 0.22 μl/mm2. No significant differences from baseline were observed at any time during the study. At the final quantitative sudomotor axon reflex test evaluation (2 yr, n = 9; 1.5 yr, n = 2), average sweat volume was 0.76 ± 0.98 μl/mm2 (P = 0.55, paired t test). Increases in sweat volume were observed in seven patients, and decreases were seen in four patients.
Safety
AE
No infusion reactions to agalsidase alfa were observed after patients were switched from EOW to weekly dosing. The AE that occurred while on weekly dosing all were expected manifestations or complications of Fabry disease. Serial routine blood tests did not suggest any agalsidase alfa–related laboratory abnormalities.
Serious AE
A total of 10 serious AE (SAE) were reported in six patients. None was deemed by the primary investigator to be related to agalsidase alfa. In addition to the excluded patient described in Patients and Demographics, who accounted for a total of five of the 10 SAE, another patient was hospitalized for chest pain and right leg pain, which resolved after 12 h and was thought possibly to represent a transient ischemic attack. Another patient had an overnight hospital admission after a syncopal episode that occurred while he was lifting weights in a gym. An additional patient was admitted overnight twice, once for nausea and vomiting and once for a pain crisis. One other patient underwent an elective mitral valve annuloplasty.
Antibody Status
Five patients had developed positive IgG anti–agalsidase alfa antibody titers with EOW dosing. No additional patients developed IgG anti–agalsidase alfa antibodies after switching to weekly dosing. In the five antibody-positive patients, there was no trend for a change in titer in either direction as a result of increasing the frequency of agalsidase alfa infusions. No IgE anti–agalsidase alfa antibodies were found with either EOW or weekly dosing.
Discussion
In this study, increasing the frequency of dosing with agalsidase alfa from EOW to weekly in a subgroup of patients with Fabry disease whose eGFR continued to decline at rapid rate while being treated EOW significantly slowed the rate of decline in eGFR. Nine (82%) of 11 patients demonstrated either a positive eGFR slope (n = 3, 27%) or a slowing in their rate of decline (i.e., less negative eGFR slope; n = 6, 55%) after switching to weekly administration of agalsidase alfa. Only two patients failed to improve their slope after switching from EOW to weekly dosing.
The original dosage and frequency of dosing of agalsidase alfa (0.2 mg/kg EOW) were chosen on the basis of a theoretical calculation of the metabolic load of Gb3 produced in the body and the specific catalytic activity of agalsidase alfa, coupled with the demonstration that a single infusion of enzyme markedly reduced Gb3 levels in the liver and urinary sediment in patients with Fabry disease (23). In addition, the EOW dosing regimen was chosen for practical reasons and was based on previous experience with ERT in Gaucher disease (25). Despite the clinical benefits reported here with EOW dosing of agalsidase alfa, this EOW frequency regimen may be suboptimal in some patients. Less than 1% of the maximal plasma concentration of agalsidase alfa is detectable 8 h after infusion (data on file, Shire Human Genetic Therapies), and the intracellular half-life of the enzyme is estimated to be 24 to 48 h (23). Therefore, little or no enzyme would be expected to be present in the targeted tissues during the second postinfusion week, leaving susceptible cells “unprotected.” This conclusion may be consistent with clinical observations by some patients who report a return of some of their Fabry disease symptoms, such as neuropathic pain and lack of sweating as well as loss of energy or vitality in the second week after their EOW infusions compared with the first postinfusion week. Some of our patients in this study also reported improvement in these symptoms with weekly infusion of enzyme.
The onset of renal insufficiency is significantly delayed in hemizygous male patients with a small but measurable residual GALA enzyme activity (e.g., 2 to 8% of normal) (10). This delay suggests that the continuous presence of even low enzyme activity levels in cells may be sufficient to reverse substantially the metabolic defect and slow or delay the progression to renal failure in male patients with Fabry disease. Therefore, more frequent weekly dosing would be expected to help maintain intracellular enzyme levels in the kidney as well as in other target organ tissues.
Complete or partial clearance of Gb3 from renal interstitial capillary endothelial cells as well as other renal cell types has been reported with both agalsidase alfa and agalsidase beta therapy (13,19). Both agalsidase alfa and agalsidase beta have also been reported to stabilize renal function in patients who had Fabry disease and participated in long-term, open-label extension studies of their respective randomized clinical trials (20,21). However, in both of those studies, the vast majority of patients enrolled had relatively normal renal function (i.e., normal eGFR) at baseline. In the large Fabry Outcome Survey (FOS), which records postmarketing experience with agalsidase alfa, multivariate analysis of data from 201 male and female patients with Fabry disease showed that the duration of treatment with agalsidase alfa was a significant factor in affecting a slowing in the rate of decline in eGFR (26). A subset of 12 patients in the FOS study who had stage 2 chronic kidney disease at the time of starting ERT had serial eGFR estimations (26). During the year before beginning agalsidase alfa, mean eGFR had dropped from 83.7 ± 3.7 to 71.9 ± 2.2 ml/min per 1.73 m2 (mean ± SEM; P < 0.05), representing an average rate of decline in GFR of 11.8 ± 2.5 ml/min per 1.73 m2/yr. During 1 yr of agalsidase alfa treatment, eGFR remained constant (mean 72.8 ± 2.5 ml/min per 1.73 m2), providing evidence of a renoprotective effect of ERT. In the subset of eight patients in the FOS study (26), who had stage 3 chronic kidney disease at baseline, eGFR had declined by 3.0 ± 2.4 ml/min per 1.73 m2 per in the year before beginning agalsidase alfa but continued to decline in the first year of ERT by a similar rate (−4.5 ± 1.5 ml/min per 1.73 m2/yr). However, it is noteworthy that this slope of decline in eGFR while on agalsidase alfa therapy was still slower than the slope (−12.2 ml/min per yr) reported in a comparable cohort of untreated stage 3 patients reported by Branton et al. (10). It is possible that these patients might also have benefited by switching from EOW to weekly dosing of agalsidase alfa. In addition, one might hypothesize that initiation of ERT at an early stage may prevent progression to severe organ dysfunction.
Further evidence of a renoprotective effect of ERT in Fabry disease comes from a recent randomized, placebo-controlled clinical trial of agalsidase beta (1.0 mg/kg, EOW) in patients with elevated serum creatinine levels (≥1.2 and <3.0 mg/dl) or reduced eGFR (<80 ml/min) at baseline (27). Although the results favored agalsidase beta in the intention-to-treat analysis of time to first clinical event (a composite of renal, cardiac, or cerebrovascular events and death) adjusted for baseline proteinuria, the trend failed to reach statistical significance (hazard ratio 0.47; 95% confidence interval 0.21 to 1.03; P = 0.06). When only the 74 protocol-adherent patients were analyzed, the reduction in risk for a clinical event with agalsidase beta was statistically significant. A post hoc secondary analysis suggested that the apparent benefit on the primary outcome was stronger in patients with baseline serum creatinine levels ≤1.5 mg/dl or with baseline eGFR >55 ml/min per 1.73 m2.
It was somewhat surprising that three patients (patients 3, 7, and 9) demonstrated an increase in eGFR after switching from EOW to weekly dosing of agalsidase alfa, because the renal pathologies are thought to be irreversible (11) and therefore the optimum response that would be expected is the stabilization of kidney function. However, these three patients had low proteinuria measurements upon switching to weekly dosing (Table 2). Proteinuria is a well-established prognostic factor for progression of kidney disease in type 2 diabetes (28) and was recently shown to be an important risk factor for progression of kidney dysfunction in Fabry disease (27). Although this observation suggests that patients with low baseline proteinuria may be better candidates for a positive response after switching to weekly dosing, five of seven patients with baseline proteinuria in excess of 1000 mg/24 h also showed a substantial slowing of the loss of eGFR after switching to weekly dosing.
Changing the dosing frequency from EOW to weekly had no substantial effect on either plasma or urine sediment Gb3 levels, which had already been maximally reduced during EOW dosing. Neither plasma Gb3 levels nor urine sediment Gb3 levels was significantly correlated with any signs and symptoms of Fabry disease (29). In addition, no studies have yet been reported to show a correlation between the magnitude of decrease in these Gb3 levels and clinical efficacy. Therefore, the decrease in plasma and urine sediment Gb3 levels that was seen in this study is an indication of in vivo enzyme activity and not evidence of clinical efficacy (29,30)
Individualized dosing of ERT has been reported and is favored in Gaucher disease (31,32), another lysosomal storage disorder, but the appropriate dosage that results in an optimal clinical response remains controversial (33,34). Individualization of ERT in Fabry disease is a paradigm often proposed, but the lack of an appropriate surrogate marker in Fabry disease by which to assess efficacy imposes practical challenges for the clinician (17). In this study, we have demonstrated the use of a true clinical outcome measure (eGFR slope) to assess individualized ERT in patients with Fabry disease.
Study Limitations
This study was exploratory in nature and was further limited because of the open-label design and the relatively small number of patients studied. The patients studied were referred to the National Institutes of Health, a tertiary biomedical research institution, and may not necessarily reflect the nonreferral Fabry population. As demonstrated in the multivariate analysis, changes in concomitant medications (ACE inhibitors or ARB) may have influenced the outcome, such that the results may not be solely due to the change in the frequency of enzyme infusions but may be due, at least in part, also to optimization of concomitant mediations. Renal biopsies may add to the assessment of the effect of increasing the dosing frequency on renal function, but this invasive procedure may hinder the ability to recruit patients into this study. In addition, neither the amount nor the type of glomerular Gb3 deposition has been correlated with degree of renal dysfunction (35). Although Fabry disease is rare and exhibits a considerable phenotypic heterogeneity, both of which make it challenging to find and study a uniformly affected population of patients with comparable renal involvement, this study provides a first step in systematically approaching the individualization of ERT in Fabry disease.
Acknowledgments
This work was supported in part by the Intramural Program of the National Institute of Neurologic Disorders and Stroke and by Shire Human Genetic Therapies.
We acknowledge the editorial assistance provided by Edward Weselcouch, PhD. We thank Dr Jeffrey Kopp for helpful suggestions.
Footnotes
Disclosures
None.
References
- 1.Brady R, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967;276:1163–1167. doi: 10.1056/NEJM196705252762101. [DOI] [PubMed] [Google Scholar]
- 2.Brady RO, Schiffmann R. Clinical features of and recent advances in therapy for Fabry disease. JAMA. 2000;284:2771–2775. doi: 10.1001/jama.284.21.2771. [DOI] [PubMed] [Google Scholar]
- 3.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 4.Ries M, Moore DF, Robinson CJ, Tifft CJ, Rosenbaum KN, Brady RO, Schiffmann R, Krasnewich D. Quantitative dys-morphology assessment in Fabry disease. Genet Med. 2006;8:96–101. doi: 10.1097/01.gim.0000200950.25118.dd. [DOI] [PubMed] [Google Scholar]
- 5.Ries M, Ramaswami U, Parini R, Lindblad B, Whybra C, Willers I, Gal A, Beck M. The early clinical phenotype of Fabry disease: A study on 35 European children and adolescents. Eur J Pediatr. 2003;162:767–772. doi: 10.1007/s00431-003-1299-3. [DOI] [PubMed] [Google Scholar]
- 6.MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38:769–775. doi: 10.1136/jmg.38.11.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cable WJ, Kolodny EH, Adams RD. Fabry disease: Impaired autonomic function. Neurology. 1982;32:498–502. doi: 10.1212/wnl.32.5.498. [DOI] [PubMed] [Google Scholar]
- 8.Ries M, Gupta S, Moore DF, Sachdev V, Quirk JM, Murray GJ, Rosing DR, Robinson C, Schaefer E, Gal A, Dambrosia JM, Garman SC, Brady RO, Schiffmann R. Pediatric Fabry disease. Pediatrics. 2005;115:e344–e355. doi: 10.1542/peds.2004-1678. [DOI] [PubMed] [Google Scholar]
- 9.MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001;38:750–760. doi: 10.1136/jmg.38.11.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, Goldfarb L, Brady RO, Balow JE, Austin HA, 3rd, Kopp JB. Natural history of Fabry renal disease: Influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore) 2002;81:122–138. doi: 10.1097/00005792-200203000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Alroy J, Sabnis S, Kopp JB. Renal pathology in Fabry disease. J Am Soc Nephrol. 2002;13(Suppl):S134–S138. [PubMed] [Google Scholar]
- 12.Ries M, Bettis KE, Choyke P, Kopp JB, Austin HA, 3rd, Brady RO, Schiffmann R. Parapelvic kidney cysts: A distinguishing feature with high prevalence in Fabry disease. Kidney Int. 2004;66:978–982. doi: 10.1111/j.1523-1755.2004.00846.x. [DOI] [PubMed] [Google Scholar]
- 13.Schiffmann R, Kopp JB, Austin HA, 3rd, Sabnis S, Moore DF, Weibel T, Balow JE, Brady RO. Enzyme replacement therapy in Fabry disease: A randomized controlled trial. JAMA. 2001;285:2743–2749. doi: 10.1001/jama.285.21.2743. [DOI] [PubMed] [Google Scholar]
- 14.Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001;345:9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- 15.Schiffmann R, Floeter MK, Dambrosia JM, Gupta S, Moore DF, Sharabi Y, Khurana RK, Brady RO. Enzyme replacement therapy improves peripheral nerve and sweat function in Fabry disease. Muscle Nerve. 2003;28:703–710. doi: 10.1002/mus.10497. [DOI] [PubMed] [Google Scholar]
- 16.Dehout F, Roland D, Treille de Granseigne S, Guillaume B, Van Maldergem L. Relief of gastrointestinal symptoms under enzyme replacement therapy in patients with Fabry disease. J Inherit Metab Dis. 2004;27:499–505. doi: 10.1023/B:BOLI.0000037342.59612.69. [DOI] [PubMed] [Google Scholar]
- 17.Ries M, Schiffmann R. Fabry disease: Angiokeratoma, biomarker, and the effect of enzyme replacement therapy on kidney function. Arch Dermatol. 2005;141:904–905. doi: 10.1001/archderm.141.7.904-b. author reply 905–906. [DOI] [PubMed] [Google Scholar]
- 18.Schiffmann R, Ries M. Fabry’s disease: An important risk factor for stroke. Lancet. 2005;366:1754–1756. doi: 10.1016/S0140-6736(05)67636-2. [DOI] [PubMed] [Google Scholar]
- 19.Thurberg BL, Rennke H, Colvin RB, Dikman S, Gordon RE, Collins AB, Desnick RJ, O’Callaghan M. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002;62:1933–1946. doi: 10.1046/j.1523-1755.2002.00675.x. [DOI] [PubMed] [Google Scholar]
- 20.Schiffmann R, Ries M, Timmons M, Flaherty JT, Brady RO. Long-term therapy with agalsidase alfa for Fabry disease: Safety and effects on renal function in a home infusion setting. Nephrol Dial Transplant. 2006;21:345–354. doi: 10.1093/ndt/gfi152. [DOI] [PubMed] [Google Scholar]
- 21.Wilcox WR, Banikazemi M, Guffon N, Waldek S, Lee P, Linthorst GE, Desnick RJ, Germain DP. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004;75:65–74. doi: 10.1086/422366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schiffmann R, Ries M, Timmons M, Flaherty JT, Brady RO. Long-term therapy with agalsidase alfa for Fabry disease: Safety and effects on renal function in a home infusion setting. Nephrol Dial Transplant. 2005;21:345–354. doi: 10.1093/ndt/gfi152. [DOI] [PubMed] [Google Scholar]
- 23.Schiffmann R, Murray GJ, Treco D, Daniel P, Sellos-Moura M, Myers M, Quirk JM, Zirzow GC, Borowski M, Loveday K, Anderson T, Gillespie F, Oliver KL, Jeffries NO, Doo E, Liang TJ, Kreps C, Gunter K, Frei K, Crutch-field K, Selden RF, Brady RO. Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci U S A. 2000;97:365–370. doi: 10.1073/pnas.97.1.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levey AS. Clinical practice. Nondiabetic kidney disease. N Engl J Med. 2002;347:1505–1511. doi: 10.1056/NEJMcp013462. [DOI] [PubMed] [Google Scholar]
- 25.Schiffmann R, Brady RO. New prospects for the treatment of lysosomal storage diseases. Drugs. 2002;62:733–742. doi: 10.2165/00003495-200262050-00002. [DOI] [PubMed] [Google Scholar]
- 26.Schwarting A, Dehout F, Feriozzi S, Beck M, Mehta A, Sunder-Plassmann G. Enzyme replacement therapy and renal function in 201 patients with Fabry disease. Clin Nephrol. 2006;66:77–84. [PubMed] [Google Scholar]
- 27.Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, McDonald M, Finkel R, Packman S, Bichet DG, Warnock DG, Desnick RJ. Agalsidase-beta therapy for advanced Fabry disease: A randomized trial. Ann Intern Med. 2007;146:77–86. doi: 10.7326/0003-4819-146-2-200701160-00148. [DOI] [PubMed] [Google Scholar]
- 28.de Zeeuw D, Remuzzi G, Parving HH, Keane WF, Zhang Z, Shahinfar S, Snapinn S, Cooper ME, Mitch WE, Brenner BM. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: Lessons from RENAAL. Kidney Int. 2004;65:2309–2320. doi: 10.1111/j.1523-1755.2004.00653.x. [DOI] [PubMed] [Google Scholar]
- 29.Vedder AC, Linthorst GE, van Breemen MJ, Groener JE, Bemelman FJ, Strijland A, Mannens MM, Aerts JM, Hollak CE. The Dutch Fabry cohort: Diversity of clinical manifestations and Gb3 levels. J Inherit Metab Dis. 2007;30:68–78. doi: 10.1007/s10545-006-0484-8. [DOI] [PubMed] [Google Scholar]
- 30.Bekri S, Lidove O, Jaussaud R, Knebelmann B, Barbey F. The role of ceramide trihexoside (globotriaosylceramide) in the diagnosis and follow-up of the efficacy of treatment of Fabry disease: A review of the literature. Cardiovasc Hematol Agents Med Chem. 2006;4:289–297. doi: 10.2174/187152506778520718. [DOI] [PubMed] [Google Scholar]
- 31.Altarescu G, Schiffmann R, Parker CC, Moore DF, Kreps C, Brady RO, Barton NW. Comparative efficacy of dose regimens in enzyme replacement therapy of type I Gaucher disease. Blood Cells Mol Dis. 2000;26:285–290. doi: 10.1006/bcmd.2000.0310. [DOI] [PubMed] [Google Scholar]
- 32.de Fost M, Hollak CE, Groener JE, Aerts JM, Maas M, Poll L, Wiersma MG, Haussinger D, Brett S, Brill N, Vom Dahl S. Superior effects of high dose enzyme replacement therapy in type 1 Gaucher disease on bone marrow involvement and chitotriosidase levels; a two center retrospective analysis. Blood. 2006;9:9. doi: 10.1182/blood-2005-12-5072. [DOI] [PubMed] [Google Scholar]
- 33.Beutler E. Dosage-response in the treatment of Gaucher disease by enzyme replacement therapy. Blood Cells Mol Dis. 2000;26:303–306. doi: 10.1006/bcmd.2000.0311. [DOI] [PubMed] [Google Scholar]
- 34.Zimran A, Elstein D, Beutler E. Low-dose therapy trumps high-dose therapy again in the treatment of Gaucher disease. Blood. 2006;25:25. doi: 10.1182/blood-2006-03-010801. [DOI] [PubMed] [Google Scholar]
- 35.Warnock DG. Fabry disease: diagnosis and management, with emphasis on the renal manifestations. Curr Opin Nephrol Hypertens. 2005;14:87–95. doi: 10.1097/00041552-200503000-00002. [DOI] [PubMed] [Google Scholar]