

The crystal structure of porcine pancreatic elastase complexed with a novel trimacrocyclic peptide inhibitor FR901451 was solved at 1.9 Å resolution.
Keywords: elastase, cyclic peptide inhibitor, protein–inhibitor complex
Abstract
Porcine pancreatic elastase (PPE) resembles the attractive drug target leukocyte elastase, which has the ability to degrade connective tissue in the body. The crystal structure of PPE complexed with a novel trimacrocyclic peptide inhibitor, FR901451, was solved at 1.9 Å resolution. The inhibitor occupied the subsites S3 through S3′ of PPE and induced conformational changes in the side chains of Arg64 and Arg226, which are located at the edges of the substrate-binding cleft. Structural comparison of five PPE–inhibitor complexes, including the FR901451 complex and non-ligated PPE, reveals that the residues forming the S2, S1, S1′ and S2′ subsites in the cleft are rigid, but the two arginine residues playing a part in the S3 and S3′ subsites are flexible. Structural comparison of PPE with human leukocyte elastase (HLE) implies that the inhibitor binds to HLE in a similar manner to the FR901451–PPE complex. This structural insight may help in the design of potent elastase inhibitors.
1. Introduction
Elastase is a serine protease classified into the chymotrypsin family and is possibly the most destructive of enzymes, having the ability to degrade virtually all of the connective components in the body. Uncontrolled proteolytic degradation by elastase has been implicated in a number of pathological conditions. Therefore, elastase is an attractive therapeutic target for pulmonary disease and many compounds have already been developed as inhibitors (Kuraki et al., 2002 ▶; Tremblay et al., 2003 ▶; Chughtai & O’Riordan, 2004 ▶).
Fujita and coworkers discovered the novel inhibitor FR901451 as a product of Flexibacter sp. No. 758, with IC50 values of 0.27 and 0.23 µM against porcine pancreatic elastase (PPE) and human leukocyte elastase (HLE), respectively (Fujita et al., 1994 ▶). This compound is a trimacrocyclic peptide consisting of 11 normal amino acids and having three non-peptide bonds (Fig. 1 ▶). In this paper, we report the 1.9 Å X-ray crystal structure of the complex of porcine pancreatic elastase (PPE) with FR901451, a comparison with other structures of inhibitor–PPE complexes and non-ligated PPE and a comparison with the HLE structure.
Figure 1.

Chemical structure of FR901451. All amino-acid residues have the l configuration; the oligopeptide is numbered in the usual way. FR901451 is a trimacrocyclic peptide. Two lactone cyclizations between Thr2 and Asp8 and between Ser7 and Asp10 are observed. An amide cyclization between Lys4 and Asp11 is also observed.
2. Materials and methods
Commercially available PPE was purchased from Worthington Biochemicals and used for crystallization without further purification. FR901451 was isolated at Fujisawa Pharmaceutical Co. Ltd (currently Astellas Pharma Inc.). The FR901451–PPE complex solution was prepared by directly dissolving the inhibitor to a final concentration of 1 mM in 20 mg ml−1 protein solution. Crystals of the complex were prepared under similar crystallization conditions to those reported previously (Kinoshita et al., 2003 ▶).
Crystals cryoprotected in Paratone-N oil (Hampton Research) were frozen at 100 K and X-ray diffraction data were collected at beamline BL38B1 of the SPring-8 facility, Japan. Data were indexed, integrated and scaled using the programs MOSFLM and SCALA (Collaborative Computational Project, Number 4, 1994 ▶).
The difference Fourier map was calculated using phases and amplitudes obtained from the apo structure (Kinoshita et al., 2003 ▶) without inhibitor, ions and solvent molecules. Model building and map fitting were performed using the programs QUANTA/X-BUILD (Accelrys Inc.) and O (Jones et al., 1991 ▶). Subsequent refinement involved iterative manual rebuilding and maximum-likelihood refinement using the program CNS (Brünger et al., 1998 ▶). Data-collection and refinement statistics of the complex are shown in Table 1 ▶. The final coordinates of the complex have been deposited in the Protein Data Bank with code 2cv3. Structural comparisons of the PPE structures were performed using the program QUANTA (Accelrys Inc.).
Table 1. Data-collection and refinement statistics of the FR901451–PPE complex.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 50.83, b = 57.35, c = 74.51 |
| Maximum resolution (Å) | 1.90 |
| Observed reflections | 62274 |
| Unique reflections | 17458 |
| Completeness (%) | 98.7 (99.9) |
| Rmerge† (%) | 9.0 (21.0) |
| I/σ(I) | 5.9 (3.3) |
| Refinement | |
| Resolution range (Å) | 19.77–1.90 |
| No. of protein non H-atoms | 1913 |
| No. of FR901451 non H-atoms | 91 |
| No. of solvent molecules | 195 |
| R‡ (%) | 16.2 |
| Rfree§ (%) | 22.4 |
| Average B factor (Å2) | |
| All atoms | 12.0 |
| Protein only | 10.4 |
| Inhibitor only | 13.0 |
| Solvent only | 23.7 |
| R.m.s.d. bond lengths (Å) | 0.018 |
| R.m.s.d. bond angles (°) | 2.0 |
R
merge = 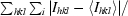
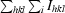 .
.
R = 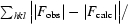
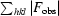 .
.
R free is equivalent to R, but is calculated using a 5% set of reflections excluded from the maximum-likelihood refinement stages.
3. Results and discussion
The overall protein structure of the FR901451–PPE complex is very similar to that of the native enzyme (Meyer et al., 1988 ▶). The inhibitor fits closely into the substrate-binding cleft of the enzyme (Fig. 2 ▶). Well defined interactions exist between the five N-terminal residues Thr1–Phe5 and the C-terminal residue Asp11 and elastase subsites S3 through S3′, which have been defined in detail (Bode et al., 1989 ▶). Thr1, Thr2, Leu3, Lys4, Phe5 and Asp11 of the inhibitor bind to the S3, S2, S1, S1′, S2′ and S3′ sites, respectively. These six residues make hydrophobic and/or van der Waals interactions with surrounding residues from the respective subsites. Leu3 of the inhibitor fully occupies the S1 subsite, which is most important for substrate recognition, and is almost completely buried from solvent access. In addition to the hydrophobic interactions, the typical hydrogen bonds of substrate or peptide inhibitors with the enzyme are conserved in the FR901451–PPE complex. The carbonyl group of Leu3 occupies the oxyanion hole, making hydrogen bonds to Gly201 NH and Ser203 NH. Thr1 of the inhibitor makes two hydrogen bonds to the backbone of Val224, which is significant for recognition of the β-strand structure of the substrate.
Figure 2.

FR901451 in the substrate-binding cleft of porcine pancreatic elastase. (a) The inhibitor is shown as magenta sticks and van der Waals mesh diagrams. The substrate-binding region of PPE is shown as a van der Waals surface. Arg64 and Arg226 are highlighted as stick models. Water molecules binding to the active site are displayed as blue spheres. (b) Surface diagram as in (a) but with the view rotated by 90°.
The other five residues Pro6–Asp10 of the inhibitor make no significant interaction with the enzyme. Of these, Trp9 of the inhibitor exists in the solvent region and no clear electron density is found for it (Fig. 3 ▶). This incompatibility between the molecular model and electron density may partially be reflected in the difference between the R and R free values. The five outside residues (Pro6–Asp10) may contribute to fixing the active conformation, as the Thr1–Lys4 part of FR901451 binds in a similar conformation to the linear substrate-like inhibitor FR136706 (Fig. 4 ▶). Generally, a cyclized inhibitor is entropically more advantageous than a flexible linear inhibitor if both types of inhibitors bind in the same conformation. In many enzymes, linear peptide inhibitors gain higher inhibitory activities by cyclization (Khan et al., 1998 ▶; Nantermet et al., 2003 ▶; Hu et al., 2003 ▶; Ghosh et al., 2005 ▶).
Figure 3.

FR901451 within the 2F o − F c electron-density map contoured at the 1.0σ level. The map around Trp9 is discontinuous and ambiguous.
Figure 4.

Superimposed structures of inhibitors in the crystals. Thr1–Lys4 of FR901451 (magenta sticks) bind to PPE in a similar conformation to the linear inhibitor FR136706 (Kinoshita et al., 2003 ▶; displayed as blue sticks).
Comparison of the structure of the FR901451 complex with PPE structures ligated with various inhibitors (Kinoshita et al., 2003 ▶; Nakanishi et al., 2000 ▶; Matern et al., 2003 ▶; Aÿ et al., 2003 ▶) indicates a very similar conformation, with only slight differences in the peptide backbones (Table 2 ▶). Most of the side chains around the substrate-binding cleft also maintain a very similar conformation. However, the side chains of Arg64 and Arg226, which are located at the edges of the substrate-binding cleft and partially form the S3 and S3′ subsites, have varying conformations (Fig. 5 ▶). The arginine residues do not contribute to the respective crystal packings. Therefore, it is presumed that the residues adjust the cleft shape according to the inhibitor binding. In all these complexes, the arginine flexibility leads to interaction with the inhibitor or avoidance of steric hindrance. In the FR901451 complex, Arg64 and Arg226 move from their respective positions in the non-ligated structure (Meyer et al., 1988 ▶) and make van der Waals contacts with Asp11 and Thr1 of the inhibitor, respectively. The flexibilities of the S3 and S3′ subsites of PPE may be the reason that PPE can recognize the various moieties using these subsites.
Table 2. PPE complexes with peptide inhibitors.
| PDB code | Compound name | No. of amino acids | Binding subsites | Structural feature | R.m.s.d. with 2cv3† (Å) | Reference |
|---|---|---|---|---|---|---|
| 2cv3 | FR901451 | 11 | S3–S3′ | Tricyclic | — | This work |
| 1mmj | FR136706 | 5 | S5–S1′ | Linear | 0.35 | Kinoshita et al. (2003 ▶) |
| 1qr3 | FR901277 | 8 | S4–S2′ | Bicyclic | 0.30 | Nakanishi et al. (2000 ▶) |
| 1okx | Scyptolin A | 8 | S4–S1 | Monocyclic | 0.50 | Matern et al. (2003 ▶) |
| 1mcv | HEI-TOE1 | 28 | S4–S3′ | Linear, three S—S bonds | 0.50 | Aÿ et al. (2003 ▶) |
Superimpositions were performed using the Cα atoms of the proteins.
Figure 5.

Flexibility of the side chains of Arg64 (right) and Arg226 (left). FR901451 (magenta) and arginine residues are extracted from the superimposed structures. The arginine residues from the FR901451 complex are shown in grey, those from the non-ligated structure in orange, those from the FR136706 complex in blue, those from the FR901277 complex in brown, those from the Scyptolin A complex in black and those from the HEI-TOE1 complex in green.
Structural comparison of PPE and HLE indicates that FR901451 binds to HLE in a similar manner to the FR901451–PPE complex. The central region of the active site of PPE including subsites S2 through S2′ can easily be overlaid onto that of HLE (Navia et al., 1989 ▶). Therefore, the interaction mode is likely to be conserved between PPE and HLE in this region. On the other hand, there are large structural differences between PPE and HLE in the S3 and S3′ subsites, based upon insertions or deletions in their amino-acid sequences. However, Thr1 and Asp11 of the inhibitor may possibly be accommodated by the S3 and S3′ subsites of HLE based upon an assumption from computer modelling. The wider S3 and S3′ subsites of HLE do not obstruct inhibitor binding and side-chain rotamers of the residues corresponding to the two arginine residues which are putatively assigned as Asn61 and Arg217 in HLE could make van der Waals contacts with the inhibitor. The structural prospect of FR901451 binding to both elastases in a similar manner is consistent with the observation that the inhibitor has similar inhibitory activities towards both PPE and HLE (Fujita et al., 1994 ▶).
In this communication, we have presented the crystal structure of the FR901451–PPE complex. FR901451 binds at the S3, S2, S1, S1′, S2′ and S3′ subsites of PPE and occupies most of the space of the substrate-binding cleft. Although the S3 and S3′ subsites of PPE are structurally distinct from those of HLE, structural comparison of the two elastases indicates that the inhibitor binds to HLE in a similar manner as in the PPE complex. This structural information may contribute to the drug discovery of novel elastase inhibitors.
Supplementary Material
PDB reference: FR901451–PPE, 2cv3, r2cv3sf
Acknowledgments
We would like to thank Dr I. Nakanishi, Graduate School of Pharmaceutical Science, Kyoto University and Dr D. Barrett, Medicinal Chemistry III, Chemical Research Laboratory, Astellas Pharma Inc. for helpful discussions and critical evaluation of the manuscript.
References
- Aÿ, J., Hilpert, K., Krauss, N., Schneider-Mergener, J. & Hohne, W. (2003). Acta Cryst. D59, 247–254. [DOI] [PubMed] [Google Scholar]
- Bode, W., Meyer, E. & Powers, J. C. (1989). Biochemistry, 28, 1951–1963. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, N. S., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Chughtai, B. & O’Riordan, T. G. (2004). J. Aerosol Med.17, 289–298. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Fujita, T., Hatanaka, H., Hayashi, K., Shigematsu, N., Takase, S., Okamoto, M. & Okuhara, M. (1994). J. Antibiot.47, 1359–1368. [DOI] [PubMed] [Google Scholar]
- Ghosh, A. K., Devasamudram, T., Hong, L., DeZutter, C., Xu, X., Weeransena, V., Koelsch, G., Blicer, G. & Tang, J. (2005). Bioorg. Med. Chem. Lett.15, 15–20. [DOI] [PubMed] [Google Scholar]
- Hu, X., Nguyen, K. T., Verlinde, C. L. M. J., Hol, W. G. J. & Pei, D. (2003). J. Med. Chem.46, 3771–3774. [DOI] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Khan, A. R., Parrish, J. C., Fraser, M. E., Smith, W. W., Bartlett, P. A. & James, M. N. G. (1998). Biochemistry, 37, 16839–16845. [DOI] [PubMed] [Google Scholar]
- Kinoshita, T., Nakanishi, I., Sato, A. & Tada, T. (2003). Bioorg. Med. Chem. Lett.13, 21–24. [DOI] [PubMed] [Google Scholar]
- Kuraki, T., Ishibashi, M., Takayama, M., Shiraishi, M. & Yoshida, M. (2002). Am. J. Respir. Crit. Care Med.166, 496–500. [DOI] [PubMed] [Google Scholar]
- Matern, U., Schleberger, C., Jelakovic, S., Weckesser, J. & Schulz, G. E. (2003). Chem. Biol.10, 997–1001. [DOI] [PubMed] [Google Scholar]
- Meyer, E., Cole, G., Radhakrishnan, R. & Epp, O. (1988). Acta Cryst. B44, 26–38. [DOI] [PubMed] [Google Scholar]
- Nakanishi, I., Kinoshita, T., Sato, A. & Tada, T. (2000). Biopolymers, 53, 434–445. [DOI] [PubMed] [Google Scholar]
- Nantermet, P. G., Barrow, J. C., Newton, C. L., Pellicore, J. M., Young, M., Lewis, S. D., Lucas, B. J., Krueger, J. A., McMasters, D. R., Yan, Y., Kuo, L. C., Vacca, J. P. & Selnick, H. G. (2003). Bioorg. Med. Chem. Lett.13, 2781–2784. [DOI] [PubMed] [Google Scholar]
- Navia, M. A., McKeever, B. M., Springer, J. P., Lin, T.-Y., Williams, H. R., Fluder, E. M., Dorn, C. P. & Hoogsteen, K. (1989). Proc. Natl Acad. Sci. USA, 86, 8–11. [DOI] [PMC free article] [PubMed]
- Tremblay, G. M., Janelle, M. F. & Bourbonnais, Y. (2003). Curr. Opin. Invest. Drugs, 4, 556–565. [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: FR901451–PPE, 2cv3, r2cv3sf