

The isomerase domain of glucosamine-6-phosphate synthase from C. albicans has been crystallized and X-ray diffraction data have been collected. Preliminary analysis of the data reveals the oligomeric structure of the eukaryotic synthase to be a ‘dimer’ of prokaryotic-like dimers.
Keywords: glucosamine-6-phosphate synthase, aldose–ketose isomerase
Abstract
Glucosamine-6-phosphate synthase (EC 2.6.1.16) catalyses the first and practically irreversible step in the hexosamine metabolism pathway, the end product of which, uridine 5′-diphospho-N-acetyl d-glucosamine, is an essential substrate for assembly of the cell wall. The isomerase domain, consisting of residues 346–712 (42 kDa), of glucosamine-6-phosphate synthase from Candida albicans has been crystallized. X-ray analysis revealed that the crystals belonged to space group I4, with unit-cell parameters a = b = 149, c = 103 Å. Diffraction data were collected to 3.8 Å. Preliminary results from molecular replacement using the homologous bacterial monomer reveal that the asymmetric unit contains two monomers that resemble a bacterial dimer. The crystal lattice consists of pairs of such symmetry-related dimers forming elongated tetramers.
1. Introduction
Glucosamine-6-phosphate synthase (l-glutamine:d-fructose-6-phosphate amidotransferase; EC 2.6.1.16) catalyses the first and practically irreversible step in the hexosamine-metabolism pathway, a branch of the glycolytic pathway (reviewed by Milewski, 2002 ▶). The end product of the hexosamine-metabolism pathway is uridine 5′-diphospho-N-acetyl-d-glucosamine (UDP-GlcNAc), an essential substrate for assembly of the cell wall in fungi and bacteria. Fungal glucosamine-6-phosphate (GlcN-6-P) synthase is of interest as a potential target in antifungal therapy and is part of our wider study aimed at improved chemotherapy against fungal pathogens by targeting their vital functions. Conditions favouring fungal pathogens occur ever more frequently as an unintentional result of the use of antibiotics or in the course of diseases that weaken the human immune system. Fungi, being eukaryotes like the organisms they infect, present a subtler challenge to medicine than bacteria. The enzyme under study in this project comes from the fungus Candida albicans, an opportunistic human pathogen classified as a yeast. Although there is a clear amino-acid sequence homology between the bacterial and yeast GlcN-6-P synthases, the enzyme architecture is more complex in eukaryotes than in prokaryotes. In Escherichia coli the protein is a homodimer of subunit molecular weight ∼70 kDa, while the eukaryotic protein is a homotetramer of 80 kDa subunits (Milewski et al., 1999 ▶). In addition, only the eukaryotic enzyme is allosterically inhibited by UDP-GlcNAc, the end product of the reaction pathway. The reaction mechanism of GlcN-6-P synthase, both prokaryotic and eukaryotic, is complex and involves amino transfer and sugar isomerization. X-ray crystallographic studies of prokaryotic GlcN-6-P synthase revealed that these two functions were performed by two distinct structural domains of the enzyme subunit (Isupov et al., 1996 ▶; Teplyakov et al., 1999 ▶, 2001 ▶). The isomerase domain is believed to be responsible for oligomerization of the C. albicans GlcN-6-P synthase (Milewski, unpublished data). For the purpose of these studies, we constructed and purified a recombinant protein comprising residues 346–712 of the native C. albicans GlcN-6-P synthase (Olchowy et al., 2004 ▶). This fragment demonstrated a very high degree of homology to the 241–608 part of the E. coli enzyme, unequivocally identified as its isomerase domain (Teplyakov et al., 1998 ▶). Here, we report the crystallization and preliminary X-ray analysis of the isomerase domain of GlcN-6-P synthase from C. albicans.
2. Methods and results
2.1. Expression and purification
The His6-N-tagged isomerase domain of GlcN-6-P synthase was overproduced in E. coli BL21(DE3)pLysS cells transformed with the pET23b-FRU expression plasmid. Overexpression was induced with isopropyl-β-d-thiogalactopyranoside at a final concentration of 0.5 mM. The overproduced protein was isolated in a three-step procedure involving preparation of crude extract, metal-chelate affinity chromatography performed on Ni2+-IDA agarose (His-Bind Resin, Novagen) and gel filtration on Superdex 200 (Olchowy et al., 2004 ▶). The protein was isolated with 81% yield. The purified protein was stored and crystallized from an aqueous solution containing 10 mM HEPES pH 7.0, 10 mM glucose-6-phosphate (Glc-6-P) and 10 mg ml−1 protein.
2.2. Crystallization
Numerous attempts to crystallize the protein failed using several commercial crystallization kits as well as home-made mixtures of commonly used salts and organic solvents. Finally, we obtained crystals using Clear Strategy Screen 1 (Molecular Dimensions). Thin needle-shaped crystals grew as hanging drops or sitting drops in vapour-diffusion setups in 20%(w/v) PEG 600 and 0.15 M KSCN at pH 8.5 (Fig. 1 ▶). Several other solutions in the kit, all containing KSCN, also gave small crystals. The crystals grew at 277 K and dissolved slowly at room temperature. The needles grew up to 0.3 mm in length, but their thickness was limited to ∼20 µm.
Figure 1.

Crystals of the isomerase domain of GlcN-6-P synthase
2.3. Data collection and analysis
X-ray diffraction data were collected at the EMBL X11 beamline at the DORIS storage ring, DESY, Hamburg from a crystal of the isomerase domain grown in the presence of Glc-6-P. The thin needle-shaped crystals were very fragile and difficult to handle; they were also difficult to cryocool. Transferring the crystals to solutions containing any of the commonly used cryoprotectants invariably caused a major loss of X-ray diffraction. The crystals could be vitrified successfully only if oil was used for cryoprotection. Light oils with low viscosity generally gave better results than heavy oils. The best turned out to be ‘Iliada’ Greek salad oil (Agrovim). Diffraction images were recorded on a MAR CCD 165 mm detector and processed with the DENZO/SCALEPACK program suite (Otwinowski & Minor, 1997 ▶). Data statistics are summarized in Table 1 ▶. The space group was determined to be I4 based on autoindexing of the diffraction spots, merging the data assuming different symmetries and analysis of the systematic absences (none were found). The V M value assuming two monomers per asymmetric unit was 3.4 Å3 Da−1, corresponding to a solvent content of 64% (Matthews, 1968 ▶).
Table 1. Data-collection statistics.
Values in parentheses are for the last resolution shell.
| Beamline | EMBL-X11 |
| Temperature (K) | 100 |
| Wavelength (Å) | 0.8128 |
| Space group | I4 |
| Unit-cell parameters (Å) | |
| a = b | 149.3 |
| c | 103.0 |
| Resolution range (Å) | 20.0–3.8 (3.87–3.80) |
| Rmerge† | 0.132 (0.308) |
| No. of raw measurements | 230399 |
| No. of unique reflections | 11183 |
| Completeness (%) | 100.0 (100.0) |
| Mosaicity (°) | 0.5 |
| Data redundancy | 4.4 (4.6) |
| 〈I/σ(I)〉 | 11.5 (5.4) |
| Reflections > 3σ (%) | 76 (62) |
R
merge = 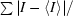
 , where 〈I〉 is the average intensity for a given measurement and the summation is over all measurements
, where 〈I〉 is the average intensity for a given measurement and the summation is over all measurements
2.4. Molecular replacement
The phase problem was solved by molecular replacement using the program PHASER v.1.3 running in automatic mode (McCoy et al., 2005 ▶) using as the search model the atomic coordinates (all protein atoms) of the isomerase domain from the GlcN-6-P synthase from E. coli (PDB code 1moq; Teplyakov et al., 1998 ▶). The rotation function contained three peaks higher than 75% of the difference between the top and the mean value. The peaks were at 4.2, 4.1 and 3.4 standard deviations above the mean (Z scores). After the translation function, the first two peaks (corresponding to the first two peaks of the rotation function) had Z scores of 14 and 16, respectively, with no steric clashes. The remaining peaks in the translation function had Z scores near 5. Although a single subunit was used as the search model, the solution obtained from molecular replacement consisted of two subunits interacting in a similar way to subunits in the bacterial dimer. The crystal lattice consisted of symmetry-related pairs of such dimers interacting end-to-end (Fig. 2 ▶). The phases obtained from molecular replacement were used in an electron-density modification procedure that included non-crystallographic symmetry averaging, solvent flattening and histogram matching using the program DM (Cowtan, 1994 ▶). The molecular envelope was determined using the automatic protocol of DM with the solvent content set at 60%. The NCS correlation between the related symmetry regions was initially 0.82 and increased to 0.97 after 100 cycles of averaging while increasing the resolution from 8.0 to 3.8 Å. The electron-density map thus obtained shows a significant improvement and departure from the initial map based on molecular replacement. The map shows interpretable density (inset in Fig. 2 ▶) for most of the protein and good surface complementarity between the pairs of subunits that form the tetramer.
Figure 2.

A tetramer of isomerase domains of the GlcN-6-P synthase obtained as the result of molecular replacement performed with the homologous domain of the E. coli synthase (Teplyakov et al., 1998 ▶) as the search model. The monomers associate pairwise, as in the bacterial dimer. Two such symmetry-related dimers (shown in different colours) associate back-to-back in the C. albicans crystal structure. The N-termini of the polypeptide chains are indicated. The inset in the bottom right corner shows an α-helical fragment of the electron density after the density-modification procedure, as described in the text. The map was contoured at the level of one root-mean-square deviation.
3. Discussion
The crystals are small and difficult to handle. Nevertheless, useful X-ray data were obtained. Despite the weak intensities and limited resolution of the data, the phase problem has been solved unambiguously and the initial electron-density map is interpretable. Much remains to be done before an atomic model is fully built and refined, but the results obtained already reveal the basic plan of the oligomeric structure of the isomerase domains in the C. albicans GlcN-6-P synthase. In prokaryotes, the isomerase domains form the core of the dimeric molecule. It is likely that in the eukaryotic enzyme the isomerase domains also form the core of the tetrameric molecule. The monomers in the crystal lattice interact in such a manner that their N-termini point outwards. It is possible that in the intact synthase molecule the glutaminase domains that form the N-terminal part of the polypeptide chains also point outward.
When the model of atomic coordinates is built and refined it should enable us to compare the isomerase domain of the eukaryotic GlcN-6-P synthase with the related prokaryotic protein. Of particular interest are the differences that account in eukaryotes for the additional binding site of the allosteric effector UDP-GlcNAc. This compound is known to inhibit eukaryotic but not prokaryotic GlcN-6-P synthase (Milewski, 2002 ▶) and our preliminary data indicate that in the C. albicans enzyme the UDP-GlcNAc binding site is located in the isomerase domain. This domain contains also the binding site for transition-state analogue inhibitors such as 2-amino-2-deoxy-d-glucitol 6-phosphate (Badet-Denisot et al., 1995 ▶), its derivatives (Bearne & Blouin, 2000 ▶; Janiak et al., 2003 ▶) and arabinose-5-phosphate oxime (Le Camus et al., 1998 ▶). We hope that structural analysis of the fungal enzyme will facilitate designing antifungal drugs, allow a deeper understanding of the key metabolic processes of amino-sugar biosynthesis in fungi and elucidate aspects of evolution of major enzymes involved in these processes.
Acknowledgments
This work was supported by a grant from the Polish State Committee for Scientific Research (KBN-3-P04A-035-25) and by the European Community Research Infrastructure Action under the F6P ‘Structuring the European Research Area Programme’ (contract No. RII3/CT/2004/5060008). RJ acknowledges support under the European Community ‘Improving the Human Research Potential and Socio-Economic Knowledge Base Programme’ Marie Curie Training Sites Scheme (contract No. HPMT-CT-20000-00174).
References
- Badet-Denisot, M.-A., Leriche, C., Massiere, F. & Badet, B. (1995). Bioorg. Med. Chem. Lett.5, 815–820.
- Bearne, S. L. & Blouin, C. (2000). J. Biol. Chem.275, 135–140. [DOI] [PubMed] [Google Scholar]
- Cowtan, K. (1994). Jnt CCP4/ESF–EACBM Newsl Protein Crystallogr.31, 34–38.
- Isupov, M. N., Obmolova, G., Butterworth, S., Badet-Denisot, M. A., Badet, B., Polikarpov, I., Littlechild, J. A. & Teplyakov, A. (1996). Structure, 4, 801–810. [DOI] [PubMed] [Google Scholar]
- Janiak, A. M., Hoffmann, M., Milewska, M. J. & Milewski, S. (2003). Bioorg. Med. Chem.11, 1653–1662. [DOI] [PubMed] [Google Scholar]
- Le Camus, C., Badet-Denisot, M.-A. & Badet, B. (1998). Tetrahedron Lett.39, 2571–2572.
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Milewski, S. (2002). Biochim. Biophys. Acta, 1597, 173–192. [DOI] [PubMed] [Google Scholar]
- Milewski, S., Kuszczak, D., Jedrzejczak, R., Smith, R. J., Brown, A. J. & Gooday, G. W. (1999). J. Biol. Chem.274, 4000–4008. [DOI] [PubMed] [Google Scholar]
- Olchowy, J., Sachadyn, P., Kur, J. & Milewski, S. (2004). Eur. J. Biochem.271, Suppl. 1, P.32-38.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Teplyakov, A., Obmolova, G., Badet, B. & Badet-Denisot, M. A. (2001). J. Mol. Biol.313, 1093–1102. [DOI] [PubMed] [Google Scholar]
- Teplyakov, A., Obmolova, G., Badet-Denisot, M. A. & Badet, B. (1999). Protein Sci.8, 596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplyakov, A., Obmolova, G., Badet-Denisot, M. A., Badet, B. & Polikarpov, I. (1998). Structure, 15, 1047–1055. [DOI] [PubMed]