

The crystallization of HLA-B*2706 in complex with two peptides is reported.
Keywords: HLA-B27 subtypes, major histocompatibility complex polymorphism, HLA-B*2706, subtype-dependent peptide-binding modes, ankylosing spondylitis
Abstract
The human leukocyte antigen (HLA) alleles HLA-B*2704 and HLA-B*2706 show an ethnically restricted distribution and are differentially associated with ankylosing spondylitis, with HLA-B*2706 lacking association with this autoimmune disease. However, the products of the two alleles differ by only two amino acids, at heavy-chain residues 114 (His in HLA-B*2704; Asp in HLA-B*2706) and 116 (Asp in HLA-B*2704; Tyr in HLA-B*2706). Both residues could be involved in contacting amino acids of a bound peptide, suggesting that peptides presented by these subtypes play a role in disease pathogenesis. Two HLA-B*2706–peptide complexes were crystallized using the hanging-drop vapour-diffusion method with PEG as precipitant. Data sets were collected to resolutions of 2.70 Å (viral peptide pLMP2, RRRWRRLTV; space group P212121) and 1.83 Å (self-peptide pVIPR, RRKWRRWHL; space group P21). Using HLA-B*2705 complexed with the pGR peptide (RRRWHRWRL) as a search model, unambiguous molecular-replacement solutions were found for both HLA-B*2706 complexes.
1. Introduction
The association of most or even all autoimmune diseases with the major histocompatibility complex (HLA complex in humans) is one of the hallmarks of this genetic region (Horton et al., 2004 ▶). However, the molecular details underlying these associations have only begun to be understood in a small number of cases (Rioux & Abbas, 2005 ▶). These difficulties are exemplified by the class I allele HLA-B27. Although it has been known since 1973 that HLA-B27 is very strongly associated with spondyloarthropathies (Brewerton et al., 1973 ▶; Schlosstein et al., 1973 ▶), there is still no generally accepted explanation that could account for all clinical, biochemical and functional aspects that characterize the relationship between HLA-B27 and these diseases. Spondyloarthropathies encompass an entire set of related diseases, with ankylosing spondylitis (AS) being one of them (Khan, 2002 ▶). A possible clue to explain the association between AS and HLA-B27 might come from a detailed functional and biophysical comparison of HLA-B27 subtypes that differ in their disease association but are nevertheless very closely related (Khan & Ball, 2002 ▶; Ramos & López de Castro, 2002 ▶; Kim et al., 2005 ▶).
HLA class I molecules consist of a highly polymorphic heavy chain (HC) that is non-covalently associated with the non-polymorphic protein β2-microglobulin (β2m). Two extracellular HC domains form a groove which carries a peptide that may be derived from self- or nonself-proteins within the cell (Madden, 1995 ▶). Structural and thermodynamic comparisons of the products of HLA-B27 subtypes in complex with several different peptides have already been carried out (see, for example, Hülsmeyer et al., 2002 ▶, 2004 ▶, 2005 ▶; Fiorillo et al., 2005 ▶; Rückert et al., 2005 ▶). In individuals with the AS-associated HLA-B27 subtype B*2705, cytotoxic T-lymphocytes with specificity for the self-antigen pVIPR [RRKWRRWHL, derived from vasoactive intestinal peptide type 1 receptor (residues 400–408)] have been observed. These cells increase in number during the development of AS (Fiorillo et al., 2000 ▶) and a substantial fraction cross-reacts with the viral pLMP2 peptide [RRRWRRLTV, derived from latent membrane protein 2 (residues 236–244) of Epstein–Barr virus; Brooks et al., 1993 ▶; Fiorillo et al., 2000 ▶]. However, pVIPR-reactive T cells were not found in individuals with the non-AS-associated B*2709 subtype (Fiorillo et al., 2000 ▶). The B*2705 and B*2709 proteins differ by only a single amino-acid substitution (Asp116His) at the floor of the binding groove, but this exchange has drastic consequences for the conformations of the pVIPR and pLMP2 peptides within the peptide-binding clefts (Hülsmeyer et al., 2004 ▶; Fiorillo et al., 2005 ▶).
The alleles B*2704 and B*2706 are also differentially associated with AS and constitute another pair of subtypes that differ only minimally from each other (Vega et al., 1986 ▶; Rudwaleit et al., 1996 ▶): B*2704 (His114, Asp116, as in B*2705) and B*2706 (Asp114, Tyr116) may also be expected to bind peptides distinguishably, thereby leading to differential T-cell responses (Sesma et al., 2002 ▶). Peptide-elution experiments have indeed shown that 12% of the peptides bound to B*2704 cannot be eluted from B*2706 molecules, while 10% of the B*2706-bound peptides are not detectable in B*2704 (Garcia et al., 1997 ▶; Sesma et al., 2002 ▶; López de Castro et al., 2004 ▶). Both subtypes are restricted to orientals, in particular individuals from China and Southeast Asia, and it was demonstrated that, like B*2705, B*2704 is associated with AS, while B*2706 (like B*2709) is not (López-Larrea et al., 1995 ▶; Nasution et al., 1997 ▶; Ren et al., 1997 ▶; Chen et al., 2002 ▶; Dhaliwal et al., 2003 ▶).
A structural comparison of B*2706 with other HLA-B27 subtypes, especially with the closely related B*2704 (Loll, Zawacka, Biesiadka, Petter et al., 2005 ▶), will contribute to a better understanding of the effect of amino-acid exchange at the floor of the peptide-binding groove on the repertoire of bound peptides. In particular, it will be interesting to investigate the possibility of differential subtype-dependent molecular mimicry (Lang et al., 2002 ▶) between pLMP2 and pVIPR. Molecular mimicry has been observed between the two peptides when displayed by B*2705, but not by B*2709, indicating that it might be connected with the distinct AS-associations of these subtypes (Fiorillo et al., 2005 ▶).
2. Materials and methods
2.1. Protein preparation
The nonapeptides pLMP2 (RRRWRRLTV) and pVIPR (RRKWRRWHL) were synthesized using standard solid-phase procedures and purified by reverse-phase HPLC (Alta Bioscience, Birmingham, England). The cDNA clone for the HC of B*2706 containing the three extracellular domains was generated by in vitro mutagenesis from a B*2704 clone. Human β2m and the HC of B*2706 were separately expressed in Escherichia coli as inclusion bodies. They were solubilized by treatment with 50%(w/v) urea. According to a previously described reconstitution protocol (Garboczi et al., 1992 ▶; Hülsmeyer et al., 2002 ▶), the HLA-B27–peptide complexes were refolded for 14 d at 277 K, starting from denatured HC (12 mg), β2m (10 mg) and 4 mg of either pVIPR or pLMP2. All three components were rapidly injected into 400 ml refolding buffer (400 mM arginine–HCl, 2 mM EDTA, 5 mM reduced glutathione, 0.5 mM oxidized glutathione, 100 mM Tris–HCl pH 7.5). Size-exclusion chromatography with Superdex 75 (Pharmacia) was then utilized to obtain highly pure intact complexes that eluted at a position corresponding to their calculated size. Fractions containing the ternary complexes were pooled and their composition (HC, β2m) surveyed by sodium dodecyl sulfate gel electrophoresis. As omission of the peptides during reconstitution invariably resulted in lack of complex formation and absence of the chromatographic peak that is characteristic of the ternary complex, no further biochemical analyses regarding its composition were carried out. For crystallization experiments the protein was concentrated by Amicon Ultra-15 to 13–15 mg ml−1 in a buffer system containing 20 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.01% sodium azide.
2.2. Crystallization and data collection
B*2706–pLMP2 and B*2706–pVIPR were crystallized by hanging-drop vapour diffusion at 291 K on a siliconized cover slip over a 1 ml reservoir containing 1 ml precipitant solution (100 mM Tris–HCl pH 7.0 or pH 8.0) and varying concentrations [18–28%(w/v)] of polyethylene glycol (PEG) 8000, PEG 6000 or PEG 4000 to optimize crystal formation. 1.5 µl protein solution was mixed with 1.5 µl precipitant solution identical to that within the respective reservoir, employing conditions that have been described previously (Hülsmeyer et al., 2002 ▶, 2004 ▶, 2005 ▶; Fiorillo et al., 2005 ▶; Loll, Zawacka, Biesiadka, Petter et al., 2005 ▶). The wells were immediately sealed, but since the initially obtained microcrystals were too small for X-ray data collection, streak-seeding was carried out after 1–1.5 h. A thick cat whisker was used to crush crystals in a crystallization drop from a previous experiment involving a similar composition of the precipitant solution and a thinner whisker was initially drawn through this drop and then through the drop with the newly prepared B*2706–pLMP2 or B*2706–pVIPR complexes. Following a second sealing of the wells, crystals of B*2706–pLMP2 were obtained after 4 d from a solution containing 24%(w/v) PEG 8000, 100 mM Tris–HCl pH 7.0 and B*2706–pVIPR crystallized using a precipitant solution composed of 20%(w/v) PEG 8000, 100 mM Tris–HCl pH 7.0. The crystals of both subtypes grew to maximum dimensions of about 80 × 20 × 10 µm in one week and had similar size and morphology. Prior to data collection, the crystals were transferred to a cryoprotectant solution by stepwise increases of the glycerol concentration to 10% and finally frozen in liquid nitrogen.
The data set from B*2706–pLMP2 was collected at beamline ID 14-2 of the European Synchrotron Radiation Facility (ESRF) in Grenoble (France) and that from B*2706–pVIPR at beamline BW6 of the Deutsches Elektronen-Synchrotron (DESY) in Hamburg (Germany). Both data sets were measured at a temperature of 100 K. The quality of B*2706–pLMP2 diffraction was slightly improved using flash-annealing (Harp et al., 1998 ▶) by shielding the cryostream for about 5 s. Integration and scaling were accomplished using the HKL suite (Otwinowski & Minor, 1997 ▶). The crystals of B*2706–pLMP2 belonged to the orthorhombic space group P212121 and diffracted to 2.70 Å, whereas crystals of B*2706–pVIPR belonged to the monoclinic space group P21 and diffracted beyond 1.83 Å (Table 1 ▶). Applying the Matthews equation (Matthews, 1968 ▶) and assuming a molecular weight of ∼44 kDa for the HC–peptide–β2m complex, solvent contents of 49.7% for B*2706–pLMP2 and 57.5% for B*2706–pVIPR were estimated with one molecule per asymmetric unit. Crystallization data and X-ray data-collection statistics are summarized in Table 1.
Table 1. Data-collection statistics of HLA-B*2706–pLMP2 and HLA-B*2706–pVIPR.
Values in parentheses refer to the highest resolution shell.
| HLA-B*2706–pLMP2 | HLA-B*2706–pVIPR | |
|---|---|---|
| Space group | P212121 | P21 |
| Unit-cell parameters (Å, °) | a = 50.7, b = 82.0, c = 108.9, α = 90, β = 90, γ = 90 | a = 50.8, b = 81.7, c = 64.43, α = 90, β = 108, γ = 90 |
| Solvent content (%) | 49.6 | 57.5 |
| Matthews coefficient† (Å3 Da−1) | 2.5 | 2.9 |
| Resolution (Å) | 30.00–2.70 (2.77–2.70) | 30.00–1.83 (1.90–1.83) |
| Unique reflections | 12253 (880) | 43847 (4069) |
| Completeness (%) | 99.0 (93.2) | 99.1 (92.2) |
| 〈I〉/〈σ(I)〉 | 16.5 (3.5) | 34.7 (6.0) |
| Rsym‡ | 0.054 (0.283) | 0.036 (0.209) |
According to Matthews (1968 ▶).
R
sym = 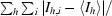
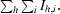
The phase problem was solved using likelihood-based molecular replacement with PHASER (Storoni et al., 2004 ▶). The atomic coordinates of the high-resolution crystal structure of B*2705–pGR (PDB code 2a83; Rückert et al., 2005 ▶) served as search model (peptide and water molecules as well as the side chains of residues distinguishing B*2705 from B*2706 were omitted). Clear solutions for both structures were found using diffraction data in the resolution range 20–3 Å. Examination of the electron-density map generated from the initial phases allowed us to identify the presence of the nonapeptides as well as polymorphic HC amino-acid residues.
3. Results and discussion
The highly pure B*2706–pLMP2 and B*2706–pVIPR complexes were crystallized and crystal formation was optimized using streak-seeding techniques, resulting in well ordered crystals that diffracted to 2.70 Å (B*2706–pLMP2) and 1.83 Å (B*2706–pVIPR) resolution at synchrotron sources (ESRF and DESY, respectively). Contrary to our expectation from experiments involving the B*2704 complexes with the pLMP2 and pVIPR peptides (Loll, Zawacka, Biesiadka, Petter et al., 2005 ▶), which both crystallized in space group P212121, the B*2706–pLMP2 and B*2706–pVIPR complexes crystallized in space groups P212121 and P21, respectively. This lack of crystal isomorphism has been previously observed in crystals of B*2703 in complex with pLMP2 and pVIPR (Loll, Zawacka, Biesiadka, Rückert et al., 2005 ▶) and might indicate that the nonapeptides adopt conformations in B*2706 similar to those in B*2703 (Loll, Zawacka, Biesiadka, Rückert et al., 2005 ▶; Zawacka et al., manuscript in preparation). However, since B*2706 differs in six amino acids from B*2703, with five of these being part of the peptide-binding groove (Ramos & López de Castro, 2002 ▶; López de Castro et al., 2004 ▶), it is impossible to predict the peptide-binding mode within the two B*2706 complexes with certainty without detailed model building.
To this end, refinement of both B*2706 crystal structures has been initiated to permit a comparison with the previously solved crystal structures of pLMP2 and pVIPR with B*2705, B*2709 and B*2703 (Hülsmeyer et al., 2004 ▶; Fiorillo et al., 2005 ▶; Loll, Zawacka, Biesiadka, Rückert et al., 2005 ▶) and in particular with the closely related B*2704 (Loll, Zawacka, Biesiadka, Petter et al., 2005 ▶).
Acknowledgments
Financial support for this work was provided by Deutsche Forschungsgemeinschaft (to BL and JB as well as to WS, BU-Z and A. Ziegler through SFB 449/B6) and by Fonds der Chemischen Industrie [to A. Zawacka (Kekulé-Fellowship) and WS]. We thank Uschi Gruber for excellent technical assistance and are grateful for allocation of beam time and support at ESRF (Grenoble) as well as DESY (Hamburg).
References
- Brewerton, D. A., Hart, F. D., Nicholls, A., Caffrey, M., James, D. C. & Sturrock, R. D. (1973). Lancet, 1, 904–907. [DOI] [PubMed] [Google Scholar]
- Brooks, J. M., Murray, R. J., Thomas, W. A., Kurilla, M. G. & Rickinson, A. B. (1993). J. Exp. Med.178, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, I. H., Yang, K. L., Lee, A., Huang, H. H., Lin, P. Y. & Lee, T. D. (2002). Eur. J. Immunogenet.29, 435–438. [DOI] [PubMed] [Google Scholar]
- Dhaliwal, J. S., Too, C. L., Lisut, M., Lee, Y. Y. & Murad, S. (2003). Tissue Antigens, 62, 330–332. [DOI] [PubMed] [Google Scholar]
- Fiorillo, M. T., Maragno, M., Butler, R., Dupuis, M. L. & Sorrentino, R. (2000). J. Clin. Invest.106, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo, M. T., Rückert, C., Hülsmeyer, M., Sorrentino, R., Saenger, W., Ziegler, A. & Uchanska-Ziegler, B. (2005). J. Biol. Chem.280, 2962–2971. [DOI] [PubMed] [Google Scholar]
- Garboczi, D. N., Hung, D. T. & Wiley, D. C. (1992). Proc. Natl Acad. Sci. USA, 89, 3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, F., Marina, A. & López de Castro, J. A. (1997). Tissue Antigens, 49, 215–221. [DOI] [PubMed]
- Harp, J. M., Timm, D. E. & Bunick, G. (1998). Acta Cryst. D54, 622–628. [DOI] [PubMed] [Google Scholar]
- Horton, R., Wilming, L., Rand, V., Lovering, R. C., Bruford, E. A., Khodiyar, V. K., Lush, M. J., Povey, S., Talbot, C. C. Jr, Wright, M. W., Wain, H. M., Trowsdale, J., Ziegler, A. & Beck, S. (2004). Nature Rev. Genet.5, 889–899. [DOI] [PubMed]
- Hülsmeyer, M., Fiorillo, M. T., Bettosini, F., Sorrentino, R., Saenger, W., Ziegler, A. & Uchanska-Ziegler, B. (2004). J. Exp. Med.199, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hülsmeyer, M., Hillig, R. C., Volz, A., Rühl, M., Schröder, W., Saenger, W., Ziegler, A. & Uchanska-Ziegler, B. (2002). J. Biol. Chem.277, 47844–47853. [DOI] [PubMed] [Google Scholar]
- Hülsmeyer, M., Welfle, K., Pöhlmann, T., Misselwitz, R., Alexiev, U., Welfle, H., Saenger, W., Uchanska-Ziegler, B. & Ziegler, A. (2005). J. Mol. Biol.346, 1367–1379. [DOI] [PubMed] [Google Scholar]
- Khan, M. A. (2002). Ann. Intern. Med.136, 896–907. [DOI] [PubMed] [Google Scholar]
- Khan, M. A. & Ball, E. J. (2002). Best Pract. Res. Clin. Rheumatol.16, 675–690. [PubMed] [Google Scholar]
- Kim, T. H., Uhm, W. S. & Inman, R. D. (2005). Curr. Opin. Rheumatol.17, 400–405. [DOI] [PubMed] [Google Scholar]
- Lang, H. L., Jacobsen, H., Ikemizu, S., Andersson, C., Harlos, K., Madsen, L., Hjorth, P., Sondergaard, L., Svejgaard, A., Wucherpfennig, K., Stuart, D. I., Bell, J. I., Jones, E. Y. & Fugger, L. (2002). Nature Immunol.3, 940–943. [DOI] [PubMed]
- Loll, B., Zawacka, A., Biesiadka, J., Petter, C., Rückert, C., Saenger, W., Uchanska-Ziegler, B. & Ziegler, A. (2005). Acta Cryst. F61, 939–941. [DOI] [PMC free article] [PubMed]
- Loll, B., Zawacka, A., Biesiadka, J., Rückert, C., Volz, A., Saenger, W., Uchanska-Ziegler, B. & Ziegler, A. (2005). Acta Cryst. F61, 372–374. [DOI] [PMC free article] [PubMed]
- López de Castro, J. A., Alvarez, I., Marcilla, M., Paradela, A., Ramos, M., Sesma, L. & Vazquez, M. (2004). Tissue Antigens, 63, 424–445. [DOI] [PubMed] [Google Scholar]
- López-Larrea, C., Sujirachato, K., Mehra, N. K., Chiewsilp, P., Isarangkura, D., Kanga, U., Dominguez, O., Coto, E., Pena, M., Setien, F. & Gonzalez-Roces, S. (1995). Tissue Antigens, 45, 169–176. [DOI] [PubMed] [Google Scholar]
- Madden, D. R. (1995). Annu. Rev. Immunol.13, 587–622. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Nasution, A. R., Mardjuadi, A., Kunmartini, S., Suryadhana, N. G., Setyohadi, B., Sudarsono, D., Lardy, N. M. & Feltkamp, T. E. (1997). J. Rheumatol.24, 1111–1114. [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Ramos, M. & López de Castro, J. A. (2002). Tissue Antigens, 60, 191–205. [DOI] [PubMed] [Google Scholar]
- Ren, E. C., Koh, W. H., Sim, D., Boey, M. L., Wee, G. B. & Chan, S. H. (1997). Tissue Antigens, 49, 67–69. [DOI] [PubMed] [Google Scholar]
- Rioux, J. D. & Abbas, A. K. (2005). Nature (London), 435, 584–589. [DOI] [PubMed] [Google Scholar]
- Rückert, C., Loll, B., Fiorillo, M. T., Moretti, R., Biesiadka, J., Saenger, W., Ziegler, A., Sorrentino, R. & Uchanska-Ziegler, B. (2005). In the press. [DOI] [PubMed]
- Rudwaleit, M., Bowness, P. & Wordsworth, P. (1996). Immunogenetics, 43, 160–162. [DOI] [PubMed] [Google Scholar]
- Schlosstein, L., Terasaki, P. I., Bluestone, R. & Pearson, C. M. (1973). N. Engl. J. Med.288, 704–706. [DOI] [PubMed] [Google Scholar]
- Sesma, L., Montserrat, V., Lamas, J. R., Marina, A., Vazquez, J. & López de Castro, J. A. (2002). J. Biol. Chem.277, 16744–16749. [DOI] [PubMed] [Google Scholar]
- Storoni, L. C., McCoy, A. J. & Read, R. J. (2004). Acta Cryst. D60, 432–438. [DOI] [PubMed] [Google Scholar]
- Vega, M. A., Bragado, R., Ivanyi, P., Pelaez, J. L. & López de Castro, J. A. (1986). J. Immunol.137, 3557–3565. [PubMed] [Google Scholar]