

Abstract
During formation of the neuromuscular junction (NMJ), agrin secreted by motor axons signals the embryonic muscle cells to organize a postsynaptic apparatus including a dense aggregate of acetylcholine receptors (AChRs). Agrin signaling at the embryonic NMJ requires the activity of nitric oxide synthase (NOS). Common downstream effectors of NOS are guanylate cyclase (GC), which synthesizes cyclic GMP, and cyclic GMP-dependent protein kinase (PKG). Here we show that GC and PKG are important for agrin signaling at the embryonic NMJ of the frog, Xenopus laevis. Inhibitors of both GC and PKG reduced endogenous AChR aggregation in embryonic muscles by 50–85%, and blocked agrin-induced AChR aggregation in cultured embryonic muscle cells. A cyclic GMP analog, 8-bromo-cyclic GMP, increased endogenous AChR aggregation in embryonic muscles to 3- to 4-fold control levels. Overexpression of either GC or PKG in embryos increased AChR aggregate area by 60–170%, whereas expression of a dominant negative form of GC inhibited endogenous aggregation by 50%. These results indicate that agrin signaling in embryonic muscle cells requires the activity of GC and PKG as well as NOS.
Keywords: neuromuscular junction, synapse formation, agrin, muscle cells, acetylcholine receptors, nitric oxide, cyclic GMP, guanylate cyclase, cyclic GMP-dependent protein kinase, Xenopus embryos
INTRODUCTION
Formation of the neuromuscular junction (NMJ), a well-studied synapse, is a complex process which occurs over a period of days to weeks in embryos of different species (Sanes and Lichtman, 1999b). Assembly of the postsynaptic apparatus, and, indirectly, the nerve terminal, is directed by agrin released by motor axons (Gautam et al., 1996; Cohen et al., 1997; Jones et al., 1997). Postsynaptic proteins, including acetylcholine receptors (AChRs), are directed by agrin to aggregate into stable structures anchored to the actin cytoskeleton (Dai et al., 2000; reviewed in Godfrey and Schwarte, 2003). Agrin acts in muscle cells through a membrane tyrosine kinase, MuSK, a component of the agrin receptor (DeChiara et al., 1996; Glass et al., 1996, 1997). However, signaling steps downstream of MuSK activation leading to aggregation of postsynaptic AChRs and other proteins are not well defined. Understanding the molecular mechanisms involved in assembly of the neuromuscular junction will provide a basis for comparison with signaling events in formation and plasticity of synapses in the central nervous system (CNS), and may also suggest therapeutic approaches for neuromuscular diseases.
Nitric oxide synthase (NOS), an enzyme that synthesizes the free radical gas nitric oxide (NO), is concentrated postsynaptically at the NMJ (Chao et al., 1997; Yang et al., 1997), and NO regulates acetylcholine release from motor nerve terminals (Wang et al., 1995). NOS is also localized postsynaptically at some CNS synapses, and NO has been implicated as a retrograde signaling molecule in long-term potentiation (Hawkins et al., 1998). NOS activity is required for agrin signaling of AChR aggregation in chicken skeletal muscle cells (Jones and Werle, 2000) and in Xenopus embryo muscles (Godfrey and Schwarte, 2003). Overexpression of NOS in Xenopus embryos greatly increases AChR aggregate area at embryonic NMJs, whereas NOS inhibitors block endogenous AChR aggregation 50–90% and completely block agrin-induced aggregation in cultured embryonic muscle cells (Schwarte and Godfrey, 2004).
Common downstream effectors of NO signaling in cells include guanylate cyclase (GC), the enzyme that synthesizes cyclic GMP (cGMP), and cyclic GMP-dependent protein kinase (PKG; Hofmann et al., 2000). Agrin-induced aggregation of AChRs into large, dense clusters in the membrane of cultured chicken muscle cells requires activity of both GC and PKG (Jones and Werle, 2004). Here we asked whether GC and PKG activities are also necessary for synaptic aggregation of AChRs during formation of the embryonic NMJ. Inhibitors of both GC and PKG reduced AChR aggregation at the NMJ, and blocked agrin-induced increases in AChR aggregation in cultured embryonic muscle cells. Conversely, overexpression of either GC or PKG increased AChR aggregate area at the NMJ, as did a cyclic GMP analog, but a dominant negative form of GC inhibited synaptic aggregation. These data strongly suggest that GC and PKG are involved in agrin signaling of postsynaptic differentiation at the embryonic NMJ and in cultured muscle cells.
MATERIALS AND METHODS
cDNAs; RNA synthesis
The cDNA encoding green fluorescent protein (GFP; S65T mutant) was obtained from Dr. Richard Dorsky (University of Utah). The cDNAs coding for rat soluble GC subunits α and β (Chinkers et al., 1989; Yuen et al., 1994) were a gift of Dr. David Garbers (University of Texas Southwestern Medical School), and a dominant negative GC α1 construct (D529A mutant; Yuen et al., 1994) was obtained from Dr. Peter Yuen (National Institutes of Health). The cDNAs encoding bovine cGMP-dependent protein kinase Iα (PKG; Wernet et al., 1989) and a constitutively active form of human PKGIα (PKG-GFP fusion protein; Browning et al., 2001) were from Dr. Bonnie Firestein (Rutgers University) and Dr. Darren Browning (Medical College of Georgia), respectively. Protein coding sequences of all cDNAs were amplified by PCR. Sense primers contained the SP6 RNA polymerase promoter and the 5′ untranslated sequence from Xenopus beta globin found in the pCS2 vector (Godfrey et al., 2000; Rupp et al., 1994). RNA was synthesized using SP6 polymerase and the mMessage Machine kit (Ambion; Godfrey et al., 1999).
RNA injection
Embryos of Xenopus laevis were obtained by in vitro fertilization (Moon and Christian, 1989) and injected with synthetic RNAs at the one-cell stage using a Nanoject II injector (Drummond) fitted with glass micropipets (20 μm diameter tip). Embryos were injected (Moon and Christian, 1989) with 4.6–9.2 nl nuclease-free water containing GFP RNA (1–2 ng) alone or combined with RNAs encoding GC or PKG (3–12 ng).
Screening embryos and labeling acetylcholine receptors
Following injection of RNAs, embryos were transfered into 0.1X modified Barth’s solution (MBS; Gurdon and Wickens, 1983) and allowed to develop to stage 31 (Nieuwkoop and Faber, 1994), then screened for GFP fluorescence. Embryos were fixed and AChRs were labeled with 1.5 μg/ml Alexa 594-α-bungarotoxin (Invitrogen) as described (Schwarte and Godfrey, 2004).
Confocal microscopy and image analysis
AChR aggregates were imaged using a confocal microscope (Zeiss LSM 510). Six stacks of images were acquired from three to six embryos for each condition, two stacks of 4 optical sections (at 1 μm intervals) from each of myotomes 4, 5 and 6 (Godfrey et al., 1999). The images were taken through a 40X objective with a 2.5-fold optical zoom (total magnification 100X). Optical sections were imaged to show innervated (medial) portions of the muscles in which AChR aggregates formed a continuous line, and were centered along the intermyotomal septa where NMJs form. Since muscle cells at this stage were about 10 μm in diameter, each 4 μm stack imaged primarily one set of cells. The area of AChR aggregates in a montage of all 4 images in each stack was measured with Metamorph image analysis software (Universal Imaging Corp.). Threshold was set to mark aggregates in each series of images, and aggregate area was converted to a percentage of the total area of the montaged images. Statistics were calculated as described (Godfrey et al., 1999).
Treatment of embryos with GC and PKG inhibitors and a cGMP analog
Embryos were exposed to the GC inhibitor 1H-[1,2,4]oxadiazolo[4,3-alpha]quinoxalin-1-one (ODQ), the PKG inhibitor Rp-8-pCPT-cGMPS (Rp-8), or 8-bromo-cyclic GMP (8-Br-cGMP) from 26 h of development (stage 24) to stage 31, a period of 18–20 h at 16–18°C. Reagents (Biomol or EMD Biosciences) were dissolved in 1X MBS. In some experiments, penetration of inhibitors into muscles was facilitated by removing the skin overlying the trunk myotomes and dissolving inhibitors in cell culture medium. AChR aggregates were labeled and their area was quantified as described above. Skin was removed from myotomes of embryos treated with Rp-8, some experiments with 8-Br-cGMP, and matching controls, but not for ODQ treatments. Removing skin did not delay development.
Cell culture studies
Myotomal muscles were removed from stage 21–23 embryos and were dissociated (Peng et al., 1991). Cells were cultured in 0.4 cm2 wells (NUNC Lab-Tek #178599; cells from 0.7–1 embryo per well), which were mounted on glass coverslips with Sylgard (Dow Corning) and coated with a basement membrane extract (E.C.L., Upstate) as described (Schwarte and Godfrey, 2004). Cultures were used for experiments after 1–3 days, when the cells had attached and spread. Cultures were pretreated with inhibitors for 2 h prior to adding 6–12 ng/ml agrin (half-maximal dose = 3.5–5 ng/ml) with or without inhibitors for 14 h. After 16–18 h at 22°C, cells were labeled 1 h with Alexa 594-α-bungarotoxin (1.5 μg/ml), rinsed twice with culture medium and fixed 10 min in 95% ethanol at −20°C. Cultures were mounted in a glycerol-based mounting medium containing n-propyl gallate to retard fading of fluorescence (Valnes and Brandtzaeg, 1985). Aggregates of AChRs (≥ 1 μm) were counted in each of 20 cells per well in duplicate wells for each condition.
RESULTS
Inhibitors of guanylate cyclase and cGMP-dependent protein kinase reduce endogenous AChR aggregation at the embryonic neuromuscular junction, but a cyclic GMP analog greatly increases AChR aggregate area in vivo
To determine whether the activities of guanylate cyclase (GC) and cGMP-dependent protein kinase (PKG) are required for endogenous AChR aggregation at the embryonic NMJ, we exposed developing myotomal muscles to inhibitors during the period when muscles become innervated by motor axons (stages 24–31). Confocal images were centered on the almost continuous line of AChR aggregates found on the ends of muscle cells at the intermyotomal septa, where NMJs form (Fig. 1A). The GC inhibitor ODQ and the PKG inhibitor Rp-8-pCPT-cGMPS reduced endogenous AChR aggregate area at neuromuscular synapses by 85% and 50% on average, respectively (Figs. 1B, 1C; Table 1). In contrast, a cell-permeable cGMP analog, 8-bromo-cGMP, increased AChR aggregate area (Fig. 1D; Table 1) to levels 3–4 times those in untreated control embryo muscles (e.g., Fig. 1A; Table 1). Despite the changes in AChR aggregate area, embryos treated with these agents appeared to develop normally, including the formation of myotomal muscles. However, treatment with ODQ, but not with Rp-8 or 8-Br-cGMP, delayed development from stage 24 to stage 31 by 1–3 h.
Figure 1.
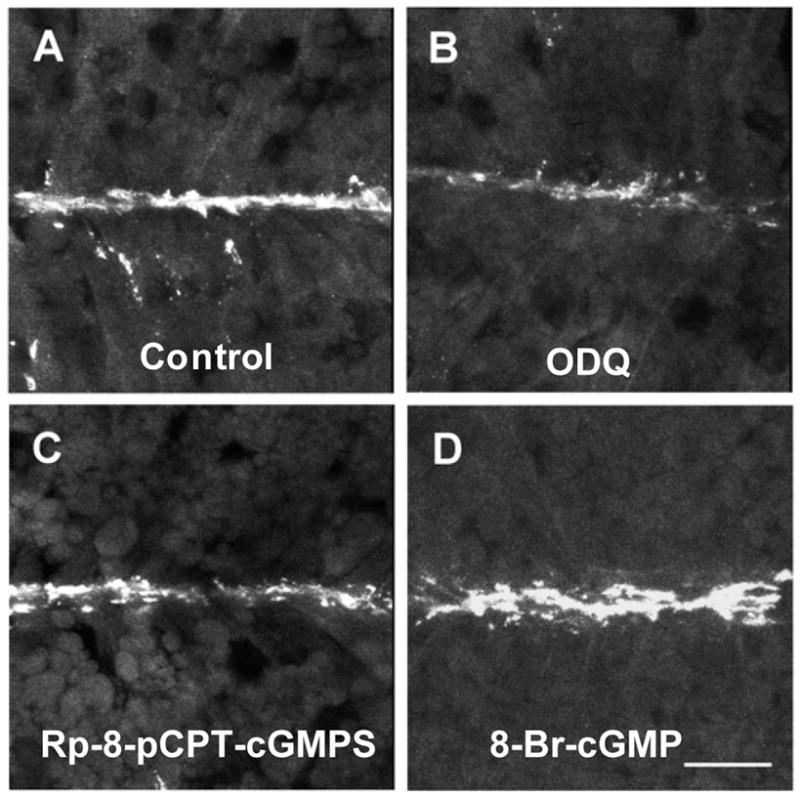
Inhibitors of GC and PKG reduce endogenous AChR aggregation at the embryonic neuromuscular junction, and a cyclic GMP analog greatly increases AChR aggregate area compared to untreated control embryos (A). Embryos were exposed to the GC inhibitor ODQ (50 μM, B), the PKG inhibitor Rp-8-pCPT-cGMPS (0.5 mM, C), or the cGMP analog 8-Br-cGMP (100 μM, D) during the period of neuromuscular junction formation (stages 24–31). In the experiments shown here, skin was removed from myotomes of embryos treated with Rp-8-pCPT-cGMPS and 8-Br-cGMP and their respective controls, but not from embryos treated with ODQ or corresponding controls (A). AChR aggregates were labeled and imaged by confocal microscopy, and area of aggregates was quantified (Table 1) with Metamorph image analysis software as described in Materials and Methods. Images shown are maximum projections of stacks of 4 images taken at 1 μm intervals. In the image stacks projected in this figure, AChR aggregate area was inhibited 86% by ODQ (B) and 65% by Rp-8-pCPT-cGMPS (C), and increased 460% by 8-Br-cGMP (D), compared to the respective untreated controls (e.g., A).
Table 1.
A cyclic GMP analog increases AChR aggregation and inhibitors of guanlylate cyclase and cGMP-dependent protein kinase reduce endogenous AChR aggregation at the embryonic neuromuscular junction.
| Experiment No. | Treatment | AChR aggregate area (% ± SEM) | Area ratio experimental: Control | P vs. Control |
|---|---|---|---|---|
| 1 | Control (unskinned) | 0.18 ± 0.07 | -- | -- |
| 8-Br-cGMP (100 μM) | 0.74 ± 0.10 | 4.0 | 0.001 | |
| 2 | Control (skinned) | 0.34 ± 0.06 | -- | -- |
| 8-Br-cGMP (100 μM) | 0.987 ± 0.20 | 2.9 | 0.004 | |
| 3 | Control (unskinned) | 1.36 ± 0.21 | -- | -- |
| ODQ (50 μM) | 0.21 ± 0.06 | 0.15 | 5 × 10−5 | |
| 4 | Control (skinned) | 0.96 ± 0.21 | -- | -- |
| Rp-8-pCPT-cGMPs(0.5 mM) | 0.48 ± 0.08 | 0.50 | 0.05 |
In experiments 2 and 4, the skin overlying trunk myotomes was removed by suction at stage 24. Embryos were exposed to 8-Br-cGMP or inhibitor in 0.1X MBS (unskinned) or 1X MBS (skinned) from stage 24 until stage 31, when they were fixed for labeling of AChRs with α-bungarotoxin and subsequent confocal microscopy. Each type of experiment was performed 2–4 times.
To determine whether inhibitors of GC and PKG acted on muscle cells in a cell-autonomous manner, we cultured embryonic myotomal muscle cells that had not yet been innervated. Treatment of these cells with the GC inhibitors ODQ (Fig. 2C) or the PKG inhibitors Rp-8-pCPT-cGMPS (Fig. 2D) or KT5823 (not shown) reduced agrin-induced AChR aggregation (Fig. 2B) up to 100% (Table 2). Application of inhibitors at the IC50 or Ki values for the mammalian enzymes (* in Table 2; Butt et al., 1994; Schrammel et al., 1996; Kase et al., 1987) reduced AChR aggregation in Xenopus embryo muscle cells by 44–70% on average, suggesting that the effects were specifically due to inhibition of GC or PKG. These results strongly suggest that activity of both GC and PKG in muscle cells is necessary for agrin signaling leading to AChR aggregation.
Figure 2.
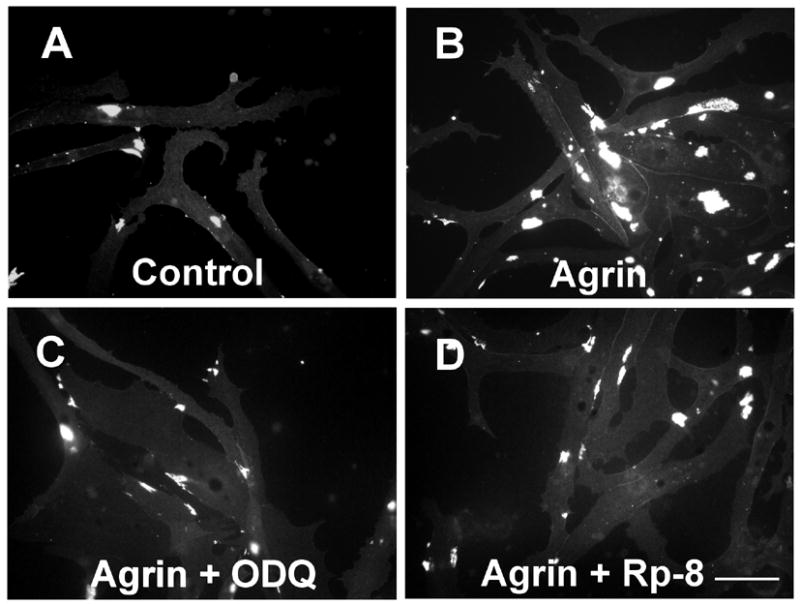
Inhibitors of GC and PKG block agrin-induced AChR aggregation on cultured Xenopus embryo myotomal muscle cells. Cells were pretreated with inhibitors 2.5 hr before 17 h incubation with 7 ng/ml agrin (half-maximal dose, 4.4 ng/ml) with or without inhibitors. AChR aggregates (white) were then labeled with fluorescent α-bungarotoxin, cells were fixed, mounted and aggregates counted as described in Materials and Methods. In the experiment from which these images were taken, the average inhibition of agrin-induced AChR aggregation was 99% with ODQ (2 μM), 75% with KT5823 (1 μM; data not shown) and 75% with Rp-8-pCPT-cGMPS (0.5 μM). The number of aggregates per cell in the images shown was 1.8 in untreated control cells (A), 3.4 after agrin treatment (B), 2.5 after agrin plus ODQ (66% inhibition, C), and 0.2 after agrin plus Rp-8-pCPT-cGMPS (88% inhibition, D).
TABLE 2.
Inhibitors of guanlylate cyclase (GC) and cGMP-dependent protein kinase (PKG) block agrin-induced AChR aggregation in cultured embryonic muscle cells
| Inhibitor | Concentration | Inhibition of agrin-induced AChR aggregation | ||
|---|---|---|---|---|
| average | range | experiments | ||
| ODQ (GC) | 20 nM* | 44% | 30–56% | 5 |
| 200 nM | 52% | 8–96% | 6 | |
| 2 μM | 64% | 49–100% | 6 | |
| 50 μM | 100% | 100–100% | 2 | |
| KT5823 (PKG) | 200–250 nM* | 64% | 53–84% | 5 |
| 1 μM | 87% | 67–100% | 4 | |
| Rp-8-pCPT-cGMPs (PKG) | 500 nM* | 70% | 34–100% | 5 |
| 5 μM | 60% | 29–100% | 7 | |
| 100 μM | 96% | 92–100% | 2 | |
Embryonic myotomal muscle cells were cultured 1–3 days as described in Materials and Methods. Cultures were pretreated with inhibitor for 2 h prior to adding 6–12 ng/ml agrin (half-maximal dose = 3.5–5 ng/ml) with inhibitor for 14 h. After 16–18 h at 22°C, cells were labeled with Alexa 594-α-bungarotoxin, fixed and mounted as described in Materials and Methods. Aggregates of AChRs (≥ 1 μm) were counted in each of 20 cells per well in duplicate wells for each condition. Values shown are averages and ranges of percent inhibition of agrin-induced AChR aggregates/cell in several different experiments. Asterisks indicate published IC50 or Ki values for each inhibitor.
Overexpression of GC or PKG increases AChR aggregation and a dominant negative form of GC reduces AChR aggregate area at the embryonic NMJ
To further assess the role of GC and PKG in postsynaptic differentiation of the embryonic NMJ, we overexpressed these enzymes by injecting RNA encoding each enzyme into one-cell stage embryos. Injection of RNA encoding both α and β subunits of GC increased AChR aggregate area by 100% over control embryos (Fig. 3A), but no increase was seen if RNA encoding only one of these GC subunits was injected (Figs. 3B, 3C; Table 3). In contrast, injection of RNA encoding a dominant negative form of the GC α subunit (D529A; Yuen et al., 1994) resulted in a 50% decrease in endogenous AChR aggregation (Fig. 3D; Table 3). When RNA encoding PKGIα was injected, AChR aggregate area increased by 60–170% (Table 4), similar to the effect of GC overexpression. Injection of RNA encoding the fusion protein PKG-GFP, a constitutively active form of PKG (Browning et al., 2001), resulted in a larger increase in AChR aggregate area than injection of PKGIα RNA (Table 4). These results complement our findings with inhibitors and 8-bromo-cGMP, and confirm the importance of both GC and PKG in agrin-induced signaling leading to AChR aggregation at the embryonic NMJ.
Figure 3.
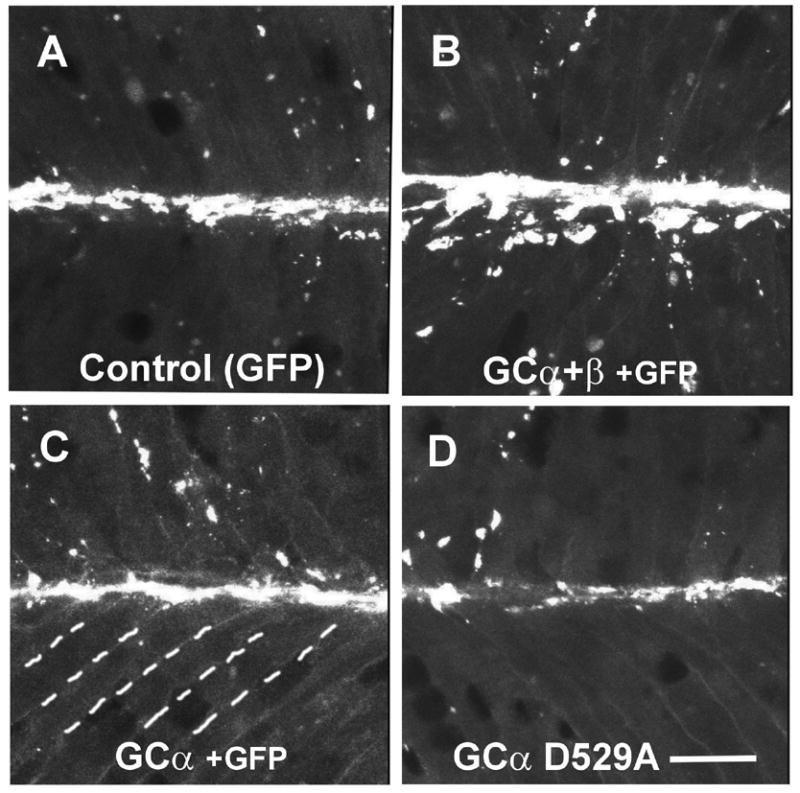
Overexpression of guanylate cyclase (GC) increases AChR aggregation in embryonic muscles, but a dominant negative GC inhibits endogenous aggregation at NMJs. Embryos were injected with RNA encoding GFP (A), GC α and β subunits (B), GC α only (C), or a dominant negative GC-α construct (D) encoding an inactive point mutant. GC α and β increased AChR aggregation 2–3 fold, GC α alone did not affect aggregation, but the dominant negative GC α reduced aggregate area by about 50%. Quantitative data from the experiments shown here are found in Table 3 (Experiment 1 for panels A, B and C; Experiment 2 for panel D). Dashed lines in panel C indicate boundaries of muscle fibers. Bar, 20 μm.
TABLE 3.
Overexpression of guanylate cyclase increases and a dominant negative form inhibits AChR aggregation at the embryonic neuromuscular junction
| Experiment No. | RNA Injected (ng/embryo) | AChR aggregate area (% ± SEM) | Area ratio GC/control | P vs. Control |
|---|---|---|---|---|
| 1 | GFP (1) | 1.11 ± 0.26 | -- | -- |
| GC α + β (6) | 2.34 ± 0.16 | 2.1 | 0.003 | |
| GC α (3) | 1.20 ± 0.26 | 1.08 | N.S. | |
| GC β (3) | 1.08 ± 0.50 | 0.97 | N.S. | |
| 2 | GFP (1) | 1.01 ± 0.14 | -- | -- |
| GC α-D529A (8) | 0.51 ± 0.14 | 0.51 | 0.028 |
RNA encoding rat guanlyate cyclase (GC) α and/or β subunits, or a dominant negative mutant form of GC α (D529A), was injected into fertilized Xenopus embryos at the one-cell stage, and embryos were fixed at stage 31 and analyzed as described in Materials and Methods. All embryos including controls were injected with 1 ng of GFP RNA as a marker for successful injection. Overexpression of GC α + β, but not α or β alone, increased AChR aggregate area at the embryonic neuromuscular junction in four different experiments to 1.8–2.2-fold control values (injection with GFP RNA). Expression of dominant negative GC α inhibited endogenous AChR aggregation by 49–65% in three separate experiments. N.S. = not significant (P values of 0.4 and 0.5).
TABLE 4.
Overexpression of cGMP-dependent protein kinase increases AChR aggregation at the embryonic neuromuscular junction
| RNA Injected (ng) | AChR aggregate area (% ± SEM) | Area ratio PKG/control | P vs. Control |
|---|---|---|---|
| GFP (1) | 0.416 + 0.11 | -- | -- |
| PKG1α (8) | 0.667 + 0.06 | 1.63 | 0.035 |
| PKG-GFP (8) | 1.02 + 0.13 | 2.45 | 0.004 |
RNA encoding human PKG1α, or a constitutively active mutant form of PKG1α (PKG-GFP), was injected into fertilized Xenopus embryos at the one-cell stage, and embryos were fixed at stage 31 and analyzed as described in Materials and Methods. All embryos including controls were also injected with 1 ng of GFP RNA. Overexpression of PKG1α increased AChR aggregate area at the embryonic neuromuscular junction in four different experiments to 1.6–2.7-fold control values (injection with GFP RNA) and PKG-GFP increased endogenous AChR aggregation to 2.5-fold control values in two experiments.
DISCUSSION
In many cell types, NO stimulates the ‘soluble’ guanylate cyclase (sGC) isoform to synthesize more cyclic GMP, which in turn activates cGMP-dependent protein kinase (PKG; Hofmann et al., 2000). Significantly, all three enzymes in this pathway, NOS, GC and PKG, are concentrated in the postsynaptic apparatus of the mammalian NMJ (Chao et al., 1997; Schoser and Behrends, 2001), suggesting that the NO-cGMP signaling pathway plays a role in the formation and/or function of this synapse. We have previously shown that NOS activity is necessary for postsynaptic differentiation of embryonic NMJs and agrin-induced AChR aggregation in cultured muscle cells (Schwarte and Godfrey, 2004). The findings presented here indicate that activity of both GC and PKG is also critical for agrin signaling at the NMJ.
The ability of inhibitors of GC and PKG to block endogenous AChR aggregation at embryonic NMJs indicates that the activity of these enzymes is important for postsynaptic differentiation in vivo. Inhibitors completely blocked agrin-induced AChR aggregation in cultured embryonic muscle cells and reduced endogenous aggregation at embryonic NMJs by 50–85%. Incomplete inhibition of postsynaptic AChR aggregation in embryos could reflect incomplete diffusion or instability of inhibitors in the embryonic muscles. Another possibility is that compensatory mechanisms allow limited agrin signaling in embryonic muscles when GC or PKG is inhibited. Although we favor the first explanation, compensatory mechanisms could involve cyclic AMP-dependent protein kinase (PKA), which has recently been shown to activate the small GTPases Rac1 and Cdc42 (O’Connor and Mercurio, 2001; Chadhi et al., 2005; Howe et al., 2005). These small GTPases are activated during agrin signaling and their activation is required for AChR aggregation (Weston et al., 2000; Luo et al., 2003). The complete inhibition of agrin-induced AChR aggregation by GC and PKG inhibitors in cultured embryonic muscle cells indicates that activity of these enzymes is essential for agrin signaling these cells. Our results are also consistent with the effects of the same inhibitors on agrin activity in cultured chick embryo myotubes (Jones and Werle, 2004), indicating that the role of GC and PKG in agrin signaling is phylogenetically conserved.
Overexpression of GC or PKG in embryos resulted in 60–170% increases in AChR aggregate area in the innervated regions of embryonic muscles, similar to the effect of overexpressing agrin and NOS in embryos (Godfrey et al., 1999; Schwarte and Godfrey, 2004). In contrast, expression of a dominant negative mutant form of GC reduced AChR aggregate area in embryonic muscles by 50%. Finally, treating embryos with the cGMP analog 8-Br-cGMP increased AChR aggregate area by 200–300%. Taken together, these results indicate that cGMP stimulates AChR aggregation at NMJs. Moreover, results with the dominant negative mutant of GC, like the inhibitor studies, suggest that GC activity is critical to agrin signaling in embryonic muscles.
Another possible interpretation of the inhibition or increase of AChR aggregation we observed in embryonic muscles and in cultured muscle cells is that the changes in aggregation reflect changes in the number of AChRs on the surface of the muscle cells. Acetylcholine receptors shuttle between intracellular membrane compartments and the plasma membrane of skeletal muscle cells (Fambrough, 1979). Recycling of receptors has also been observed at the NMJ in vivo (Bruneau et al., 2005; Bruneau et al., 2006). Thus, changes in AChR aggregation on the surface of muscle cells could reflect changes in insertion and/or removal of AChRs from the plasma membrane. However, the number of surface AChRs in cultured chick embryo myotubes was not significantly changed by treatment with agrin, 8-Br-cGMP, or the GC inhibitor ODQ in combination with agrin (Jones and Werle, 2004). Thus, it is possible but unlikely that the changes in AChR aggregation we observed with the same agents in cultured frog embryo muscle cells and embryonic muscles were due to changes in the number of AChRs on the surface of the muscle cells.
Intracellular signaling molecules can be categorized as mediators or modulators (Sanes and Lichtman, 1999a). Mediators are essential molecules required for signaling to occur. Studies with null mutant mice have established that postsynaptic AChR aggregation at the NMJ requires agrin, its receptor MuSK, and rapsyn, which links clustered AChRs to the cytoskeleton (Sanes and Lichtman, 2001). Modulators are molecules that enhance or inhibit signaling but are less essential. Nitric oxide and cyclic GMP are clearly important for agrin signaling, but are they essential mediators or modulators? The evidence to date suggests that NO and cGMP mediate agrin signaling in frog embryos and chick embryo muscle cells. First, inhibitors of NOS and GC completely blocked agrin-induced AChR aggregation in cultured muscle cells from both organisms (Jones and Werle, 2000; Schwarte and Godfrey, 2004; Jones and Werle, 2004; this report). Second, inhibitors of NOS, GC, and PKG blocked up to 90% of endogenous AChR aggregation at embryonic frog NMJs (Schwarte and Godfrey, 2004; this report). Third, both NO donors and a cGMP analog mimicked agrin by aggregating AChRs in both cultured muscle cells (including those from mouse) and embryonic muscles, a result not expected if NO and cGMP are modulators of agrin signaling (Jones and Werle, 2000; Schwarte and Godfrey, 2004; Jones and Werle, 2004; M. Parasa, R.C. Schwarte and E.W. Godfrey, unpublished results). Fourth, 8-Br-cGMP does not potentiate agrin signaling at a saturating dose of agrin, indicating that cGMP acts within the agrin pathway (Jones and Werle, 2004). Determining the extent and manner by which NO and cGMP mediate agrin signaling will require further studies to understand how they are regulated during this process and to identify their downstream targets.
Which effectors of agrin signaling could be activated by GC and PKG? Recent findings show that agrin-induced polymerization of actin microfilaments is required for clustering of AChRs and other associated proteins (Dai et al., 2000). Two Rho GTPases, Rac and Rho, which control the organization of the actin cytoskeleton (Mackay and Hall, 1998), are necessary and, when expressed together, sufficient for aggregation of AChRs in myotubes (Weston et al., 2000; Weston et al., 2003). Agrin activates both Rac and Rho in myotubes (Weston et al., 2000; Weston et al., 2003), as well as p21-activated kinase (PAK1), a known target of Rac1 (Luo et al., 2003). These findings suggest that Rac and Rho are essential components for agrin signaling in muscle cells.
Activity of NOS and PKG may in turn regulate Rac in agrin signaling. Our preliminary results suggest that agrin-induced Rac activation in mouse myotubes requires NOS activity (R.C. Schwarte and E.W. Godfrey, unpublished results). Furthermore, brief treatment of myotubes with NO donors or 8-bromo-cyclic GMP induces AChR aggregation and activates Rac (R.C. Schwarte, M. Parasa, and E.W. Godfrey, unpublished results). Thus, Rac activation appears to be downstream of NOS (and possibly GC) activity during agrin signaling.
A potential target of the NO/cGMP pathway is one or more of the guanine-nucleotide exchange factors (GEFs), which activate Rho GTPases by stimulating GTP exchange (Hall, 1998; Jaffe and Hall, 2005; Overbeck et al., 1995). Phosphorylation of GEFs activates Rho GTPases (Aghazadeh et al., 2000; Kiyono et al., 2000). For example, PKG could phosphorylate a Rac-specific GEF during agrin signaling. Consistent with this notion, PKG activates Rac1 indirectly in HEK-293 cells (Hou et al., 2004). Thus, it will be important to determine whether GEFs are phosphorylated by PKG during agrin signaling, and if so, whether the phosphorylated GEFs activate Rac and/or Rho in response to agrin.
Acknowledgments
We thank Madhuri Parasa for technical assistance, Drs. David Garbers and Peter Yuen for GC cDNAs, and Dr. Bonnie Firestein for PKG cDNA. Figure 3 was previously published in a review article (Godfrey and Schwarte, 2003) and is reproduced here by permission of Kluwer Academic Publishers. This work was funded by grants from the National Institutes of Health (MH57545), the Jeffress Memorial Trust and the Muscular Dystrophy Association to E.W.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aghazadeh B, Lowry WE, Huang XY, Rosen MK. Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell. 2000;102:625–633. doi: 10.1016/s0092-8674(00)00085-4. [DOI] [PubMed] [Google Scholar]
- Browning DD, McShane M, Marty C, Ye RD. Functional analysis of type 1 alpha cGMP-dependent protein kinase using green fluorescent fusion proteins. J Biol Chem. 2001;276:13039–13048. doi: 10.1074/jbc.M009187200. [DOI] [PubMed] [Google Scholar]
- Bruneau EG, Akaaboune M. The dynamics of recycled acetylcholine receptors at the neuromuscular junction in vivo. Development. 2006;133:4485–4493. doi: 10.1242/dev.02619. [DOI] [PubMed] [Google Scholar]
- Bruneau EG, Sutter D, Hume RI, Akaaboune M. Identification of nicotinic acetylcholine receptor recycling and its role in maintaining receptor density at the neuromuscular junction in vivo. J Neurosci. 2005;25:9949–9959. doi: 10.1523/JNEUROSCI.3169-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt E, Eigenthaler M, Genieser H-G. (Rp)-8-pCPT-cGMPS, a novel cGMP-dependent protein kinase inhibitor. Eur J Pharmacol. 1994;269:265–268. doi: 10.1016/0922-4106(94)90095-7. [DOI] [PubMed] [Google Scholar]
- Chadhi A, Miller B, Sorokin A. Endothelin 1 induces beta 1 Pix translocation via protein kinase A-dependent pathway. J Biol Chem. 2005;280:578–584. doi: 10.1074/jbc.M411130200. [DOI] [PubMed] [Google Scholar]
- Chinkers M, Garbers DL, Chang MS, Lowe DG, Chin HM, Goeddel DV, Schulz S. A membrane form of guanylate cyclase is an atrial natriuretic peptide receptor. Nature. 1989;338:78–83. doi: 10.1038/338078a0. [DOI] [PubMed] [Google Scholar]
- Chao DS, Silvagno F, Xia H, Cornwell TL, Lincoln TM, Bredt DS. Nitric oxide synthase and cyclic GMP-dependent protein kinase concentrated at the neuromuscular endplate. Neuroscience. 1997;76:665–672. doi: 10.1016/s0306-4522(96)00367-3. [DOI] [PubMed] [Google Scholar]
- Cohen I, Rimer M, Lomo T, McMahan UJ. Agrin-induced postsynaptic-like apparatus in skeletal muscle fibers in vivo. Mol Cell Neurosci. 1997;10:237–253. doi: 10.1006/mcne.1997.0623. [DOI] [PubMed] [Google Scholar]
- Dai Z, Luo X, Xie H, Peng HB. The actin-driven movement and formation of acetylcholine receptor clusters. J Cell Biol. 2000;150:1321–1334. doi: 10.1083/jcb.150.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- Fambrough DM. Control of acetylcholine receptors in skeletal muscle. Physiol Rev. 1979;59:165–227. doi: 10.1152/physrev.1979.59.1.165. [DOI] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- Glass DJ, Apel ED, Shah S, Bowen DC, DeChiara TM, Stitt TN, Sanes JR, Yancopoulos GD. Kinase domain of the muscle-specific receptor tyrosine kinase (MuSK) is sufficient for phosphorylation but not clustering of acetylcholine receptors: required role for the MuSK ectodomain? Proc Natl Acad Sci U S A. 1997;94:8848–8853. doi: 10.1073/pnas.94.16.8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey EW, Roe J, Heathcote RD. Overexpression of agrin isoforms in Xenopus embryos alters the distribution of synaptic acetylcholine receptors during development of the neuromuscular junction. Dev Biol. 1999;205:22–32. doi: 10.1006/dbio.1998.9104. [DOI] [PubMed] [Google Scholar]
- Godfrey EW, Roe J, Heathcote RD. Agrin fragments differentially induce ectopic aggregation of acetylcholine receptors in myotomal muscles of Xenopus embryos. J Neurobiol. 2000;44:436–445. [PubMed] [Google Scholar]
- Godfrey EW, Schwarte RC. The role of nitric oxide signaling in the formation of the neuromuscular junction. J Neurocytol. 2003;32:591–602. doi: 10.1023/B:NEUR.0000020612.87729.98. [DOI] [PubMed] [Google Scholar]
- Gurdon JB, Wickens MP. The use of Xenopus oocytes for the expression of cloned genes. Methods Enzymol. 1983;101:370–86. doi: 10.1016/0076-6879(83)01028-9. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hawkins RD, Son H, Arancio O. Nitric oxide as a retrograde messenger during long-term potentiation in hippocampus. Prog Brain Res. 1998;118:155–172. doi: 10.1016/s0079-6123(08)63206-9. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Ammendola A, Schlossmann J. Rising behind NO: cGMP-dependent protein kinases. J Cell Sci. 2000;113:1671–1676. doi: 10.1242/jcs.113.10.1671. [DOI] [PubMed] [Google Scholar]
- Hou Y, Ye RD, Browning DD. Activation of the small GTPase Rac1 by cGMP-dependent protein kinase. Cell Signal. 2004;16:1061–1069. doi: 10.1016/j.cellsig.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Howe AK, Baldor LC, Hogan BP. Spatial regulation of the cAMP-dependent protein kinase during chemotactic cell migration. Proc Natl Acad Sci U S A. 2005;102:14320–14325. doi: 10.1073/pnas.0507072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Ann Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Jones G, Meier T, Lichtensteiner M, Witzemann V, Sakmann B, Brenner HR. Induction by agrin of ectopic and functional postsynaptic-like membrane in innervated muscle. Proc Natl Acad Sci U S A. 1997;94:2654–2659. doi: 10.1073/pnas.94.6.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MA, Werle MJ. Nitric oxide is a downstream mediator of agrin-induced acetylcholine receptor aggregation. Mol Cell Neurosci. 2000;16:649–660. doi: 10.1006/mcne.2000.0901. [DOI] [PubMed] [Google Scholar]
- Jones MA, Werle MJ. Agrin-induced AChR aggregate formation requires cGMP and aggregate maturation requires activation of cGMP-dependent protein kinase. Mol Cell Neurosci. 2004;25:195–204. doi: 10.1016/j.mcn.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun. 1987;142:436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- Kiyono M, Kaziro Y, Satoh T. Induction of rac-guanine nucleotide exchange activity of Ras-GRF1/CDC25(Mm) following phosphorylation by the nonreceptor tyrosine kinase Src. J Biol Chem. 2000;275:5441–5446. doi: 10.1074/jbc.275.8.5441. [DOI] [PubMed] [Google Scholar]
- Luo ZG, Je HS, Wang Q, Yang F, Dobbins GC, Yang ZH, Xiong WC, Lu B, Mei L. Implication of geranylgeranyltransferase I in synapse formation. Neuron. 2003;40:703–717. doi: 10.1016/s0896-6273(03)00695-0. [DOI] [PubMed] [Google Scholar]
- Mackay DJ, Hall A. Rho GTPases. J Biol Chem. 1998;273:20685–20688. doi: 10.1074/jbc.273.33.20685. [DOI] [PubMed] [Google Scholar]
- Moon RT, Christian JL. Microinjection and expression of synthetic mRNAs in Xenopus embryos. Technique. 1989;1:76–89. [Google Scholar]
- Nieuwkoop PD, Faber J. Normal Table of Xenopus laevis(Daudin) 2. Garland; New York: 1994. (Originally Published in 1967, North Hollond, Amsterdam) [Google Scholar]
- O’Connor KL, Mercurio AM. Protein kinase A regulates Rac and is required for the growth factor-stimulated migration of carcinoma cells. J Biol Chem. 2001;276:47895–47900. doi: 10.1074/jbc.M107235200. [DOI] [PubMed] [Google Scholar]
- Overbeck AF, Brtva TR, Cox AD, Graham SM, Huff SY, Khosravi-Far R, Quilliam LA, Solski PA, Der CJ. Guanine nucleotide exchange factors: activators of Ras superfamily proteins. Mol Reprod Dev. 1995;42:468–476. doi: 10.1002/mrd.1080420415. [DOI] [PubMed] [Google Scholar]
- Rupp RA, Snider L, Weintraub H. Xenopus embryos regulate the nuclear localization of XMyoD. Genes Dev. 1994;8:1311–1323. doi: 10.1101/gad.8.11.1311. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Can molecules explain long-term potentiation? Nature Neurosci. 1999a;2:597–604. doi: 10.1038/10154. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999b;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Induction, maturation and maintenance of a postsynaptic apparatus. Nature Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Schoser BG, Behrends S. Soluble guanylyl cyclase is localized at the neuromuscular junction in human skeletal muscle. Neuroreport. 2001;12:979–81. doi: 10.1097/00001756-200104170-00023. [DOI] [PubMed] [Google Scholar]
- Schrammel A, Behrends S, Schmidt K, Koesling D, Mayer B. Characterization of 1H-[1,2,4]oxadiazolp[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- Schwarte RC, Godfrey EW. Nitric oxide synthase activity is required for postsynaptic differentiation of the embryonic neuromuscular junction. Dev Biol. 2004;273:276–284. doi: 10.1016/j.ydbio.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Valnes K, Brandtzaeg P. Retardation of immunofluorescence fading during microscopy. J Histochem Cytochem. 1985;33:755–761. doi: 10.1177/33.8.3926864. [DOI] [PubMed] [Google Scholar]
- Wang T, Xie Z, Lu B. Nitric oxide mediates activity-dependent synaptic suppression at developing neuromuscular synapses. Nature. 1995;374:262–266. doi: 10.1038/374262a0. [DOI] [PubMed] [Google Scholar]
- Weston C, Gordon C, Teressa G, Hod E, Ren XD, Prives J. Cooperative regulation by Rac and Rho of agrin-induced acetylcholine receptor clustering in muscle cells. J Biol Chem. 2003;278:6450–6455. doi: 10.1074/jbc.M210249200. [DOI] [PubMed] [Google Scholar]
- Weston C, Yee B, Hod E, Prives J. Agrin-induced acetylcholine receptor clustering is mediated by the small guanosine triphosphatases Rac and Cdc42. J Cell Biol. 2000;150:205–212. doi: 10.1083/jcb.150.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernet W, Flockerzi V, Hofmann F. The cDNA of the two isoforms of bovine cGMP-dependent protein kinase. FEBS Lett. 1989;17:191–196. doi: 10.1016/0014-5793(89)81453-x. [DOI] [PubMed] [Google Scholar]
- Yang CC, Alvarez RB, Engel WK, Haun CK, Askanas V. Immunolocalization of nitric oxide synthases at the postsynaptic domain of human and rat neuromuscular junctions – light and electron microscopic studies. Exp Neurol. 1997;148:34–44. doi: 10.1006/exnr.1997.6663. [DOI] [PubMed] [Google Scholar]
- Yuen PST, Doolittle LK, Garbers DL. Dominant negative mutants of nitric oxide-sensitive guanylyl cyclase. J Biol Chem. 1994;269:791–793. [PubMed] [Google Scholar]