

Mouse MHC class I H-2Db in complex with human β2m and the LCMV-derived peptide gp33 has been produced and crystallized. Resolution of the structure of this complex combined with the structural comparison with the previously solved crystal structure of H-2Db/mβ2m/gp33 should lead to a better understanding of how the β2m subunit affects the overall conformation of MHC complexes as well as the stability of the presented peptides.
Keywords: MHC class I H-2Db, β2-microglobulin
Abstract
β2-Microglobulin (β2m) is non-covalently linked to the major histocompatibility (MHC) class I heavy chain and interacts with CD8 and Ly49 receptors. Murine MHC class I can bind human β2m (hβ2m) and such hybrid molecules are often used in structural and functional studies. The replacement of mouse β2m (mβ2m) by hβ2m has important functional consequences for MHC class I complex stability and specificity, but the structural basis for this is unknown. To investigate the impact of species-specific β2m subunits on MHC class I conformation, murine MHC class I H-2Db in complex with hβ2m and the peptide gp33 derived from lymphocytic choriomeningitis virus (LCMV) has been expressed, refolded in vitro and crystallized. Crystals containing two complexes per asymmetric unit and belonging to the space group P21, with unit-cell parameters a = 68.1, b = 65.2, c = 101.9 Å, β = 102.4°, were obtained.
1. Introduction
Class I major histocompatibility complex (MHC) molecules transport peptides derived from intracellular proteins to the cell surface and present them to CD8+ T cells and natural killer (NK) cells, respectively. The MHC class I complex consists of a polymorphic membrane-anchored heavy chain, a non-covalently bound light chain β2-microglobulin (β2m) and a peptide. β2m is a relatively conserved protein that displays 69% identity at the amino-acid level between mouse and human (Gates et al., 1981 ▶), allowing cross-species association of β2m with the MHC class I heavy chain (Bernabeu et al., 1984 ▶; Kubota, 1984 ▶).
The impact of changes in β2m species on the stability as well as the function of MHC class I molecules in the context of recognition by antibodies, T cells and NK cells have been probed in a number of studies (Matsumoto et al., 2001 ▶; Michaelsson et al., 2001 ▶; Shields et al., 1999 ▶; Wang et al., 2002 ▶; Mitsuki et al., 2004 ▶). The interactions between the three constituents of MHC class I molecules are interdependent, so that peptide binding is affected by the β2m subunit and, conversely, the binding of β2m to the heavy chain is influenced by the peptide (Shields, Kubota et al., 1998 ▶; Parker et al., 1992 ▶; Pedersen et al., 1994 ▶, 1995 ▶). Consequently, β2m has an impact on both the stability and the conformation of MHC class I complexes, as well as on peptide-exchange capacity (Pedersen et al., 1994 ▶, 1995 ▶; Shields et al., 1999 ▶; Smith et al., 1993 ▶; Schultz et al., 1998 ▶). It has been previously demonstrated that human β2m (hβ2m) in complex with mouse MHC class I heavy chains enhances the stability of the complex and its peptide-exchange capacity compared with mouse β2m (mβ2m) (Shields, Moffat et al., 1998 ▶; Pedersen et al., 1995 ▶). However, the underlying structural mechanisms have not yet been determined. An understanding of these mechanisms would facilitate the development of mutated β2m that would result in better stabilization of peptide binding.
During the last decade, a large number of inhibitory MHC class I-specific receptors expressed by NK cells and subsets of T cells have been characterized. Using soluble tetrameric MHC constructs, we and others have demonstrated that H-2Db binds to both NK receptors Ly49A and Ly49C (Hanke et al., 1999 ▶; Michaelsson et al., 2000 ▶) and that such an interaction is strongly affected by the β2m subunit (Dam et al., 2003 ▶; Michaelsson et al., 2001 ▶; Matsumoto et al., 2001 ▶; Mitsuki et al., 2004 ▶; Wang et al., 2002 ▶). Murine MHC class I molecules in complex with hβ2m are poorly recognized by these Ly49 receptors in binding and functional studies (Matsumoto et al., 2001 ▶; Michaelsson et al., 2001 ▶).
The molecular and mechanistic basis underlying the capacity of the co-receptor CD8 to enhance T-cell recognition is still not fully understood. There is a general correlation between T-cell stimulation and the affinity of the T-cell receptor (TCR) for the MHC–peptide complex and the properties of the bound peptides affect activation of the TCR (Holler & Kranz, 2003 ▶). It is known that CD8 enhances T-cell antigen recognition by binding to the α2, α3 and β2m domains of MHC class I molecules during recognition of peptide-MHC class I antigens on the surface of target cells (Holler & Kranz, 2003 ▶; Gao et al., 1997 ▶; Kern et al., 1998 ▶; Liu et al., 2003 ▶; Luescher et al., 1995 ▶). Murine CD8αα makes contact with at least four residues in mβ2m, but the contribution of these residues to CD8 binding has not been fully investigated.
To investigate the impact of species-specific β2m subunits on MHC class I conformation in relation to the aspects reviewed above, we will compare the crystal structures of complexes of H-2Db and gp33, a peptide derived from lymphocytic choriomeningitis virus (LCMV), with mβ2m on one hand and hβ2m on the other. The structures of H-2Db–hβ2m–gp33 and of the previously solved H-2Db–mβ2m–gp33 complexes (Velloso et al., 2004 ▶; Achour et al., 2002 ▶) only differ in the β2m subunit. Such comparison should thus allow a more accurate identification of the structural differences imposed by substituting one β2m subunit for another.
2. Materials and methods
2.1. Generation of soluble H-2Db–hβ2m–gp33 MHC complexes
The H-2Db cDNA sequence coding for amino acids 1–276 (GPHSMRYFETAVSRPGLEEPRYISVGYVDNKEFVRFDSDAENPRYEPRAPWMEQEGPEYWERETQKAKGQEQWFRVSLRNLLGYYNQSAGGSHTLQQMSGCDLGSDWRLLRGYLQFAYEGRDYIALNEDLKTWTAADMAAQITRRKWEQSGAAEHYKAYLEGECVEWLHRYLKNGNATLLRTDSPKAHVTHHPRSKGEVTLRCWALGFYPADITLTWQLNGEELTQDMELVETRPAGDGTFQKWASVVVPLGKEQNYTCRVYHEGLPEPLTLRWEP) was amplified by RT-PCR. The sequenced PCR product was cloned in the pET-17b expression vector (Novagen) and transformed into BL21(DE3)pLysS (Novagen). The human β2m sequence encoding amino acids 1–99 (IQRTPKIQVYSRHPAENGKSNFLNCYVSGFHPSDIEVDLLKNGERIEKVEHSDLSFSKDWSFYLLYYTEFTPTEKDEYACRVNHVTLSQPKIVKWDRDM) cloned in the pHN1 plasmid vector was kindly provided by Dr E. Y. Jones (Oxford Centre for Molecular Science, Oxford, England). An additional methionine residue was inserted at the N-terminal of the hβ2m recombinant protein. No tags were used in any of the constructs. Peptide gp33 (KAVYNFATM) was from Research Genetics (Huntsville, AL, USA). Protein expression of both H-2Db heavy chain and hβ2m was induced by IPTG and the products were purified separately as inclusion bodies using previously described protocols (Achour et al., 1999 ▶, 2002 ▶). H-2Db heavy chain and hβ2m were produced independently from each other. The proteins were extracted from inclusion bodies in 8 M urea. The concentrations of the final products were determined spectrophotometrically and the purity of the products was assessed by SDS–PAGE under denaturing conditions (Laemmli, 1970 ▶). Most often, we obtained an average of 60–100 mg ml−1 H-2Db or hβ2m protein per litre of IPTG-induced bacterial culture. All of the three components that form the MHC complex (heavy chain, β2m and peptide) are required for the proper refolding of MHC class I complexes. Refolding was performed by dilution at 277 K using a molar ratio of heavy chain:β2m:peptide of 1:2:10 (Garboczi et al., 1992 ▶; Reid et al., 1996 ▶). The refolding solution comprised 0.4 M l-arginine, 20 mM Tris–HCl pH 7.5, 1 mM EDTA, 5 mM GSH (oxidized glutathione), 0.5 mM GSSG (reduced glutathione) and 100 mM PMSF. The β2m subunit can be refolded alone and is very stable. This molecule is commonly added to the refolding solution and used as a preliminary template in order to increase refolded MHC complex ratios. The refolding of the complex was thus induced by the addition of 3 mg refolded hβ2m to the refolding solution, which was kept cold at 277 K. A combination of 10 mg peptide, 17 mg hβ2m and 20 mg H-2Db was added to the solution and stirred for 48 h. The refolding was pulsed four times with H-2Db heavy chain at intervals of 8 h with 5 mg H-2Db in urea. After 48 h, the refolding mixture was concentrated using Amicon concentration devices (Millipore). Refolded H-2Db–hβ2m–gp33 complexes were purified and buffer-exchanged into 20 mM Tris–HCl pH 7.5 by FPLC using a Superdex 75 gel-filtration column (Amersham Biosciences, Uppsala, Sweden). The yield of refolded complexes was in most cases about 10% of the original material.
2.2. Crystallization of H-2Db in complex with hβ2m and LCMV-derived gp33
Crystals were obtained in hanging drops by vapour diffusion. Crystal screens (Hampton Research, Laguna Niguel, CA, USA) were used to establish initial crystallization conditions, which were then refined in a finer grid. The best crystals were obtained in 20% PEG 6K, 100 mM Tris–HCl pH 8.0 at room temperature. Typically, 2 µl 6 mg ml−1 protein solution in 20 mM Tris–HCl pH 7.5 was mixed at a 1:1 ratio with the crystallization reservoir solution (20% PEG 6 K, 100 mM Tris–HCl pH 8.0).
2.3. Data collection and processing
Data collection was performed under cryogenic conditions (T = 100 K) at beamline I711 in MAX-lab (Lund, Sweden) (λ = 0.9831 Å) using a MAR 345 image plate. X-ray data for the H-2Db–gp33–hβ2m complex were collected to 2.7 Å resolution. Crystals were first soaked in a cryoprotectant solution containing 25% glycerol in reservoir solution (20% PEG 6K, 100 mM Tris–HCl pH 8.0) before data collection. The diffraction data were processed using the HKL program package (Otwinowski, 1993 ▶). Data-collection statistics for the data set used in the final refinement are presented in Table 1 ▶. Crystals of the H-2Db–gp33–hβ2m complexes belong to space group P21, with unit-cell parameters a = 68.1, b = 65.2, c = 101.9 Å, β = 102.4°.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell (2.75–2.7 Å).
| X-ray source | I711 |
| Wavelength (Å) | 1.029 |
| Resolution (Å) | 25.0–2.7 |
| Space group | P21 |
| Unit-cell parameters | a = 68.1, b = 65.2, c = 101.9, α = 90, β = 102.4, γ = 90 |
| Total No. of unique reflections | 22481 |
| No. of observed reflections | 82092 |
| Completeness (%) | 91.8 (51.4) |
| Rmerge† | 4.9 (31.5) |
| 〈I/σ(I)〉 | 20.2 (2.4) |
| Mosaicity (°) | 0.77 |
R
merge = 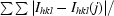
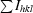 , where I
hkl(j) is the observed intensity and I
hkl is the average value of the intensity.
, where I
hkl(j) is the observed intensity and I
hkl is the average value of the intensity.
3. Results and discussion
H-2Db heavy chain and β2m were expressed to high levels (80 ± 10 mg l−1) with a final purity greater than 90%. The H-2Db molecules were refolded in the presence of hβ2m and peptide. Chromatographic elution profiles were completely reproducible, demonstrating three peaks comprising aggregated high-molecular-weight complexes (peak 1), the refolded complex (peak 2) and uncomplexed β2m (peak 3) (Fig. 1 ▶ a). The refolding resulted in yields of approximately 12–15% of MHC complexes, which could be purified to homogeneity by FPLC gel filtration (Fig. 1 ▶ b). As complex formation was limited by availability of the heavy chain, there was always an excess of β2m, as represented by peak 3 (Fig. 1 ▶ a), which was recycled in subsequent refolding procedures.
Figure 1.

(a) Typical chromatogram depicting the elution profile of FPLC Superdex 75 gel-filtrated refolding products. The load rate was 0.5 ml min−1, the sample volume was 200 µl and elution was monitored at 280 nm. The first void peak (1) contains non-native aggregated products, peak 2 represents correctly refolded monomeric H-2Db complexes and peak 3 represents hβ2m. (b) Proteins separated by SDS–PAGE (15%) under reducing conditions stained with Coomassie brilliant blue. Lane 1, molecular-weight markers (protein size in kDa is indicated to the left); lane 2, purified H-2Db–hβ2m–gp33 MHC complex.
The best crystals appeared after a few days in 20% PEG 6K, 100 mM Tris–HCl pH 8.0 at room temperature (Fig. 2 ▶). The H-2Db–hβ2m–gp33 complex crystallized in space group P21, with unit-cell parameters a = 68.1, b = 65.2, c = 101.9 Å, β = 102.4°. A Matthews coefficient calculation, determined assuming two MHC complexes per asymmetric unit, resulted in a V M value of 2.6 Å3 Da−1, corresponding to 52% solvent content. For three MHC complexes in the asymmetric unit the V M value was 1.7 Å3 Da−1, corresponding to only 27% solvent content in the crystal. Thus, the presence of three MHC complexes in the asymmetric unit is not very probable, but cannot be ruled out. A self-rotation analysis showed no evidence of either twofold or threefold non-crystallographic symmetry. However, a native Patterson map revealed the presence of a strong peak (27% of origin peak) at x = 0, y = 0.5 and z = 0.5, implying a pure translational relationship between two complexes in the asymmetric unit. The statistics of the data set are listed in Table 1 ▶. The average B factor as derived from a Wilson plot was 74 Å2. The native data set to 2.7 Å resolution is 91.8% complete and has an R merge of 4.9% and 〈I/σ(I)〉 = 20.2. The structure determination of the H-2Db–hβ2m–gp33 complex is currently under way using molecular replacement.
Figure 2.

Photograph of a typical crystal of H-2Db–hβ2m–gp33. Crystal dimensions are 0.4 × 0.2 × 0.1 mm.
4. Conclusions
We have produced and crystallized mouse MHC class I H-2Db in complex with human β2m and the LCMV-derived peptide gp33. Resolution of the structure of this complex combined with the structural comparison with the previously solved crystal structure of H-2Db–mβ2m–gp33 (Achour et al., 2002 ▶; Tissot et al., 2000 ▶) should lead to a better understanding of how the β2m subunit affects the overall conformation of MHC complexes as well as the stability of the presented peptides. Since these two MHC complexes only differ in the nature of the β2m species, their comparison should also provide us with important information regarding the conformation of the presented peptide, the role of specific β2m amino-acid residues in the previously established better stabilization of peptides by hβ2m and the interaction of MHC class I molecules with T-cell and NK-cell receptors.
Acknowledgments
We gratefully acknowledge access to synchrotron radiation at beamline 711 at MAX-lab, Lund University, Sweden. This work was supported by grants from the Swedish Foundation for Strategic Research, the Network for Inflammation Research, the Swedish Cancer Society, the Swedish Research Council, the Åke Wiberg, Alex and Ewa Wallström, Magnus Bergwalls and the Grochinsky Foundations.
References
- Achour, A., Harris, R. A., Persson, K., Sundback, J., Sentman, C. L., Schneider, G., Lindqvist, Y. & Karre, K. (1999). Acta Cryst. D55, 260–262. [DOI] [PubMed] [Google Scholar]
- Achour, A., Michaelsson, J., Harris, R. A., Odeberg, J., Grufman, P., Sandberg, J. K., Levitsky, V., Karre, K., Sandalova, T. & Schneider, G. (2002). Immunity, 17, 757–768. [DOI] [PubMed] [Google Scholar]
- Bernabeu, C., van de Rijn, M., Lerch, P. G. & Terhorst, C. P. (1984). Nature (London), 308, 642–645. [DOI] [PubMed] [Google Scholar]
- Dam, J., Guan, R., Natarajan, K., Dimasi, N., Chlewicki, L. K., Kranz, D. M., Schuck, P., Margulies, D. H. & Mariuzza, R. A. (2003). Nature Immunol.4, 1213–1222. [DOI] [PubMed]
- Gao, G. F., Tormo, J., Gerth, U. C., Wyer, J. R., McMichael, A. J., Stuart, D. I., Bell, J. I., Jones, E. Y. & Jakobsen, B. K. (1997). Nature (London), 387, 630–634. [DOI] [PubMed] [Google Scholar]
- Garboczi, D. N., Hung, D. T. & Wiley, D. C. (1992). Proc. Natl Acad. Sci. USA, 89, 3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates, F. T. III, Coligan, J. E. & Kindt, T. J. (1981). Proc. Natl Acad. Sci. USA, 78, 554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke, T., Takizawa, H., McMahon, C. W., Busch, D. H., Pamer, E. G., Miller, J. D., Altman, J. D., Liu, Y., Cado, D., Lemonnier, F. A., Bjorkman, P. J. & Raulet, D. H. (1999). Immunity, 11, 67–77. [DOI] [PubMed] [Google Scholar]
- Holler, P. D. & Kranz, D. M. (2003). Immunity, 18, 255–264. [DOI] [PubMed] [Google Scholar]
- Kern, P. S., Teng, M. K., Smolyar, A., Liu, J. H., Liu, J., Hussey, R. E., Spoerl, R., Chang, H. C., Reinherz, E. L. & Wang, J. H. (1998). Immunity, 9, 519–530. [DOI] [PubMed] [Google Scholar]
- Kubota, K. (1984). J. Immunol.133, 3203–3210. [PubMed] [Google Scholar]
- Laemmli, U. K. (1970). Nature (London), 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Liu, Y., Xiong, Y., Naidenko, O. V., Liu, J. H., Zhang, R., Joachimiak, A., Kronenberg, M., Cheroutre, H., Reinherz, E. L. & Wang, J. H. (2003). Immunity, 18, 205–215. [DOI] [PubMed] [Google Scholar]
- Luescher, I. F., Vivier, E., Layer, A., Mahiou, J., Godeau, F., Malissen, B. & Romero, P. (1995). Nature (London), 373, 353–356. [DOI] [PubMed] [Google Scholar]
- Matsumoto, N., Mitsuki, M., Tajima, K., Yokoyama, W. M. & Yamamoto, K. (2001). J. Exp. Med.193, 147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelsson, J., Achour, A., Rolle, A. & Karre, K. (2001). J. Immunol.166, 7327–7334. [DOI] [PubMed] [Google Scholar]
- Michaelsson, J., Achour, A., Salcedo, M., Kase-Sjostrom, A., Sundback, J., Harris, R. A. & Karre, K. (2000). Eur. J. Immunol.30, 300–307. [DOI] [PubMed] [Google Scholar]
- Mitsuki, M., Matsumoto, N. & Yamamoto, K. (2004). Int. Immunol.16, 197–204. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. (1993). Proceedings of the CCP4 Study Weekend. Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 56–62. Warrington: Daresbury Laboratory.
- Parker, K. C., DiBrino, M., Hull, L. & Coligan, J. E. (1992). J. Immunol.149, 1896–1904. [PubMed] [Google Scholar]
- Pedersen, L. O., Hansen, A. S., Olsen, A. C., Gerwien, J., Nissen, M. H. & Buus, S. (1994). Scand. J. Immunol.39, 64–72. [DOI] [PubMed] [Google Scholar]
- Pedersen, L. O., Stryhn, A., Holter, T. L., Etzerodt, M., Gerwien, J., Nissen, M. H., Thogersen, H. C. & Buus, S. (1995). Eur. J. Immunol.25, 1609–1616. [DOI] [PubMed] [Google Scholar]
- Reid, S. W., Smith, K. J., Jakobsen, B. K., O’Callaghan, C. A., Reyburn, H., Harlos, K., Stuart, D. I., McMichael, A. J., Bell, J. I. & Jones, E. Y. (1996). FEBS Lett.383, 119–123. [DOI] [PubMed] [Google Scholar]
- Schultz, C. S., Rodriguez, R. A., Chew, E. A., Dimaano, C., Li, F. M., Santos, M. D. & Nieto, M. C. (1998). Immunogenetics, 48, 273–282. [DOI] [PubMed] [Google Scholar]
- Shields, M. J., Hodgson, W. & Ribaudo, R. K. (1999). Mol. Immunol.36, 561–573. [DOI] [PubMed] [Google Scholar]
- Shields, M. J., Kubota, R., Hodgson, W., Jacobson, S., Biddison, W. E. & Ribaudo, R. K. (1998). J. Biol. Chem.273, 28010–28018. [DOI] [PubMed] [Google Scholar]
- Shields, M. J., Moffat, L. E. & Ribaudo, R. K. (1998). Mol. Immunol.35, 919–928. [DOI] [PubMed] [Google Scholar]
- Smith, M. J., Basora, T., Kieran, J. E. & Nieto, M. C. (1993). Immunol. Cell Biol.71, 145–149. [DOI] [PubMed] [Google Scholar]
- Tissot, A. C., Ciatto, C., Mittl, P. R., Grutter, M. G. & Pluckthun, A. (2000). J. Mol. Biol.302, 873–885. [DOI] [PubMed] [Google Scholar]
- Velloso, L. M., Michaelsson, J., Ljunggren, H. G., Schneider, G. & Achour, A. (2004). J. Immunol.172, 5504–5511. [DOI] [PubMed] [Google Scholar]
- Wang, J., Whitman, M. C., Natarajan, K., Tormo, J., Mariuzza, R. A. & Margulies, D. H. (2002). J. Biol. Chem.277, 1433–1442. [DOI] [PubMed] [Google Scholar]