

The two C-terminal domains of the cellulase ctCel9D-Cel44A from C. thermocellum cellulosome have been crystallized in tetragonal space group P43212 and X-ray diffraction data have been collected to 2.1 and 2.8 Å from native and seleno-l-methionine-derivative crystals, respectively.
Keywords: cellulase, carbohydrate-binding module, glycoside hydrolase, Clostridium thermocellum
Abstract
Clostridium thermocellum produces a highly organized multi-enzyme complex of cellulases and hemicellulases for the hydrolysis of plant cell-wall polysaccharides, which is termed the cellulosome. The bifunctional multi-modular cellulase ctCel9D-Cel44A is one of the largest components of the C. thermocellum cellulosome. The enzyme contains two internal catalytic domains belonging to glycoside hydrolase families 9 and 44. The C-terminus of this cellulase, comprising a polycystic kidney-disease module (PKD) and a carbohydrate-binding module (CBM44), has been crystallized. The crystals belong to the tetragonal space group P43212, containing a single molecule in the asymmetric unit. Native and seleno-l-methionine-derivative crystals diffracted to 2.1 and 2.8 Å, respectively.
1. Introduction
Life on Earth is possible because nature has evolved mechanisms for recycling the carbon stored in organic compounds. Recalcitrant polysaccharides organized in the plant cell wall pose numerous obstacles to hydrolysis through the development of complex chemical structures and also by becoming inaccessible to biocatalysts in the intricacy of this macromolecular structure. Anaerobic bacteria and fungi have developed an extremely complex and dynamic extracellular macromolecular assembly of cellulases and hemicellulases, termed the cellulosome, which is used for the production of soluble sugars from plant cell-wall polysaccharides (for reviews, see Bayer et al., 2004 ▶; Doi & Kosugi, 2004 ▶). The cellulosome of the thermophilic bacterium Clostridium thermocellum has been extensively studied (Beguin & Lemaire, 1996 ▶; Bayer et al., 1998 ▶; Carvalho et al., 2003 ▶). In this thermostable complex, the multifunctional scaffoldin (CipA) contains nine reiterated ‘cohesin’ domains that individually bind to a 23-residue tandemly repeated complementary module, called the dockerin, present in the various cellulosomal catalytic subunits. Therefore, the dockerins of each cellulosomal enzyme bind specifically to one of the nine cohesin domains in CipA and thus the cellulosome comprises one molecule of CipA and nine discrete enzymes. The draft genome of C. thermocellum suggests that there are at least 71 cellulosomal proteins (http://www.jgi.doe.gov). Since dockerins cannot discriminate between the individual cohesins, it is believed that the enzymatic composition of each individual cellulosome is primarily regulated at the expression level (Bayer et al., 2004 ▶). A second type of cohesin–dockerin interaction is responsible for the integration of cellulosomes into supramacromolecular structures referred to as polycellulosomes.
The largest catalytic component of C. thermocellum cellulosome is the bifunctional cellulase CelJ (Ahsan et al., 1996 ▶), designated ctCel9D-Cel44A in this study to reflect the modern nomenclature of glycoside hydrolases (Henrissat, 1998 ▶). This enzyme comprises, sequentially from the N-terminus, a family 30 carbohydrate-binding module (CBM30), internal glycoside hydrolase (GH) family 9 and 44 catalytic domains, a type I dockerin, a polycystic kidney-disease (PKD) module and a C-terminal family 44 carbohydrate-binding module (CBM44). CBM30 and CBM44 direct the associated catalytic domains to the proximity of their target cellulosic substrates, therefore maximizing catalytic activity by promoting an intimate and prolonged interaction between enzyme and substrate (Boraston et al., 2004 ▶). Interestingly, the CBMs from ctCel9D-Cel44A recognize with equal efficiency undecorated and highly branched β-1,4-glucosidic ligands, such as cellulose and xyloglucans, respectively (C. Fontes, unpublished results), although the structural determinants that may allow the binding of CBMs, at a single binding site, to such different polysaccharides remain unknown. In order to gain insights into the structural properties that govern the promiscuity in ligand recognition revealed by these CBMs, we aim to determine the crystal structures of C. thermocellum CBM30 and CBM44. In the present communication, we describe the overexpression, purification, crystallization and preliminary X-ray analysis of the C. thermocellum ctCel9D-Cel44A C-terminal region comprising both the PKD and CBM44 modules.
2. Protein expression and purification
To express PKD-CBM44 in Escherichia coli, the region of the ctCel9D-Cel44A gene encoding PKD and CBM44 was amplified by PCR from C. thermocellum YS genomic DNA, using the thermostable DNA polymerase Pfu Turbo (Stratagene). The primers used, 5′-CTCGCTAGCAGTGCCGAAACAGTTGCT-3′ and 5′-CACCTCGAGCTTGATTGCAGGAGCGGA-3′, contained NheI and XhoI restriction sites, which are depicted in bold. The PCR product was cloned into pGEM T-easy (Promega) and sequenced to ensure that no mutations had occurred during the polymerase chain reaction. The recombinant pGEM T-easy derivative was digested with NheI and XhoI and the excised clostridial gene was cloned into the similarly restricted expression vector pET21a (Novagen) to generate pCG1. PKD-CBM44 encoded by pCG1 contains a C-terminal His6 tag. To express the recombinant protein, E. coli BL21 or the methionine auxotroph B834(DE3) transformed with pCG1 were cultured in 12 l Luria broth containing 100 µg ml−1 ampicillin or in 4 l liquid growth medium prepared as described by Carvalho et al. (2004 ▶), respectively, at 310 K to mid-exponential phase (A 550 = 0.6). At this point, isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM and the cultures were incubated for a further 5 h. Levels of recombinant protein expression were in both cases approximately 10 mg of PKD-CBM44 per litre of medium. The His6-tagged recombinant protein was purified from cell-free extracts by immobilized metal-ion affinity chromatography (IMAC) as described previously (Dias et al., 2004 ▶). Purified PKD-CBM44 protein was buffer-exchanged, using PD-10 Sephadex G-25M gel-filtration columns (Amersham Biosciences), into 50 mM HEPES buffer pH 7.5 containing 200 mM NaCl (buffer A), concentrated to 20 mg ml−1 with Amicon 10 kDa molecular-weight centrifugation membranes and subjected to gel filtration using a HiLoad 16/60 Superdex 75 column (Amersham Biosciences) with protein eluted at 1 ml min−1 in buffer A. The recombinant protein was concentrated as described previously and washed three times with 5 mM dl-dithiothreitol (DTT) in water (for the SeMet protein) or water (for the native protein), using the same centrifugal membrane, and the final protein concentration was adjusted to 60 mg ml−1. The purity of the protein, which was found to be nearly homogenous, was estimated using SDS–PAGE (Fig. 1 ▶).
Figure 1.

A Coomassie Brilliant Blue-stained 14% SDS–PAGE gel evaluation of protein purity during purification. Lane M, molecular-weight markers (kDa); lane 1, cell debris after sonication; lane 2, cell-free extract after sonication; lane 3, eluate from the column after sample loading; lane 4, purified PKD-CBM44 protein after Ni-affinity column chromatography.
3. Crystallization
The crystallization conditions were screened by the hanging-drop vapour-phase diffusion method using an in-house-modified version of the sparse-matrix method of Jancarik & Kim (1991 ▶) and the commercial Crystal Screen, Crystal Screen 2 and PEG/Ion Screen from Hampton Research (California, USA). Drops consisting of 1 µl 13, 30, 50 and 60 mg ml−1 PKD-CBM44 and 1 µl reservoir solution were prepared at 277 and 293 K. Clusters of tiny bipyramidal protein crystals (maximum dimension ∼50 µm) grew at 277 K within a week in the following three conditions: (i) 0.2 M MgCl2, 0.1 M HEPES pH 7.0, 30% ethanol; (ii) 0.2 M CaCl2, 0.1 M sodium acetate pH 4.5 and 30% ethanol and (iii) 0.2 M CaCl2, 0.1 M sodium acetate pH 4.5 and 20% 2-propanol. These crystals were very unstable and difficult to handle owing to the volatility of the precipitants. Crystals did not diffract using the in-house source and only diffracted to 3.5 Å at the ESRF. These crystals were improved on by fine-screening and by using Hampton Additive Screens I and II. Glycerol [6%(v/v)], CdCl2 (10–30 mM), xylitol [3–25%(v/v)] and SrCl2 (10 mM) produced fewer but larger and better diffracting crystals (0.1 × 0.1 × 0.2 mm) over a period of 1–8 weeks at 277 K (Fig. 2 ▶). These crystals were stabilized by adding 1 µl cryoprotectant solutions containing 30%(v/v) ethylene glycol or glycerol to the crystallization buffer stepwise over a few days, allowing them to equilibrate between each addition. The crystals were flash-frozen in liquid nitrogen after soaking in the cryoprotectant [either 30%(v/v) ethylene glycol or glycerol added to the crystallization buffer] for a few seconds.
Figure 2.

Crystals of PKD-CBM44 protein obtained by hanging-drop vapour diffusion in the presence of 0.2 M CaCl2, 0.1 M sodium acetate pH 4.5 and 30% ethanol with 6% glycerol as additive. The largest crystals are approximately 0.1 × 0.1 × 0.2 mm in size.
Crystals of seleno-l-methionine-containing protein were also obtained by vapour-phase diffusion using the hanging-drop method with equal volumes (1 µl) of protein solution (30 or 60 mg ml−1 in 5 mM DTT) and reservoir solution from a fine screen based around the above three successful conditions for the native crystals. Crystals grew at 277 K over a period of one week in two different conditions: 0.2 M CaCl2, 0.1 M sodium acetate pH 4.5 and 22.5% ethanol and condition (ii) above plus 10 mM SrCl2. The crystals were treated in the same way as the native crystals for stabilization and flash-freezing.
4. Data collection and processing
Initial data sets were collected on a home source with Cu Kα X-ray radiation (Enraf-Nonius rotating-anode generator operated at 5 kW, 100 mA equipped with a MAR imaging plate). A native data set (at λ = 1.2915 Å) and SeMet-labelled (at the selenium edge for MAD) data were collected on beamline ID14-EH4 at the ESRF (Grenoble, France) using a Quantum 4 charge-coupled device detector (ADSC) with the crystal cooled at 100 K using a Cryostream (Oxford Cryosystems Ltd). All data sets were processed using the programs MOSFLM (Leslie, 1992 ▶) and SCALA (Kabsch, 1988 ▶) from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). All crystals belong to the tetragonal space group. The Matthews coefficient (V M = 3.6 Å3 Da−1) indicated the presence of one molecule in the asymmetric unit and a solvent content of 65% (Matthews, 1968 ▶).
At the home source, the improved crystals diffracted to 3.05 Å. A complete and highly redundant data set was collected for a native crystal grown in crystallization buffer (ii) with 6%(v/v) glycerol. A total of 360° of data were collected with 1° oscillations over approximately 36 h. Only the first 256 images were used in processing as the crystal suffered radiation damage. Attempts to solve the native structure by molecular replacement using an ensemble of PKD domains (PDB codes 1b4r, 1ctn and 1loq; sequence identities <30%) and of various CBM (sequence identities <20%) structures were unsuccessful. This data was also not good enough to give a sufficient signal from the S atoms for a SAD experiment.
On beamline ID14-EH4 at the ESRF, the SeMet crystal diffracted to a resolution of 2.8 Å and the native crystal to 2.1 Å. Location of anomalous scatterers, phasing and density modification were carried out using SHELXD and SHELXE (Sheldrick & Schneider, 1997 ▶). The SeMet crystal suffered severe radiation damage during the edge data collection. Thus, only the SeMet peak data was used to determine the positions of 4/5 Se atoms. The correlation coefficient output by SHELXD was 40%. The pseudo-free correlation coefficient, contrast and connectivity figures of merit given by SHELXE for the correct heavy-atom enantiomer (P43212) were 48.55%, 0.807 and 0.908, respectively, as opposed to 24.85%, 0.148 and 0.789, respectively for the wrong hand (P41212). The final statistics for data collection and processing are summarized in Table 1 ▶. Structure determination is ongoing.
Table 1. Crystallization and data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Data set | Native 1 | SeMet (peak) | Native 2 |
|---|---|---|---|
| Main precipitant† | 30% ethanol + 6% glycerol | 22.5% ethanol | 20% 2-propanol + 10 mM CdCl2 |
| Cryoprotection | 30% ethylene glycol | 30% glycerol | 30% ethylene glycol |
| X-ray source | Cu Kα rotating anode, in-house | ID14-EH4, ESRF | ID14-EH4, ESRF |
| Wavelength (Å) | 1.5418 | 0.97934 | 1.2915 |
| Space group | P43212 | P43212 | P43212 |
| Unit-cell parameters (Å) | |||
| a = b | 87.21 | 87.10 | 87.47 |
| c | 106.14 | 107.67 | 103.19 |
| Resolution limits (Å) | 45–3.05 | 67.7–2.8 | 61.9–2.1 |
| No. of observations | 180868 (18185) | 127725 (17834) | 145323 (10261) |
| No. of unique observations | 8203 (1101) | 19840 (2884) | 23914 (3311) |
| Multiplicity | 22.0 (16.5) | 6.4 (6.2) | 6.1 (3.1) |
| Completeness (%) | 99.1 (94.5) | 99.8 (99.7) | 99.5 (96.7) |
| 〈I/σ(I)〉 | 20.8 (3.2) | 20.3 (2.6) | 15.1 (1.3) |
| Rmerge‡ | 20.7 (91.3) | 9.1 (56.0) | 6.7 (72.5) |
Plus 0.1 M sodium acetate pH 4.5 and 0.2 M CaCl2 to make up the crystallization buffer.
R
merge = 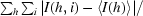
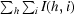 , where I(h, i) is the intensity of the measurement of reflection h and 〈I(h)〉 is the mean value of I(h, i) for all i measurements.
, where I(h, i) is the intensity of the measurement of reflection h and 〈I(h)〉 is the mean value of I(h, i) for all i measurements.
Acknowledgments
This work was supported in part by Fundação para a Ciência e a Tecnologia (Lisbon, Portugal) through grant POCI/BIA-PRO/59118/2004 and individual grants SFRH/BD/16731/2004 (CG) and SFRH/BPD/20357/2004 (SN). The authors would like to thank Dr Elspeth Gordon for her help during synchrotron data collection at beamline ID14-EH4 at the ESRF (Grenoble, France).
References
- Ahsan, M. M., Kimura, T., Karita, S., Sakka, K. & Ohmiya, K. (1996). J. Bacteriol.178, 5732–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer, E. A., Belaich, J.-P., Shoham, Y. & Lamed, R. (2004). Annu. Rev. Microbiol.58, 521–554. [DOI] [PubMed] [Google Scholar]
- Bayer, E. A., Shimon, L. J., Shoham, Y. & Lamed, R. (1998). J. Struct. Biol.124, 221–234. [DOI] [PubMed] [Google Scholar]
- Beguin, P. & Lemaire, M. (1996). Mol. Biol.31, 201–236. [DOI] [PubMed]
- Boraston, A. B., Bolam, D. N., Gilbert, H. J. & Davies, G. J. (2004). Biochem. J.382, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho, A. L., Dias, F., Prates, J. A. M., Nagy, T., Gilbert, H. J., Davies, G. J., Ferreira, L. M., Romão, M. J. & Fontes, C. M. G. A. (2003). Proc. Natl Acad. Sci. USA, 100, 13809–13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho, A. L., Goyal, A., Prates, J. A. M., Bolam, D. N., Gilbert, H. J., Pires, V. M. R., Ferreira, L. M. A., Romão, M. J. & Fontes, C. M. G. A. (2004). J. Biol. Chem.279, 34785–34793. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Dias, F. M. V., Vincent, F., Pell, G., Prates, J. A. M., Centeno, M. S. J., Tailford, L. E., Ferreira, L. M. A., Fontes, C. M. G. A., Davies, G. J. & Gilbert, H. J. (2004). J. Biol. Chem.279, 25517–25526. [DOI] [PubMed] [Google Scholar]
- Doi, R. H. & Kosugi, A. (2004). Nature Rev. Microbiol.2, 541–551. [DOI] [PubMed]
- Henrissat, B. (1998). Biochem. Soc. Trans.2, 153–156. [DOI] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Kabsch, W. (1988). J. Appl. Cryst.21, 916–924. [Google Scholar]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Sheldrick, G. M. & Schneider, T. R. (1997). Methods Enzymol.277, 319–343. [PubMed]