

The ybeY protein from E. coli is reported at a 2.7 Å resolution with a metal ion.
Keywords: Protein Structure Initiative, metalloproteins, nickel, UPF0054 family
Abstract
The three-dimensional crystallographic structure of the ybeY protein from Escherichia coli (SwissProt entry P77385) is reported at 2.7 Å resolution. YbeY is a hypothetical protein that belongs to the UPF0054 family. The structure reveals that the protein binds a metal ion in a tetrahedral geometry. Three coordination sites are provided by histidine residues, while the fourth might be a water molecule that is not seen in the diffraction map because of its relatively low resolution. X-ray fluorescence analysis of the purified protein suggests that the metal is a nickel ion. The structure of ybeY and its sequence similarity to a number of predicted metal-dependent hydrolases provides a functional assignment for this protein family. The figures and tables of this paper were prepared using semi-automated tools, termed the Autopublish server, developed by the New York Structural GenomiX Research Consortium, with the goal of facilitating the rapid publication of crystallographic structures that emanate from worldwide Structural Genomics efforts, including the NIH-funded Protein Structure Initiative.
1. Introduction
The Escherichia coli hypothetical protein ybeY (SwissProt entry P77385; MW = 17 526 Da; 155 amino-acid residues) is the product of the gene ybeY and a member of the UPF0054 family (Bateman et al., 2004 ▶). The sequence similarity of this protein to a number of predicted metal-dependent hydrolases would suggest a potential hydrolytic function (Tatusov et al., 2001 ▶). Here, we report the structure of the ybeY protein determined by the multiple anomalous dispersion method (MAD) at a resolution of 2.7 Å (PDB code 1xm5). The structure provides new insights that help to functionally annotate this previously uncharacterized protein.
The figures and tables of this paper were prepared using semi-automated tools developed by the New York Structural GenomiX Research Consortium. This paper illustrates the ability of these tools to facilitate rapid publication of crystallographic structures that emanate from worldwide Structural Genomics efforts, including the NIH-funded Protein Structure Initiative.
2. Materials and methods
The coding sequence from the E. coli ybeY gene was cloned into the pet26b vector. A six-His tag was encoded at the C-terminus and was removed by proteolysis. The native protein and the selenomethionine-substituted protein were expressed in E. coli BL21 (DE3) cells and E. coli B834 cells, respectively. The proteins were purified to homogeneity by affinity chromatography on nickel-chelating Sepharose and subsequent gel filtrations were performed on a preparative Superdex-200 column. The E. coli protein (native and SeMet derivative) was concentrated to 15 mg ml−1 in the following buffer: 10 mM HEPES, 150 mM NaCl, 10 mM methionine, 10% glycerol, 1 mM DTT. Initial screening for crystallization conditions was performed by the hanging-drop vapour-diffusion method. Hampton matrix Index (Jancarik & Kim, 1991 ▶) was used and condition No. 82 yielded initial crystals. The reservoir (0.7 ml) for refined crystallization contained 20% PEG 3350, 0.2 M MgCl2, 0.1 M Bis-Tris pH 6.5 and crystals were grown at 278 K. The protein drop (2 µl) was mixed with an identical volume from the reservoir. The crystals appeared after 2–3 d and reached a final size of 0.2 × 0.2 × 0.2 mm after one week. The crystallization conditions for the selenomethionine-substituted protein were the same as for the native protein.
Diffraction data for the native protein were collected to 2.7 Å resolution at the Advanced Photon Source beamline 19BM. Prior to data collection, crystals were transferred to a solution of mother liquor supplemented with 20% glycerol and flash-cooled to 100 K. The data were processed using the HKL2000 software suite (Otwinowski & Minor, 1997 ▶). The crystals exhibit diffraction consistent with space group P212121. There are four molecules in the asymmetric unit, with unit-cell parameters a = 46.1, b = 119.6, c = 132.9 Å, a V M value of 2.6 Å3 Da−1 and a solvent content of 51.1%. In each molecule there are six selenomethionines with a mean phase error of about 20°.
A three-wavelength SeMet MAD experiment (0.97408, 0.97938 and 0.97919 Å) was performed at National Synchrotron Light Source beamline X29 and the structure was solved with the program SOLVE (Terwilliger & Berendzen, 1999 ▶). An initial model was automatically traced using the program RESOLVE (Terwilliger, 1999 ▶). The rest of the model was built manually with O (Jones et al., 1991 ▶) and NCS restraints were used in the refinement by the program CNS (Brünger et al., 1998 ▶). The final model contains 604 residues and four metal ions. The final data-collection statistics are shown in Table 1 ▶, phasing statistics in Table 2 ▶ and refinement statistics in Table 3 ▶. Atomic coordinates and structure factors have been deposited in the PDB and are available under accession code 1xm5.
Table 1. Data-collection statistics.
Statistics in parentheses are for the highest resolution bin (2.7–2.8 Å).
| SeMet replacement | ||||
|---|---|---|---|---|
| Native | Edge | Peak | Remote | |
| Data collection | APS-19BM | NSLS-X29 | NSLS-X29 | NSLS-X29 |
| Wavelength used (Å) | 0.97919 | 0.97938 | 0.97919 | 0.96408 |
| Data-collection temperature (K) | 110 | 110 | 110 | 110 |
| Space group | P212121 | P212121 | ||
| Unit-cell parameters (Å, °) | a = 46.07, b = 119.61, c = 132.90, α = β = γ = 90 | a = 45.70, b = 119.05, c = 132.51, α = β = γ = 90 | ||
| Resolution range (Å) | 25.0–2.7 | 30.0–3.0 | 30.0–3.0 | 30.0–3.0 |
| No. of unique reflections | 19520 | 15084 | 15136 | 15109 |
| Total No. of reflections | 144379 | 89659 | 124244 | 108891 |
| Redundancy | 6.9 | 5.1 | 6.5 | 5.8 |
| Completeness (%) | 97.9 (99.5) | 99.3 (94.3) | 99.7 (97.8) | 99.4 (94.3) |
| Rmerge† | 0.059 (0.379) | 0.117 (0.335) | 0.098 (0.237) | 0.120 (0.332) |
| 〈I/σ(I)〉 | 24.9 (4.2) | 12.8 (3.8) | 14.8 (5.2) | 13.4 (4.1) |
R
merge = 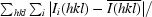
 .
.
Table 2. Phasing statistics from the program SOLVE .
| λ1 | λ2 | λ3 | |
| R.m.s. anomalous FH (%) | 4.3 | 4.8 | 3.2 |
| R.m.s. anomalous FH/E | 1.5 | 1.7 | 1.5 |
| λ1versus λ2 | λ1versus λ3 | λ2versus λ3 | |
| R.m.s. dispersive FH (%) | 3.5 | 6.7 | 3.2 |
| R.m.s. dispersive FH/E | 1.1 | 1.7 | 0.9 |
| FOM | 0.65 (SOLVE), 0.79 (RESOLVE) |
Table 3. Refinement statistics.
| Refinement | |
| Resolution range (Å) | 25.0–2.7 |
| No. of reflections used for refinement | 19520 |
| No. of reflections used for Rfree | 894 |
| R factor† | 0.234 (0.358) |
| Rfree‡ | 0.273 (0.363) |
| R.m.s. bonds (Å) | 0.007 |
| R.m.s. angles (°) | 1.2 |
| No. of non-H atoms | 4777 plus 4 Ni2+ ions |
| Average B factor (Å2) | 73.2 |
| R.m.s.d. B factor for bonded main-chain atoms (Å2) | 6.3 |
| R.m.s.d. B factor for bonded side-chain atoms (Å2) | 9.3 |
| R.m.s.d. B factor for angle main-chain atoms (Å2) | 10.2 |
| R.m.s.d. B factor for angle side-chain atoms (Å2) | 13.9 |
| Cruickshank DPI (Å) | 0.40 |
| Ramachandran plot (PROCHECK) | |
| Residues in most favored region | 467 (87.8%) |
| Residues in additional allowed regions | 59 (11.1%) |
| Disallowed regions | 3 (0.6%) |
R = 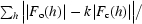
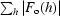 .
.
R free was calculated using a 5% randomly selected subset of the total number of reflections.
X-ray fluorescence analysis was performed to detect the transition-metal content of the protein at National Synchrotron Light Source beamline X9B according to established procedures (Chance et al., 2004 ▶; Shi et al., 2005 ▶). As outlined in the results, Ni was detected in the purified protein preparation and corresponding electron density was also observed in the crystal structure.
The figures and tables were prepared using the NYSGXRC AutoPublish web server.
3. Results and discussion
The structure of E. coli ybeY includes four identical proteins in an asymmetric unit. Quaternary structure prediction (Henrick & Thornton, 1998 ▶) suggests that this protein might exist as a homodimer. However, residue conservation was not observed on the hypothetical dimer interface to support this prediction. Thus, the oligomeric state of the ybeY protein cannot be confirmed without further experiments. The overall protein structure consists of six α-helices and four β-strands in a β-α-α-β-α-β-β-α-α-α fold (Fig. 1 ▶). The protein contains a conserved domain from the functionally uncharacterized protein family UPF0054 (pfam02130; Bateman et al., 2004 ▶). There is a metal ion, most probably an Ni2+ ion, coordinated by NE2 atoms from residues His114, His118 and His124 within each protein subunit. The fourth ion-coordination site might be a water molecule, but this cannot be confirmed because of the relatively low resolution of the data. The nickel content of the purified protein used for crystallization was analyzed using quantitative X-ray fluorescence analysis (Chance et al., 2004 ▶) and indicated a nickel:protein subunit stoichiometry of 0.5 ± 0.2 (data not shown). Previous experiments (Chance et al., 2004 ▶; Shi et al., 2005 ▶) have demonstrated that His-tagged proteins appear to show no more tendency to coordinate metal ions than do their non-His-tagged analogs. However, tetrahedral geometry with His-type ligands for Ni binding is uncommon, thus this metal-atom identification is tentative. Also, the occupancy is less than 100%.
Figure 1.

(a) Tertiary structure of the ybeY protein (chain A in asymmetric unit) prepared using MOLSCRIPT (Kraulis, 1991 ▶). (b) show a pair of stereo figures of the Cα trace with every tenth residue numbered. It is in the same orientation as (a).
A BLAST search (Altschul et al., 1990 ▶) for proteins related to E. coli ybeY reveals homologous sequences from 132 unique species (with BLAST cutoff score higher than 100), all of which are of bacterial origin. Most of the related proteins are annotated as hypothetical UPF0054 protein (Bateman et al., 2004 ▶) or predicted metal-dependent hydrolases (COG0319; Tatusov et al., 2001 ▶). The structure around the metal-ion site supports a predicted function of metal-dependent hydrolase for this protein. 14 homologs of E. coli ybeY with identity ranging from 39 to 75% were selected and are presented in a multiple sequence alignment (Fig. 2 ▶). Conserved and identical positions are marked by green and red colors, respectively. A residue was considered conserved if at least six out of ten possible physicochemical properties (polar, small, proline, tiny, aromatic, aliphatic, positive, negative, charged, hydrophobic) are shared in a given sequential position (Livingstone & Barton, 1993 ▶). This is a rather strict definition of residue conservation. The metal-binding motif is entirely conserved in these sequences and is marked by black arrows in Fig. 2 ▶.
Figure 2.

Alignment prepared by CLUSTALW (Thompson et al., 1994 ▶), AMAS (Livingstone & Barton, 1993 ▶) and ALSCRIPT (Barton, 1993 ▶). Identical and conserved regions are marked in red and green, respectively (see definition in text). The secondary structure of 1xm5 was assigned using DSSP (Kabsch & Sander, 1983 ▶) and is indicated below each block. Cylinders stands for α-helix and arrows for β-strand. Metal-binding residues are indicated by black arrows. The species for each sequence is listed following their accession ID: 1XM5 (ybeY), Escherichia coli; ZP_00092890.1, Azotobacter vinelandii; ZP_00205591, Pseudomonas syringae; YP_151278.1, Salmonella enterica; AAL19619.1, Salmonella typhimurium; YP_049423.1, Erwinia carotovora; Q7N748, Photorhabdus luminescens; CAH20351.1, Yersinia pseudotuberculosis; YP_131025.1, Photobacterium profundum; Q9JRS8, Actinobacillus actinomycetemcomitans; P57517, Buchnera aphidicola; Q8K9C1, Buchnera aphidicola; YP_113914.1, Methylococcus capsulatus; NP_879826.1, Bordetella pertussis; 1OZ9 (AQ_1354), Aquifex aeolicus.
Using the data from Fig. 2 ▶, the solvent-accessible surface was mapped and colored by degree of conservation (Fig. 3 ▶). The protein depicted in Fig. 3 ▶ shows a cleft lined with conserved and identical residues in green and red colors, respectively. These include the three histidines that coordinate the metal ion (colored blue). Positions with lower physicochemical conservation scores are colored yellow. The putative fourth coordination site for the metal is located within a cleft, as indicated by a star in Fig. 3 ▶. The red-colored patch seen below the metal ion includes many of the entirely conserved residues seen in the stretch of residues from Asn55 to Asp85 in Fig. 2 ▶.
Figure 3.

Surface-mapping representation prepared using PyMol (DeLano, 2002 ▶). Identical, conserved and less conserved residues are marked red, green and yellow, respectively. The Ni2+ ion is represented by a blue sphere. The approximate location of the putative fourth coordination site for Ni2+ ion is shown by a star.
The metal-binding site and the adjacent cleft are shown in Fig. 4 ▶. The sides of the helices that face the cleft are more conserved than the sides opposite the cleft. In addition to the three histidines coordinating the metal, Arg59, Lys61, Asn66 and Ser69 are either identical or have only very few substitutions in the multiple sequence alignment (Fig. 2 ▶). These residues might be important for the functional activity of the protein.
Figure 4.

Cleft structure prepared by PyMol (DeLano, 2002 ▶). The cleft structure is shown in secondary structure colored by conservation score as in Fig. 3 ▶. Conserved residues in the cleft that are possibly involved in function are represented by sticks. Blue sphere is the Ni2+ ion. A side view and a top view are shown in (a) and (b), respectively.
A search for structurally similar proteins was carried out using the DALI program (Holm & Sander, 1998 ▶). The closest protein structure is a hypothetical protein AQ_1354 from Aquifex aeolicus (PDB code 1oz9), which also belongs to the UPF0054 family (DALI Z score 19.6, r.m.s.d. 1.8 Å, 137 matched residues, 29.6% sequence identity for global alignment). All the other protein structures identified in the DALI search have Z scores less than 3.4. AQ_1354 is included in the sequence alignment of Fig. 2 ▶. It is predicted that AQ_1354 might be a monomer (Henrick & Thornton, 1998 ▶). The high structural similarity between AQ_1354 and ybeY implies that the ybeY protein might also be momomeric. The metal-binding site as well as some other relevant residues in the cleft are identical in ybeY and AQ_1354; however, AQ_1354 does not contain any metal atom in the structure. It was suggested that AQ_1354 might contain a metal in the binding motif and that the loss of metal might be the result of using 0.1 mM EDTA and 0.1 mM DTT in the protein purification and crystallization conditions (Oganesyan et al., 2003 ▶).
The data in this paper, which include X-ray fluorescence analysis of purified ybeY protein in solution, the crystal structure of the protein and bioinformatics analysis, provides support for the hypothesis that the ybeY protein and its closely related structure and sequence homologs contain a functional metal atom. In addition, metal binding and potential hydrolase functions may be a common feature of members of the UPF0054 protein family.
Supplementary Material
PDB reference: ybeY, 1xm5, r1xm5sf
Acknowledgments
This research is supported by the Protein Structure Initiative of the National Institute for General Medical Sciences under U54-GM74945 and the Biomedical Technology Resource Program of the National Institute for Biomedical Imaging and Bioengineering under P41-EB-01979.
References
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990). J. Mol. Biol.215, 403–410. [DOI] [PubMed] [Google Scholar]
- Barton, G. J. (1993). Protein Eng.6, 37–40. [DOI] [PubMed] [Google Scholar]
- Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E. L., Studholme, D. J., Yeats, C. & Eddy, S. R. (2004). Nucleic Acids Res.32, D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Chance, M. R., Fiser, A., Sali, A., Pieper, U., Eswar, N., Xu, G., Fajardo, J. E., Radhakannan, T. & Marinkovic, N. (2004). Genome Res.14, 2145–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA.
- Henrick, K. & Thornton, J. M. (1998). Trends Biochem. Sci.23, 358–361. [DOI] [PubMed] [Google Scholar]
- Holm, L. & Sander, C. (1998). Nucleic Acids Res, 26, 316–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. & Sander, C. (1983). Biopolymers, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kraulis, P. J. (1991). J. Appl. Cryst.24, 946–950. [Google Scholar]
- Livingstone, C. D. & Barton, G. J. (1993). Comput. Appl. Biosci.9, 745–756. [DOI] [PubMed] [Google Scholar]
- Oganesyan, V., Busso, D., Brandsen, J., Chen, S., Jancarik, J., Kim, R. & Kim, S.-H. (2003). Acta Cryst. D59, 1219–1223. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Shi, W., Zhan, C., Ignatov, A., Manjasetty, B. A., Marinkovic, N., Sullivan, M., Huang, R. & Chance, M. R. (2005). Submitted. [DOI] [PubMed]
- Tatusov, R. L., Natale, D. A., Garkavtsev, I. V., Tatusova, T. A., Shankavaram, U. T., Rao, B. S., Kiryutin, B., Galperin, M. Y., Fedorova, N. D. & Koonin, E. V. (2001). Nucleic Acids Res.29, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T. C. (1999). Acta Cryst. D55, 1863–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res.22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: ybeY, 1xm5, r1xm5sf