

Abstract
Vitamin D-receptor interacting protein (DRIP150) coactivates estrogen receptor α (ERα)-mediated transactivation in breast cancer cell lines transfected with a construct (pERE3) containing three estrogen responsive elements (EREs). In this study, we show that DRIP150 also coactivates ERα/Sp1-mediated transactivation in ZR-75, MCF-7 and MDA-MB-231 breast cancer cells transfected with a construct (pSp13) containing three consensus GC-rich motifs. Studies on coactivation of wild-type and variant ERα/Sp1 by DRIP150 indicates that the DNA-binding domain and helix 12 in the ligand binding domain of ERα are required and the coactivation response is squelched by overexpressing an NR-box peptide that contains two LXXLL motifs from GRIP2. In contrast, coactivation of ERα/Sp1 by wild-type and mutant DRIP150 expression plasmids show that coactivation of ERα/Sp1 by DRIP150 is independent of the NR-boxes. Deletion analysis of DRIP150 demonstrates that coactivation requires an α-helical NIFSEVRVYN (amino acids 795-804) motif within twenty-three amino acid sequence (789-811) in the central region of DRIP150 and similar results were obtained for coactivation of ERα by DRIP150. Thus, although different domains of ERα are required for hormone-dependent activation of ERα and ERα/Sp1, coactivation of these transcription factors by DRIP150 requires the α-helical amino acids 795-804. This is the first report of a coactivator that enhances ERα/Sp1-mediated transactivation in breast cancer cells.
Keywords: DRIP150, ERα/Sp1, ZR-75 cells, coactivation, NR box-independent
INTRODUCTION
Estrogen receptor α (ERα) and ERβ are members of the nuclear receptor superfamily of transcription factors that include steroid and thyroid hormone receptors, vitamin D and retinoid receptors and several orphan receptors [1-6]. Most nuclear receptors contain several domains (A - F) which exhibit common structural and functional characteristics. For example, both ERα and ERβ contain N-terminal A/B and C-terminal E/F domains which exhibit activation function-1 (AF-1) and AF-2, respectively, a DNA-binding domain (DBD) (C), and a hinge region (D). The DBD of human ERα and ERβ exhibit a 96% degree of homology [7] and the endogenous ligand 17β-estradiol (E2) and other estrogenic compounds induce homo- and heterodimerization of ERα/ERβ binding to cognate estrogen responsive elements (EREs) and activation of E2-dependent genes [8-13]. Despite these similarities in ligand-dependent activation of ERα and ERβ, there are significant differences in their function [1; 3; 14-17] and this is related to, in part, structural variability in their ligand binding domains (LBDs)/AF-2 and AF-1 [7; 18]. E2 and many other estrogenic compounds activate both ERα and ERβ; however, ligand structure-dependent interactions of ERα subtypes have also been reported [19-22]. Studies in this laboratory have shown that 1,1'-bis(p-hydroxyphenyl)-2,2'-dichloroethylene exhibits ERα agonist and ERβ antagonist activities [22]. The C-terminal A/B domains of ERα and ERβ exhibit only 30% homology and there is evidence that this also contributes to the functional differences between these proteins [7; 18].
E2 and other estrogenic compounds also induce or repress genes through interactions of ERα and ERβ with other DNA-bound transcription factors such as the c-jun component of activating protein 1 (AP1), specificity protein 1 (Sp1), or Sp3 [23-28]. Estrogens and antiestrogens induce transactivation in cells transfected with constructs containing AP1 motifs and activation of ER/AP1 is dependent on ligand structure, ER-subtype and cell context [26-28]. For example, estrogenic compounds activated ERα/AP1-dependent transactivation in several different cell lines, whereas antiestrogens preferentially activated ERβ/AP1 in breast/endometrial cancer cells, and cotreatment with E2 inhibited antiestrogen activation of ERβ/AP1 [26]. Hormone-dependent activation of ERα/Sp1 through interaction with selected GC-rich sites in target gene promoters is also dependent on cell context, ligand structure, and ER-subtype [25]. Both estrogens and antiestrogens activate GC-rich constructs containing 1 or 3 (pSp13) consensus Sp1 binding sites, whereas antiestrogens inhibit E2-induced transactivation in cells transfected with GC-rich constructs containing hormone-responsive gene promoters. In contrast to ERβ/AP1, neither estrogens nor antiestrogens activate ERβ/Sp1 [24].
The classical pathway for hormone-dependent activation of ER involves formation of DNA-bound heterodimers which, in turn, recruit nuclear coactivator and coregulatory proteins that facilitate interaction with the basal transcriptional machinery [29-35]. The p160 steroid receptor coactivators (SRCs) were among the first coactivators described [36; 37], and ongoing studies have identified many different classes of coactivators that exhibit receptor-, ligand-, and cell context-dependent activities. Some coactivators such as SRCs, coactivators associated arginine methyl transferase and Brahma-related gene 1, exhibit histone acetyl transferase, methyl transferase, and ATP-dependent remodeling activities which modify chromatin structure and facilitate protein-DNA interactions critical for activation of gene expression [30; 34; 38-40]. The mammalian mediator complex of proteins including the vitamin D receptor interacting proteins (DRIPs) are also coactivators of NR-mediated transactivation [41-47]. There is evidence from some studies that DRIP205 anchors the DRIP complex to nuclear receptors (NRs) and these interactions are ligand-dependent [44; 48]. Previous studies have not identified coactivators of ERα/Sp1 and their mode of action [49]. We now show that DRIP150 coactivates ERα/Sp1-mediated transactivation in breast cancer cell lines transfected with a GC-rich construct (pSp13). Coactivation of ERα/Sp1 by DRIP150 is complex and ligand-dependent and requires multiple domains of ERα. Analysis of DRIP150 shows that coactivation of ERα/Sp1 is NR-box-independent and requires the 23 amino acid sequence (789-811) containing the α-helical amino acid 795-804 region, which is also required for coactivation of ERα [50].
MATERIALS AND METHODS
Cell Lines, Chemicals and Biochemicals
ZR-75 and MCF-7 human breast cancer cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA), and cells were cultured in RPMI-1640 (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS) (Summit Biotechnology, Fort Collins, CO). Medium was further supplemented with sodium bicarbonate, glucose, Hepes, sodium pyruvate and antibiotic/antimycotic solution (Sigma). MDA-MB-231 cells were obtained from ATCC and maintained in DME-F12 (Dulbecco's modified eagle's medium/F-12) supplemented with FBS and antibiotic/antimycotic solution. MCF-7 cells were maintained in MEM supplemented with 10% FBS, sodium bicarbonate, antibiotic/antimycotic solution, and insulin at 37°C with a humidified CO2:air (5:95) mixture. Phenol-free DME-F-12, phosphate-buffered saline, and E2 were also obtained from Sigma. [γ-32P]ATP (3000 Ci/mmol) was purchased form PerkinElmer Sciences (Boston, MA) and poly [d(I-C)] from Roche Molecular Biochemicals (Indianapolis, IN). Restriction enzymes, 5X luciferase lysis buffer, luciferin, and TNT7 in vitro translation kit were purchased from Promega (Madison, WI). Reagents for the β-galactosidase assay were obtained from Tropix (Bedford, MA). Sp1 antibody for gel mobility shift assays, ER antibody, and Sp1 antibody conjugated with agarose beads for coimmunoprecipitation assays and ProteinG-plus Agarose were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All other chemicals and biochemicals were obtained from commercial sources at the highest quality available.
Oligonucleotides and Plasmids
The consensus GC-rich element (Sp1) probe and the mutant probe used in gel mobility shift assays was synthesized by the Gene Technologies Laboratory (College Station, TX) and the sequence was 5'-AGC TTA TTC GAT CGG GGC CGG GCG AGC G-3' and 5'-AGC TTA TTC GAT CGA AGC GGG GCG AGC G-3'. ERα expression plasmid was kindly provided by Dr. Ming-Jer Tsai (Baylor College of Medicine, Houston, TX). Expression plasmids for ERα mutants with deletion of amino acid 1-178 (HE19), ERαTAF1 containing D538N, E542Q, and D545N mutations, and the DNA binding domain deletion mutant HE11 were kindly provided by Dr. Pierre Chambon (Institut de Genetique et de Biologie Moleculaire Cellulaire, Illkirch, France) and Dr. Donald McDonnell (Duke University, Durham, NC). cDNA encoding DRIP150 was kindly provided by Dr. Leonard P. Freedman (Merck Research Laboratories, West Point, PA). The expression plasmid for Sp1 was prepared in this laboratory by excising the Sp1 cDNA from pPacSp1 (generously supplied by Dr. Robert Tjian, University of California, Berkeley, CA) and cloned into pcDNA3.1 expression vector with oligonucleotide modification [51]. The expression plasmid for the GRIP-1 NR-box polypeptide GAL4 fusion protein was also provided by Dr. Donald McDonnell (Duke University). The expression plasmid for the AF1 polypeptide was generated in this laboratory by cloning amino acids 1-180 of ERα into NheI/EcoRV site of pcDNA3.0. The pSp13 reporter containing three tandem consensus Sp1 sites linked to a luciferase gene was created by cloning an oligonucleotide with three Sp1 elements into HindIII-BamHI cut pXP-1 plasmids as previously described [49]. DNA binding deletion mutants ERαΔZF1 and ERαΔZF2 constructs were prepared in this laboratory as previously described [49].
Cloning of DRIP150 Mutants
DRIP150 m1, m2, m3, m11, m12, pM23, pM23A792P, pM23R801P, and pM23A792P/R801P constructs were made in this laboratory as previously described [50]. pET28b(+)-131 aa was generated by cloning 131 amino acids 755-885 region in DRIP150 into BamHI/-XhoI site of pET28b(+) vector. For PCR amplification, the upper primer used for the cloning was 5'-AAA GGA TCC GAC CGC CGC CAT GGA GCC TGT TGG TGG TAG AAA GGT GGT TGA A-3' and the lower primer used for the cloning was 5'-AAA CTC GAG GAG TTT GTT GAT GGC ATT TA-3'.
Transient Transfection Assays
Cells were seeded in 12-well plates in phenol-free DME/F-12 supplemented with 2.5% charcoal-stripped FBS. After 18 h, cells were transfected by the calcium phosphate method with 1 μg of pSp13 reporter plasmid, 0.25 μg of a CMV β-gal expression plasmid, the different amounts of ERα, HE11, HE19, or ERαTAF1 expression plasmids, and the appropriate amount of wild-type or mutant DRIP150 expression plasmid. To maintain the same amount of DNA in each sample, pcDNA3.0 was also used. After 6 - 8 h, cells were shocked with 25% glycerol in phosphate-buffered saline (PBS) for 75 sec, rinsed once with PBS, and treated with either DMSO or 10 nM E2 in DME/F-12 plus 2.5% charcoal-stripped FBS for 36 h.
For Lipofectamine2000 transfection, 400 ng of pSp13 reporter plasmid, 100 ng of CMV β-gal expression plasmid, 500 ng of ERα, and the appropriate amounts of wild-type or mutant expression plasmid were used. pcDNA3.0 was also used to maintain the same amount of transfected DNA. Cells were initially seeded in phenol-free DME/F-12 supplemented with 2.5% charcoal-stripped FBS, then washed with DME/F-12 without antibiotics and serum, and 800 μl of DME/F-12 without antibiotics and serum was added to each well in 12-well plates. DNA and lipofectamine (2 μl) were added to each 100 μl of DME/F-12 without antibiotics and serum and incubated at room temperature for 5 min, then 100 μl DME/F-12 containing DNA and lipofectamine (2 μl) were mixed and incubated at room temperature for 20 min. The mixture (200 μl) was then added to each well, incubated for 6 h, the medium was removed, and cells were treated with DMSO or 10 nM E2 in DME/F-12 with 2.5% charcoal-stripped FBS and antibiotic/antimycotic solution for 36 h.
Cells were then harvested by scraping the plates in 100 μl of 1X lysis buffer (Promega), and 35 μl of the cell lysate was used for performing luciferase and β-galactosidase assays on a Lumicount Luminometer (Packard Instrument Co.). Normalized luciferase values were calculated by dividing the luciferase by the β-gal activities for a given sample. Results are expressed as means ± SE for at least 3 separate experiments for each treatment group and compared with the DMSO control group (arbitrarily set at 1) for each set of experiments.
Gel Electrophoretic Mobility Shift Assays
Five picomoles of GC-rich oligonucleotide was labeled at the 5' end using T4-polynucleotide kinase and [γ-32P]ATP. Plasmids containing the DRIP150, ERα, Sp1 and pcDNA3.0 cDNAs were used to in vitro transcribe and translate the corresponding protein in a rabbit reticulocyte lysate system (Promega). Three μl of in vitro translated ERα was mixed with 3 μl of in vitro translated Sp1 protein and treated with E2 to give a final concentration of 2.5 × 10−8M on ice for 15 min. Different amounts of in vitro translated DRIP150 were added to the mixture and incubated on ice for 5 min. To balance the volume, in vitro translated pcDNA3.0 was also added. For samples not containing ERα, only in vitro translated Sp1 protein was mixed with in vitro translated DRIP150 on ice for 15 min. For supershift and unlabeled oligonucleotide competition experiments, 2 μl of normal IgG, Sp1 antibody, 4 μl of unlabelled wild-type (1 pmol/μl), or mutant Sp1 probe (1 pmol/μl) were added to the mixture after coincubation with DRIP150 and then incubated on ice for an additional 15 min. [32P]-Labeled Sp1 probe (120,000 cpm, 5 μl) was added to the reaction mixture, giving a final volume of 25 μl, and incubated at 20°C for 15 min. Samples were then loaded onto 5% polyacrylamide gel and run at 110 V in 0.09 M Tris, 0.09 M borate, 2 mM EDTA (pH 8.3) for 2.5 h. The gel was dried, exposed to a phosphorscreen for 12 h, and protein-DNA binding was visualized by autoradiography using a Storm PhosphoImager (Molecular Dynamics, Sunnyvale, CA).
Coimmunoprecipitation Assays
Coimmunoprecipitation experiments were carried out to study interactions between DRIP150 and Sp1, DRIP150 and ERα/Sp1, Sp1 and pET28b(+)-131aa and ERα and pET28b(+)-131aa. For each coimmunoprecipitation assay, two in vitro translated [35S]-methionine labeled proteins (2-10 μl) were mixed and incubated on ice for 15 min. For interaction assays between DRIP150 and ERα/Sp1 and ERα and pET28b(+)-131aa, two proteins were mixed and E2 was added to give a final concentration of 100 nM; 10 μl of Sp1 or ERα antibody conjugated with agarose beads was added to the above mixture and incubated for 3 h on ice with shaking every 30 min. After incubation for 3 h on ice with shaking every 30 min, PBS (1 ml) was then added to each sample, shaken for 30 sec and centrifuged at 1,500 g for 5 min. After centrifugation, the supernatant was discarded and the pelleted fraction (100 μl) was mixed with 20 μl of 1X sample buffer (50 mM Tric-HCl pH 6.8, 2% SDS, 0.1% bromophenol blue, 10% glycerol and 100 mM DTT) containing β-mercaptoethanol. The sample was then boiled for 5 min, loaded onto SDS-polyacrylamide gel and run at 150 V for 4 h. The gel was dried, exposed to a phosphorscreen for 3 days, and proteins were visualized by autoradiography using a Storm PhosphoImager (Molecular Dynamics, Sunnyvale, CA).
Statistical Analysis
Statistical differences between different treatment groups were determined using ANOVA (Fisher's Protected LSD-least significance difference) and the levels of significance were noted (p < 0.05). The results were expressed as mean ± SE for at least 3 replicate determinations for each experiment.
RESULTS
DRIP150 Coactivation of Wild-type and Variant ERα/Sp1
Previous studies in this laboratory showed that DRIP150 coactivated ERα-mediated transactivation in breast cancer cells transfected with pERE3 [50]. The major objective of this study is to determine whether DRIP150 also coactivates ERα/Sp1 in breast cancer cells transfected with a GC-rich construct (pSp13) and also to investigate and compare domains of DRIP 150 required for coactivation of ERα/Sp1 and ERα [50]. Results in Figure 1A show that in ZR-75 cells cotransfected with pSp13 and ERα expression plasmid (150 ng), 10 nM E2 significantly induced luciferase activity (3-fold), and cotransfection with DRIP150 (2.5- 7.5 ng) expression plasmid enhanced hormone-induced transactivation by approximately 3-fold. Figure 1B illustrates that DRIP150 enhanced luciferase activity in cells transfected with pSp13 indicating that DRIP150 coactivated Sp1- and ERα/Sp1-dependent transactivation. Coactivation of ERα/Sp1 by DRIP150 is expressed as fold-enhancement of the hormone-induced response (Fig. 1A) and is therefore not dependent on increased basal (hormone-independent) activity. MCF-7 (Fig. 1C) and MDA-MB-231 (Fig. 1D) breast cancer cells were transfected with pSp13, ERα and DRIP150, and the results showed that DRIP150 enhanced the fold-induction response in both cell lines.
Figure 1.
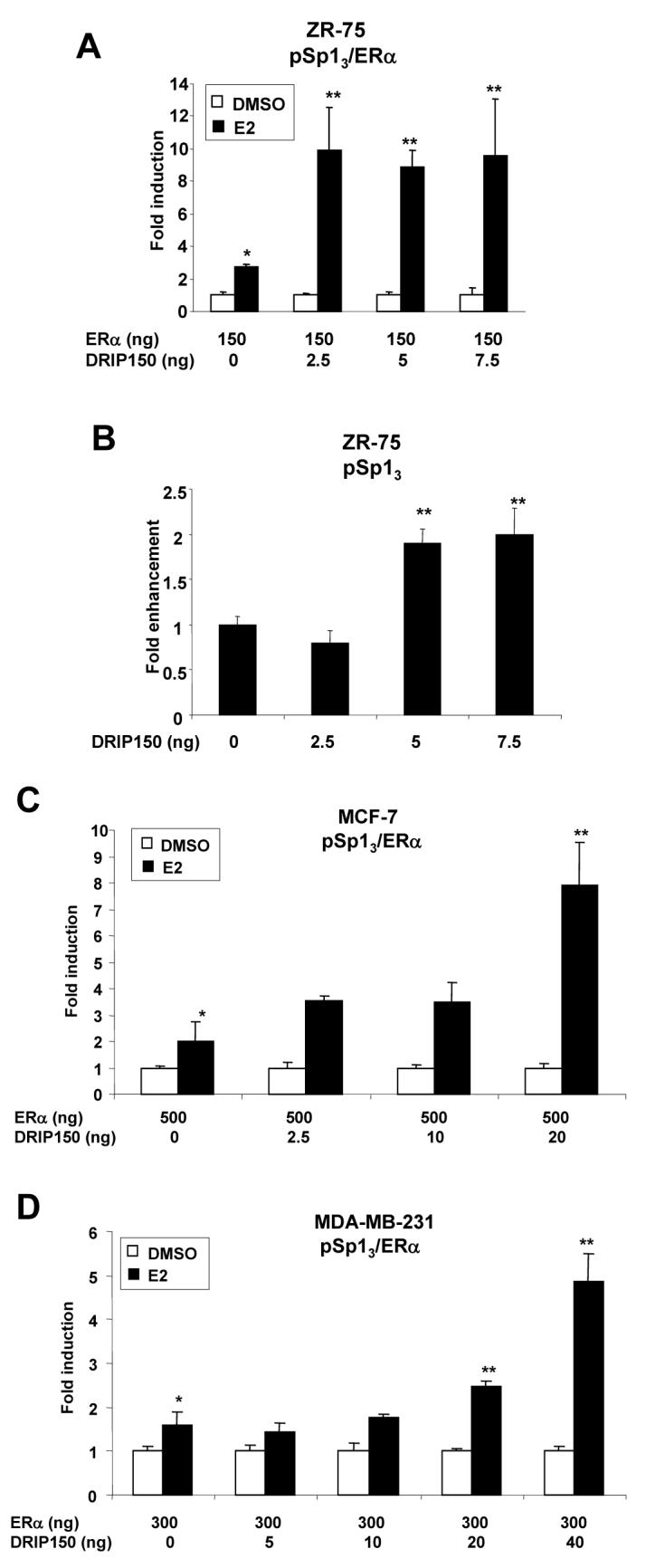
Coactivation of ERα/Sp1 in ZR-75 (A, B), MCF-7 (C) and MDA-MB-231 (D) cells. Cells were transfected with pSp13 and different amounts of ERα expression plasmid (A, C and D) or pSp13 alone (B), treated with DMSO or 10 nM E2, and luciferase activity was determined as described in the Materials and Methods. Significant (p < 0.05) induction or activation of Sp1 by E2 (*) and coactivation by DRIP150 (**) are indicated. Transfection experiments illustrated in this and all other figures are results of at least three replicate experiments for each treatment group and are expressed as means ± SE. All coactivation experiments have been confirmed in two or more experiments.
ERα/Sp1-dependent activation of pSp13 is also observed in breast cancer cells transfected with the DBD deletion mutant [23; 25; 49] and in this study, cells were transfected with ERα mutants containing deletions of the DNA binding domain (DBD) (HE11C), zinc finger 2 of the DBD (ERαΔZF2), or zinc finger 1 (ERαΔZF1) [49]. The results in Figures 2A, 2B and 2C confirm that E2 induces transactivation in ZR-75, MCF-7 and MDA-MB-231 cells transfected with HE11C. The effects of DRIP150 on coactivation of HE11C/Sp1 were cell context-dependent; enhanced fold induction was observed only in MDA-MB-231 cells but not in MCF-7 or ZR-75 cells. These results suggest that in MCF-7 and ZR-75 cells, the DBD of ERα is important for coactivation by DRIP150. A previous report showed that variant ERα constructs containing deletions of zinc finger 1 (ERαΔZF1) or zinc finger 2 (ERαΔZF2) also induced luciferase activity in cells transfected with pSp13 [49], and the potential effects of these zinc finger deletions on coacitvation by DRIP150 were further investigated. The results (Figs. 2D and 2E) show that E2 induced activity in ZR-75 cells transfected with ERαΔZF1 and ERαΔZF2, and DRIP150 either did not enhance the fold-induction (ERαΔZF1) or decrease transactivation (ERαΔZF2). Thus, both ZFs of ERα are required for coacitvation of ERα/Sp1 by DRIP150.
Figure 2.

Coactivation of HE11/Sp1 by DRIP150 in ZR-75 (A), MCF-7 (B) and MDA-MB-231 (C) cells and ERΔZF1/Sp1 (D) and ERΔZF2/Sp1 (E) in ZR-75 cells. Cells were transfected with pSp13 and ERα deletion mutants, treated with DMSO or E2, and luciferase activity was determined as described in the Materials and Methods. Significant coactivation by DRIP150 is indicated (**). Coactivation with lower concentration of DRIP150 (i.e. 2.5 and 5.0 ng) were not observed in experiments summarized in Figures 2A and 2C (data not shown).
The effects of DRIP150 on coactivation of ERα/Sp1 was also determined in MDA-MB-231, MCF-7 and ZR-75 cells transfected with the mutant ERαTAF1 that contains amino acid mutations in helix 12 of ERα [52]. E2 induced transactivation in all three cell lines transfected with pSp13 and ERαTAF1, indicating that helix 12 sites that interact with nuclear receptors (NR) boxes (LXXLL) are not required for transactivation (Figs. 3A, 3B and 3C). However, it was also apparent that DRIP150 did not coactivate ERαTAF1 in the three breast cancer cell lines; in some cells, hormone-induced activity was repressed and higher amounts of DRIP150 decreased transactivation in all three cell lines. These results suggest that an intact helix 12 is also required for coactivation of ERα by DRIP150 indicating that multiple domains of ERα are required for coactivation of ERα/Sp1 by DRIP150. Results in Figure 3D show that E2 does not activate HE19/Sp1; however, DRIP 150 coactivates HE19/Sp1 suggesting that the coactivator restores E2-responsiveness to this complex.
Figure 3.

Coactivation of ERαTAF1/Sp1 by DRIP150 in ZR-75 (A), MCF-7 (B) and MDAMB-231 (C) cells and coactivation of HE19/Sp1 (D) in ZR-75 cells. Cells were transfected with pSp13 and variant ERα expression plasmids, treated with DMSO or E2, and luciferase activity was determined as described in the Materials and Methods. Significant (p < 0.05) coactivation by DRIP150 is indicated (**).
Squelching of ERα/Sp1 and Effects on Coactivation
Overexpression of the NR-box peptide containing two LXXLL motifs from GRIP1 decreased ERα-mediated transactivation and coactivation by DRIP150 in ZR-75 cells transfected with pERE3 [50]. The NR box peptide did not affect hormonal activation of ERα/Sp1 (Fig. 4A); however, overexpression of the NR-box peptide significantly inhibited coactivation of ERα/Sp1 by DRIP150 (Fig. 4B). In contrast, overexpression of an AF-1 peptide (amino acids 1-180 of ERα) decreased ERα/Sp1-mediated transactivation (Fig. 4C) as previously reported [51]. The AF-1 peptide expression also decreased the magnitude of the coactivation response (data not shown) and this could be related, in part, to decreased hormone-dependent transactivation. The results illustrated in Figure 4D show that the decreased induction response associated with overexpression of AF-1 peptide can be reversed by DRIP150. This suggests that among the NR-box and AF-1 peptides, the former preferentially squelched DRIP150 coactivation of ERα/Sp1, whereas the latter inhibited ERα/Sp1-mediated transactivation.
Figure 4.
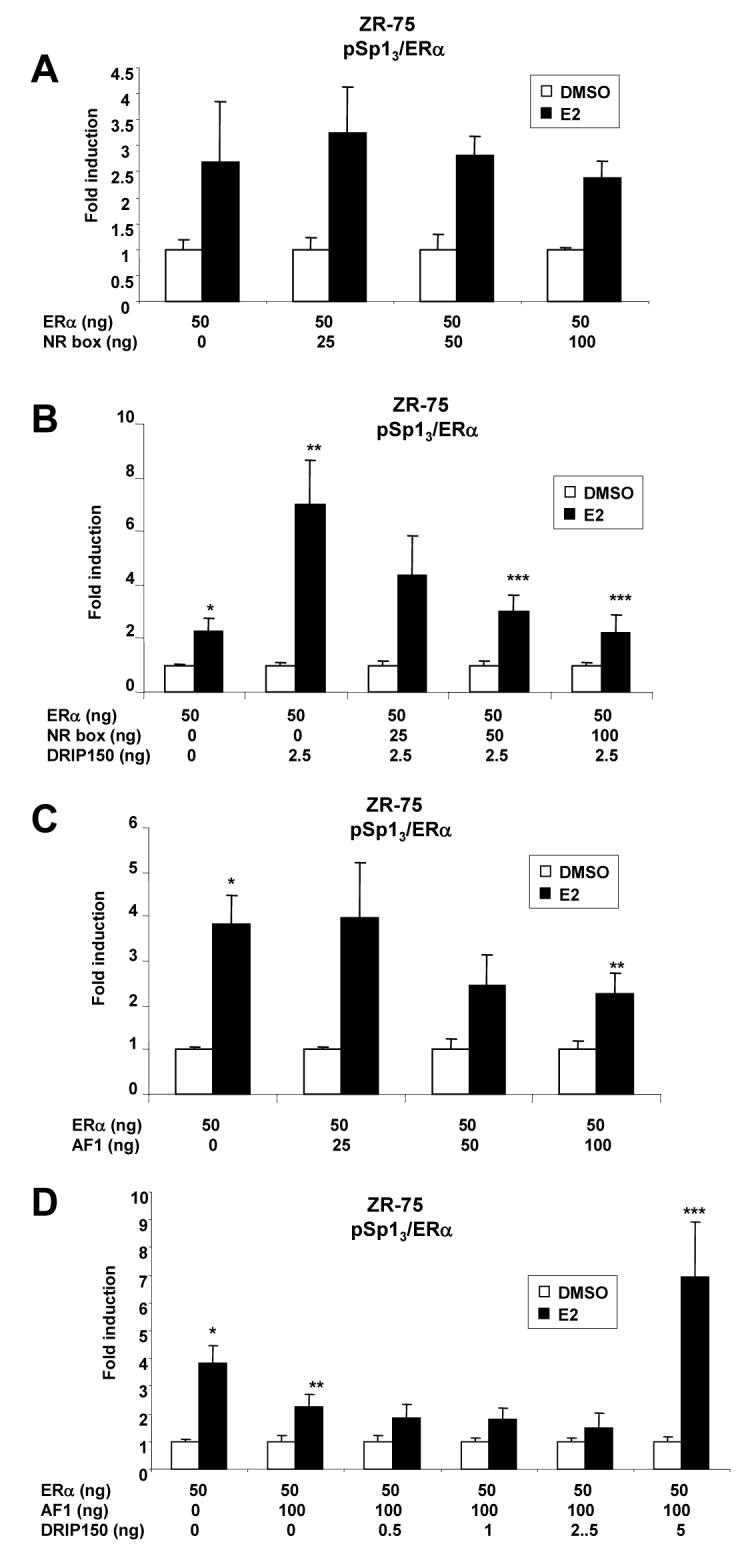
NR box and AF1 squelching of E2-induced transactivation in cells transfected with pSp13. NR box inhibition of ERα/Sp1-mediated transactivation (A) and coactivation of ERα/Sp1 by DRIP150 (B) in ZR-75 cells. Cells were transfected with pSp13 and ERα alone or in combination with DRIP150, treated with DMSO or 10 nM E2, and luciferase activity was determined as described in the Materials and Methods. NR box squelching of DRIP150-mediated coactivation of ERα/Sp1 was determined by cotransfection of different amounts of the NR box expression plasmid (0 - 100 ng). Significant (p < 0.05) induction by E2 (*) and inhibition by NR box peptide expression are indicated (***). AF1 peptide inhibition of ERα/Sp1 (C, D). Cells were transfected and treated as described in A/B. Squelching of DRIP150 coactivation of ERα/Sp1 was determined by cotransfection with different amounts (0 - 100 ng) of an expression plasmid expressing the AF1 domain of ERα [49]. Significant (p < 0.05) inhibition of ERα/Sp1-mediated transactivation by the AF1 expression plasmid is indicated (**). Significant (p < 0.05) reversal of AF1-dependent inhibition by DRIP150 (D) is also indicated (***).
DRIP150 Interactions With ERα/Sp1
Interactions of DRIP150 with ERα and Sp1 were investigated in immunoprecipitation experiments with in vitro-translated 35S-labeled proteins (Fig. 5A). In vitro expressed 35S-DRIP150, Sp1 and empty vector (lanes 1 - 3, respectively) are shown; Sp1 antibody immunoprecipitates 35S-Sp1 (lane 4) but not 35S-pcDNA3.0 (lane 5) (empty vector) and coimmunoprecipitates Sp1 and DRIP150 (lane 6). Thus, DRIP150 alone interacts with Sp1. 35S-Labeled DRIP150, Sp1, ERα and pcDNA3.0 (empty vector) are shown in Figure 5B, lanes 1 - 4, respectively. Sp1 antibody does not immunoprecipitate radioactivity after coincubation with 35S-pcDNA3.0 alone (lane 6) or in combination with ERα (lane 8). However, this antibody immunoprecipitates 35S-Sp1 (lane 5) and coimmunoprecipitates Sp1 plus ERα (lane 7) and Sp1 plus ERα plus DRIP150 (lane 9). Thus, DRIP150 forms a complex with ERα and Sp1 and directly interacts with both proteins.
Figure 5.

Interactions of DRIP150, ERα and Sp1. (A, B) Coimmunoprecipitation of DRIP150 and Sp1 alone (A) or plus ERα (B). 35S-Labeled protein or empty vector (35SpcDNA3.0) were coincubated with various antibodies and immunoprecipitates were analyzed by SDS-PAGE as described in the Materials and methods. (C) Gel mobility shift assay. 32P-Labeled Sp1 oligonucleotide was incubated with one or more proteins (ERα, Sp1 and DRIP150), Sp1 or non-specific IgG antibodies and oligonucletides and analyzed by gel mobility shift assays as described in the Materials and Methods. The specifically-bound Sp1-DNA complex and the antibody supershifted complex (SS) are indicated.
Interactions of ERα, Sp1 and DRIP150 were also determined in gel mobility shift assays using a radiolabeled GC-rich oligonucleotide (32P-Sp1) and in vitro translated Sp1, ERα and DRIP150 proteins (Fig. 5C). Sp1 plus 32P-Sp1 formed a retarded band (lane 12), and increasing amounts of DRIP150 (lanes 6 and 7) or ERα enhanced the retarded band intensity. DRIP150 also enhanced the retarded band intensity formed after coincubation with ERα plus Sp1 (lanes 8-10). The retarded band formed after coincubation with DRIP150, ERα and Sp1 was decreased in intensity after coincubation with excess (300-fold) unlabeled Sp1 oligonucleotide, but not affected by Ig G antibodies or mutant Sp1 oligonucleotide. Sp1 antibody supershifted the retarded band (lane 3). Thus, the intensity of the Sp1-DNA complex was enhanced by ERα, DRIP150 or their combination, but a ternary or quaternary supershifted complex was not detected. These results are consistent with previous studies showing that Sp1-DNA interactions are enhanced but not supershifted after coincubation with ERα and other transcription factors. Moreover, we also showed that DRIP150 also enhanced ERα-DNA binding but did not form a ternary supershifted complex [10].
Coactivation of ERα/Sp1 by DRIP150 is NR Box-independent
Results in Figure 6A show that wild-type DRIP150 and DRIP150 m1 coactivate ERα/Sp1, whereas minimal enhancement was observed for DRIP150 m2 and DRIP150 m3. This indicates that the C-terminal NR box (deleted from DRIP150 m1) was not necessary for coactivation of ERα/Sp1 by DRIP150. Moreover, in a separate experiment, DRIP150 m12 (in which both the N- and C-terminal NR boxes have been deleted) also coactivated ERα/Sp1 (Fig. 6B) showing that the NR boxes were not necessary for coactivation of ERα/Sp1 by DRIP150. These responses were similar to results of studies on DRIP150 coactivation of ERα in cells transfected with pERE3 where it was shown that a 23 amino acid sequence from 789-811 in the central core region of DRIP150 was required for coactivation [50]. The results in Figure 6C compare the coactivation of ERα/Sp1 by wild-type DRIP150 and deletion mutants that express amino acids 1-811 (DRIP150 m11) and 1-788 (DRIP150 m2), and only the former deletion mutant was active. This indicates that coactivation of ERα/Sp1 was dependent on amino acids 789-811 which contain two putative α-helical sequences at 789-794 and 795-804, and similar results were reported for coactivation of ERα [50]. Using 35S-labeled in vitro translated Sp1 (lane 2), ERα (lane 3), empty vector (lane 4), and Sp1 antibody, we investigated interactions of amino acids 755-885 in DRIP150 (pET28b(+)-131aa, lane 1) with Sp1 (lane 7) (Fig. 6D). The Sp1 antibody immunoprecipitated Sp1 (lane 7) and ERα/Sp1 (lane 8) in the presence and absence of 35S-pET28b(+)-131, respectively; however, the latter protein was not detected in the immunoprecipitates (lane 7). This results indicate that minimal association of this core region of DRIP150 with Sp1; however, using ERα antibody (Fig. 6E), 35S-pET28b(+)-131aa could be coimmunoprecipitated with ERα (lane 6), indicating a preferential interaction of this region of DRIP150 with ERα and not Sp1.
Figure 6.

Coactivation of ERα/Sp1 by DRIP150 mutants and interactions of pET28b(+)-131 aa with ERα and Sp1. Coactivation of ERα/Sp1 by DRIP150 deletion mutants (A - C). ZR-75 cells were transfected with pSp13, ERα, wild-type and variant DRIP150 expression plasmids, treated with DMSO or 10 nM E2, and luciferase activity was determined as described in the Materials and Methods. Significant (p < 0.05) coactivation by wild-type or variant DRIP150 constructs are indicated (**). Coimmunoprecipitation of 35S-pET28b(+)-131aa with ERα and Sp1 (D, E). 35SLabled proteins were in vitro expressed, coincubated, immunoprecipitated with Sp1 or ERα antibodies, and immunoprecipitates were analyzed by SDS-PAGE as described in the Materials and Methods.
The role of amino acids 789-811 of DRIP150 in mediating coactivation of ERα/Sp1 was investigated in squelching experiments using chimeric-GAL4 chimeras fused to amino acids 789-811 (pM23) and sequences mutated in the 5'-(pM23 A792P), 3'-(pM23R801P) and both (pM23A792P/R801P) helical regions (Fig. 7A). Wild-type pM23 squelches coactivation of ERα/Sp1 by DRIP150 (Fig. 7B); similar squelching was observed for pM23A792P (Fig. 7C) but not for pM23R801P (Fig. 7D) or pM23A792P/R801P (Fig. 7E). These results show that the 795-804 helical region of DRIP150 was required for squelching, suggesting that this sequence is critical for coactivation of ERα/Sp1 by DRIP150. This sequence is also important for coactivation of ERα [50] and the squelching and coimmunoprecipitation (Figs. 6D and 6E) results suggest that DRIP150 primarily coactivates ERα/Sp1 through interactions with ERα and the coactivation is independent of the LXXLL motifs in DRIP150.
Figure 7.

Squelching of DRIP150 coactivation of ERα/Sp1. (A) Wild-type and mutant GAL4-pM23 construct used in squelching experiments. Overexpression of pM23 (B), pM23 A792P (C), pM23R801P(D) and pM23A792P/R801P (E) on coactivation of ERα/Sp1 by DRIP150. ZR-75 cells were transfected with pSp13/ERα, different amounts of pM23-derived constructs, treated with DMSO or 10 nM E2, and luciferase activity was determined as described in the Materials and Methods. Significant (p < 0.05)induction by E2 (*), coactivation by DRIP150 (**), and squelching of this response by wild-type and mutant pM23 peptides (***) are indicated.
DISCUSSION
ERα/Sp1-mediated transactivation has been linked to hormone activation of several genes involved in cell cycle progression, DNA synthesis, and metabolism of purines and pyrimidines [12]. Moreover, in RNA interference studies, knockdown of Sp1 inhibits hormone-induced cell cycle progression and partially reverses E2 induced G1 to S phase progression [53]. The molecular determinants for hormonal activation of ERα/Sp1-dependent gene expression have been investigated and demonstrate that this is a novel pathway of estrogen action. ERα and Sp1 interact in the presence or absence of ligand, and ERα specifically interacts with C-terminal DNA-binding domain of Sp1 which also binds many other nuclear proteins [25]. The AF1 (and not AF2) domain of ERα is also a critical element for ERα/Sp1 action [49] and, in domain swapping experiments between ERα and ERβ, it was shown that inactivity of ERβ was due to the AF1 domain [24]. For example, a chimeric ERα/β protein containing N-terminal (A/B) domain of ERα and the C-F domain of ERβ activate GC-rich constructs in breast cancer cells [24]. Despite the critical importance of the A/B (AF1) domain, hormonal activation of ERα/Sp1 is also dependent on the hinge region (D) [49], and the C-terminal F domain but did not require the DBD [49]. It has been also reported that p160 coactivators and the AF1-dependent p68 helicase coactivator did not enhance ERα/Sp1-mediated transactivation in breast cancer cells transfected with pSp13 [49].
DRIP150 is a component of the DRIP mediator-like complex which coactivates NR-mediated transactivation. Several studies show ligand-dependent interaction of DRIP complex proteins with NRs, and DRIP205 anchors this complex through direct interactions with these receptors [44; 54-64]. However, it has also been reported that mediator complexes can act in the absence of DRIP205 [65] and one of these complexes, DRIP150, directly interacts with several NRs including ERα and ERβ [59-61]. Recently, we showed that DRIP150 coactivated ERα-mediated transcription in breast cancer cells transfected with pERE3 and this response was dependent on many factors including domains of ERα, DRIP150 and cell context [50]. DRIP150 also coactivated ERα/Sp1-mediated transactivation in ZR-75, MCF-7 and MDA-MB-231 breast cancer cells (Fig. 1), and the former cell line was the most sensitive to coactivation by transfected DRIP150. DRIP150 also enhanced Sp1-mediated transactivation in ZR-75 cells (Fig. 1B); however, this was not a necessary for coactivation of ERα/Sp1 since this response was observed using amounts of DRIP150 (i.e. 2.5 ng) that did not affect transactivation in cells transfected with pSp13 alone.
Hormone-dependent activation of ERα and ERα/Sp1 can also be observed in cells transfected with ERα variants ERαTAF1 (helix 12 mutations) [49], and DRIP150 does not enhance transactivation in cells transfected with ERα-TAF1 and pERE3 [50] or pSp13 (Figs. 3A - 3C). In contrast, E2 induces transactivation in cells transfected with pERE3 and the AF1 deletion mutant HE19 [50], whereas HE19/Sp1 is minimally responsive to E2; however, DRIP150 coactivates HE19 and HE19/Sp1 [50] (Fig. 3D). With the exception of results in MDA-MB-231 cells, DRIP150 does not coactivate HE11/Sp1 and the DBD deletion mutant of ERα is inactive in cells transfected with pERE3 [50]. Thus, coactivation of ERα and ERα/Sp1 by DRIP150 requires similar domains of ERα suggesting that DRIP150 coactivation of ERα/Sp1 may be due to primarily to preferential interactions with ERα. However, this does not exclude a role for Sp1 in this process since DRIP150 interacts with both ERα and Sp1 proteins in coimmunoprecipitation studies (Figs. 5A and 5B), and both ERα and DRIP150 enhance Sp1-DNA complex formation in gel mobility shift assays (Fig. 5C). The paradox between the interactions of ERα, DRIP150 and Sp1 in coimmunoprecipitation studies (Figs. 5A and 5C) and the formation of only a binary Sp1-DNA complex in gel mobility shift assays has previously been observed for interactions of proteins with other DNA-bound transcription factors [66-68]. This may be due to limitations of the assay and the relative low binding affinities of ERα and DRIP150 for Sp1 compared to the higher affinity interactions of Sp1 for GC-rich motifs.
ERα-dependent transactivation and coactivation of this response by DRIP150 are squelched by overexpression of a NR-box protein containing two GRIP NR box sequences [50], whereas overexpression of AF1 (amino acids 1-180 from ERα) did not affect hormone-induced transactivation [49]. In contrast, AF1 but not NR-box overexpression inhibits ERα/Sp1-mediated transactivation (Figs. 4A and 4C); however, the NR-box peptide inhibits coactivation of ERα/Sp1 by DRIP150 (Fig. 4C). These results suggest that the AF2 region of ERα is critical for coactivation of ERα/Sp1 by DRIP150.
Previous studies on DRIP150 coactivation of ERα show that this response is independent of the C- and N-terminal NR-boxes [50] and we have shown in this study that the DRIP150m12 NR-box deletion mutant also coactivates ERα/Sp1 (Fig. 6B). Moreover, the activity of several DRIP150 deletion mutants are similar for their coactivation of ERα [50], and ERα/Sp1 and amino acids 789-811 are critical determinants for coactivation by DRIP150 (Fig. 6). Based on crystal structure database, there are two potential α-helical motifs in this central core region of DRIP150; the NIFSEVRVYN (amino acids 795-804) sequence is homologous to the α-helical amino acids 69-78 in hepatocyte nuclear factor-1 [69], and the DIPAHL sequence (amino acids 789-794) corresponds to α-helical structures identified in Lanuginosa lipase and histamine Nmethyltransferase [70; 71]. Squelching experiments with the wild-type 23 amino acid sequence (pM23) and proline mutants of the 789-794 and 795-804 sequence clearly show that the 795-804 region of DRIP150 is required for inhibitory coactivation of ERα/Sp1 by DRIP150 (Fig. 7), and comparable results were obtained for inhibition of ERα coactivation by DRIP150 using these same wild-type/mutant pM23 constructs [50]. Coimmunoprecipitation studies using an in vitro expressed protein that encompasses the 755-885 central region of DRIP150 (pET28b(+)-131aa) also demonstrate that this protein which contains the critical “coactivating region” of DRIP150 preferentially interacts with ERα and not Sp1 (Figs. 6D and 6E). This interaction of ERα with the central core region of DRIP150 may explain why there is a strong parallel between the specific regions of DRIP150 required for coactivation of ERα [50] and ERα/Sp1 (Fig. 6).
In summary, this paper identifies DRIP150 as a coactivator of ERα/Sp1-mediated activation of GC-rich promoters. Although there are major differences in contributions of different domains of ERα for E2-dependent transactivation in cells transfected with pERE3 (ERα) and pSp13 (ERα/Sp1), the mechanisms of coactivation by DRIP150 are similar and primarily targeted to ERα. Interestingly, coactivation of ERα/Sp1 and ERα by DRIP150 requires an intact helix 12 in the AF2 domain of ERα [50] (Figs. 3A - 3C). Moreover, the NR-box peptide and pM23 inhibit coactivation of ERα/Sp1 by DRIP150 (Figs. 4B and 7B) and coactivation of ERα by DRIP150 [50], demonstrating comparable squelching activity by the NR-boxes and the α-helical motif in pM23 (amino acids 795-804). Thus although the LXXLL and NIFSEVRVYN sequences are different, their roles in mediating coactivation of ERα and ERα/Sp1 may be complementary due to their common and α-helical structure. This may also explain the NR-box-independent coactivation of ERα and other NRs by diverse coactivators [72; 73] which may also contain α-helical or other structural features required for linking NRs to critical nuclear factors and the basal transcriptional machinery. Current studies are investigating the role of DRIP150 and other mediator proteins as coactivators of ERα/Sp1 and their role as coactivators of ER/Sp proteins on specific E2-responsive gene promoters in breast cancer cells.
ACKNOWLEDGEMENTS
The financial assistance of the National Institutes of Health (ES09106 and CA104116) and the Texas Agricultural Experiment Station is gratefully acknowledged. S. Safe is a Sid Kyle Professor of Toxicology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Nilsson S, Gustafsson JA. Crit.Rev.Biochem.Mol.Biol. 2002;37:1–28. doi: 10.1080/10409230290771438. [DOI] [PubMed] [Google Scholar]
- 2.Olefsky JM. J.Biol.Chem. 2001;276:36863–36864. doi: 10.1074/jbc.R100047200. [DOI] [PubMed] [Google Scholar]
- 3.Hall JM, Couse JF, Korach KS. J.Biol.Chem. 2001;276:36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- 4.Chambon P. Mol.Endocrinol. 2005;19:1418–1428. doi: 10.1210/me.2005-0125. [DOI] [PubMed] [Google Scholar]
- 5.Evans RM. Mol.Endocrinol. 2005;19:1429–1438. doi: 10.1210/me.2005-0046. [DOI] [PubMed] [Google Scholar]
- 6.O'Malley BW. Mol.Endocrinol. 2005;19:1402–1411. doi: 10.1210/me.2004-0480. [DOI] [PubMed] [Google Scholar]
- 7.Ogawa S, Inouse S, Watanabe T, Hiroi H, Orimo A, Hosoi T, Ouchi Y, Muramatsu M. Biochem.Biophys.Res.Commun. 1998;243:122–126. doi: 10.1006/bbrc.1997.7893. [DOI] [PubMed] [Google Scholar]
- 8.Cowley SM, Hoare S, Mosselman S, Parker MG. J.Biol.Chem. 1997;272:19858–19862. doi: 10.1074/jbc.272.32.19858. [DOI] [PubMed] [Google Scholar]
- 9.Pace P, Taylor J, Suntharalingam S, Coombes RC, Ali S. J.Biol.Chem. 1997;272:25832–25838. doi: 10.1074/jbc.272.41.25832. [DOI] [PubMed] [Google Scholar]
- 10.Tremblay A, Tremblay GB, Labrie C, Labrie F, Giguere V. Endocrinology. 1998;139:111–118. doi: 10.1210/endo.139.1.5702. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe T, Inoue S, Ogawa S, Ishii Y, Hiroi H, Ikeda K, Orimo A, Muramatsu M. Biochem.Biophys.Res.Commun. 1997;236:140–145. doi: 10.1006/bbrc.1997.6915. [DOI] [PubMed] [Google Scholar]
- 12.Pettersson K, Brandien K, Kuiper GG, Gustafsson J-Å. Mol.Endocrinol. 1997;11:1486–1496. doi: 10.1210/mend.11.10.9989. [DOI] [PubMed] [Google Scholar]
- 13.Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe S, Van der Saag PT, Van der Burg B, Gustafsson J-Å. Endocrinology. 1998;139:4252–4263. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 14.Liu MM, Albanese C, Anderson CM, Hilty K, Webb P, Uht RM, Price RH, Jr., Pestell RG, Kushner PJ. J.Biol.Chem. 2002;277:24353–24360. doi: 10.1074/jbc.M201829200. [DOI] [PubMed] [Google Scholar]
- 15.Lazennec G, Bresson D, Lucas A, Chauveau C, Vignon F. Endocrinology. 2001;142:4120–4130. doi: 10.1210/endo.142.9.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. Cancer Res. 2004;64:423–428. doi: 10.1158/0008-5472.can-03-2446. [DOI] [PubMed] [Google Scholar]
- 17.Lindberg MK, Moverare S, Skrtic S, Gao H, hlman-Wright K, Gustafsson JA, Ohlsson C. Mol.Endocrinol. 2003;17:203–208. doi: 10.1210/me.2002-0206. [DOI] [PubMed] [Google Scholar]
- 18.Warnmark A, Treuter E, Gustafsson JA, Hubbard RE, Brzozowski AM, Pike AC. J.Biol.Chem. 2002;277:21862–21868. doi: 10.1074/jbc.M200764200. [DOI] [PubMed] [Google Scholar]
- 19.Harrington WR, Sheng S, Barnett DH, Petz LN, Katzenellenbogen JA, Katzenellenbogen BS. Mol.Cell Endocrinol. 2003;206:13–22. doi: 10.1016/s0303-7207(03)00255-7. [DOI] [PubMed] [Google Scholar]
- 20.Mueller SO, Hall JM, Swope DL, Pedersen LC, Korach KS. J.Biol.Chem. 2003;278:12255–12262. doi: 10.1074/jbc.M203578200. [DOI] [PubMed] [Google Scholar]
- 21.Sun J, Huang YR, Harrington WR, Sheng S, Katzenellenbogen JA, Katzenellenbogen BS. Endocrinology. 2002;143:941–947. doi: 10.1210/endo.143.3.8704. [DOI] [PubMed] [Google Scholar]
- 22.Gaido KW, Maness SC, McDonnell DP, Dehal SS, Kupfer D, Safe S. Mol.Pharmacol. 2000;58:852–858. [PubMed] [Google Scholar]
- 23.Porter W, Saville B, Hoivik D, Safe S. Mol.Endocrinol. 1997;11:1569–1580. doi: 10.1210/mend.11.11.9916. [DOI] [PubMed] [Google Scholar]
- 24.Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson J-A, Safe S. J.Biol.Chem. 2000;275:5379–5387. doi: 10.1074/jbc.275.8.5379. [DOI] [PubMed] [Google Scholar]
- 25.Safe S, Kim K. Prog.Nucleic Acid Res.Mol.Biol. 2004;77:1–36. doi: 10.1016/S0079-6603(04)77001-4. [DOI] [PubMed] [Google Scholar]
- 26.Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 27.Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, Katzenellenbogen BS, Enmark E, Gustafsson J-Å, Nilsson S, Kushner PJ. Mol.Endocrinol. 1999;13:1672–1685. doi: 10.1210/mend.13.10.0357. [DOI] [PubMed] [Google Scholar]
- 28.Webb P, Lopez GN, Uht RM, Kushner PJ. Mol.Endocrinol. 1995;9:443–456. doi: 10.1210/mend.9.4.7659088. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Falasca M, Schlessinger J, Malstrom S, Tsichlis P, Settleman J, Hu W, Lim B, Prywes R. Cell Growth Differ. 1998;9:513–522. [PubMed] [Google Scholar]
- 30.Lemon BD, Freedman LP. Current Opinion in Genetics & Development. 1999;9:499–504. doi: 10.1016/s0959-437x(99)00010-6. [DOI] [PubMed] [Google Scholar]
- 31.Klinge CM. Steroids. 2000;65:227–251. doi: 10.1016/s0039-128x(99)00107-5. [DOI] [PubMed] [Google Scholar]
- 32.Smith CL, O'Malley BW. Endocr.Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 33.Edwards DP. Vitam.Horm. 1999;55:165–218. doi: 10.1016/s0083-6729(08)60936-x. [DOI] [PubMed] [Google Scholar]
- 34.Näär AM, Lemon BD, Tjian R. Annu.Rev.Biochem. 2001;70:475–501. doi: 10.1146/annurev.biochem.70.1.475. [DOI] [PubMed] [Google Scholar]
- 35.Rosenfeld MG, Glass CK. J.Biol.Chem. 2001;276:36865–36868. doi: 10.1074/jbc.R100041200. [DOI] [PubMed] [Google Scholar]
- 36.Halachmi S, Marden E, Martin G, MacKay H, Abbondanza C, Brown M. Science. 1994;264:1455–1458. doi: 10.1126/science.8197458. [DOI] [PubMed] [Google Scholar]
- 37.Onate SA, Tsai SY, Tsai MJ, O'Malley BW. Science. 1995;270:1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 38.Dilworth FJ, Chambon P. Oncogene. 2001;20:3047–3054. doi: 10.1038/sj.onc.1204329. [DOI] [PubMed] [Google Scholar]
- 39.Featherstone M. Curr.Opin.Genet.Dev. 2002;12:149–155. doi: 10.1016/s0959-437x(02)00280-0. [DOI] [PubMed] [Google Scholar]
- 40.Freiman RN, Tjian R. Cell. 2003;112:11–17. doi: 10.1016/s0092-8674(02)01278-3. [DOI] [PubMed] [Google Scholar]
- 41.Näär AM, Beaurang PA, Zhou S, Abraham S, Solomon W, Tjian R. Nature. 1999;398:828–832. doi: 10.1038/19789. [DOI] [PubMed] [Google Scholar]
- 42.Ito M, Yuan CX, Malik S, Gu W, Fondell JD, Yamamura S, Fu ZY, Zhang X, Qin J, Roeder RG. Mol.Cell. 1999;3:361–370. doi: 10.1016/s1097-2765(00)80463-3. [DOI] [PubMed] [Google Scholar]
- 43.Rachez C, Lemon BD, Suldan Z, Bromleigh V, Gamble M, Näär AM, Erdjument-Bromage H, Tempst P, Freedman LP. Nature. 1999;398:824–828. doi: 10.1038/19783. [DOI] [PubMed] [Google Scholar]
- 44.Yuan CX, Ito M, Fondell JD, Fu ZY, Roeder RG. Proc.Natl.Acad.Sci.U.S.A. 1998;95:7939–7944. doi: 10.1073/pnas.95.14.7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rachez C, Suldan Z, Ward J, Chang CP, Burakov D, Erdjument-Bromage H, Tempst P, Freedman LP. Genes Dev. 1998;12:1787–1800. doi: 10.1101/gad.12.12.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun X, Zhang Y, Cho H, Rickert P, Lees E, Lane W, Reinberg D. Mol.Cell. 1998;2:213–222. doi: 10.1016/s1097-2765(00)80131-8. [DOI] [PubMed] [Google Scholar]
- 47.Jiang YW, Veschambre P, Erdjument-Bromage H, Tempst P, Conaway JW, Conaway RC, Kornberg RD. Proc.Natl.Acad.Sci.U.S.A. 1998;95:8538–8543. doi: 10.1073/pnas.95.15.8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang YK, Guermah M, Yuan CX, Roeder RG. Proc.Natl.Acad.Sci.U.S.A. 2002;99:2642–2647. doi: 10.1073/pnas.261715899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim K, Nguyen T, Saville B, Safe S. Mol.Endocrinol. 2003;17:804–817. doi: 10.1210/me.2002-0406. [DOI] [PubMed] [Google Scholar]
- 50.Lee JE, Kim K, Sacchettini JC, Smith CV, Safe S. J.Biol.Chem. 2005;280:8819–8830. doi: 10.1074/jbc.M413184200. [DOI] [PubMed] [Google Scholar]
- 51.Stoner M, Wang F, Wormke M, Nguyen T, Samudio I, Vyhlidal C, Marme D, Finkenzeller G, Safe S. J.Biol.Chem. 2000;275:22769–22779. doi: 10.1074/jbc.M002188200. [DOI] [PubMed] [Google Scholar]
- 52.Tzukerman MT, Esty A, Santiso-Mere D, Danielian P, Parker MG, Stein RG, Pike JW, McDonnell DP. Mol.Endocrinol. 1994;8:21–30. doi: 10.1210/mend.8.1.8152428. [DOI] [PubMed] [Google Scholar]
- 53.Abdelrahim M, Samudio I, Smith R, Burghardt R, Safe S. J.Biol.Chem. 2002;277:28815–28822. doi: 10.1074/jbc.M203828200. [DOI] [PubMed] [Google Scholar]
- 54.Ren Y, Behre E, Ren Z, Zhang J, Wang Q, Fondell JD. Mol.Cell.Biol. 2000;20:5433–5446. doi: 10.1128/mcb.20.15.5433-5446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burakov D, Wong CW, Rachez C, Cheskis BJ, Freedman LP. J.Biol.Chem. 2000;275:20928–20934. doi: 10.1074/jbc.M002013200. [DOI] [PubMed] [Google Scholar]
- 56.Coulthard VH, Matsuda S, Heery DM. J.Biol.Chem. 2003;278:10942–10951. doi: 10.1074/jbc.M212950200. [DOI] [PubMed] [Google Scholar]
- 57.Rachez C, Gamble M, Chang CP, Atkins GB, Lazar MA, Freedman LP. Mol.Cell.Biol. 2000;20:2718–2726. doi: 10.1128/mcb.20.8.2718-2726.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Warnmark A, Almlof T, Leers J, Gustafsson JA, Treuter E. J.Biol.Chem. 2001;276:23397–23404. doi: 10.1074/jbc.M011651200. [DOI] [PubMed] [Google Scholar]
- 59.Acevedo ML, Kraus WL. Mol.Cell.Biol. 2003;23:335–348. doi: 10.1128/MCB.23.1.335-348.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burakov D, Crofts LA, Chang CPB, Freedman LP. J.Biol.Chem. 2002;277:14359–14362. doi: 10.1074/jbc.C200099200. [DOI] [PubMed] [Google Scholar]
- 61.Treuter E, Johansson L, Thomsen JS, Warnmark A, Leers J, Pelto-Huikko M, Sjoberg M, Wright AP, Spyrou G, Gustafsson JA. J.Biol.Chem. 1999;274:6667–6677. doi: 10.1074/jbc.274.10.6667. [DOI] [PubMed] [Google Scholar]
- 62.Barletta F, Freedman LP, Christakos S. Mol.Endocrinol. 2002;16:301–314. doi: 10.1210/mend.16.2.0764. [DOI] [PubMed] [Google Scholar]
- 63.Wang Q, Sharma D, Ren Y, Fondell JD. J.Biol.Chem. 2002;277:42852–42858. doi: 10.1074/jbc.M206061200. [DOI] [PubMed] [Google Scholar]
- 64.Yang W, Freedman LP. J.Biol.Chem. 1999;274:16838–16845. doi: 10.1074/jbc.274.24.16838. [DOI] [PubMed] [Google Scholar]
- 65.Taatjes DJ, Tjian R. Mol.Cell. 2004;14:675–683. doi: 10.1016/j.molcel.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 66.Wagner SA, Green MR. Science. 1993;266:395–399. doi: 10.1126/science.8211160. [DOI] [PubMed] [Google Scholar]
- 67.Sanchez HB, Yieh L, Osborne TF. J.Biol.Chem. 1995;270:1161–1169. doi: 10.1074/jbc.270.3.1161. [DOI] [PubMed] [Google Scholar]
- 68.Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. Cell. 1997;88:405–415. doi: 10.1016/s0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]
- 69.Ceska TA, Lamers M, Monaci P, Nicosia A, Cortese R, Suck D. EMBO J. 1993;12:1805–1810. doi: 10.2210/pdb1lfb/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brzozowski AM, Savage H, Verma CS, Turkenburg JP, Lawson DM, Svendsen A, Patkar S. Biochemistry. 2000;39:15071–15082. doi: 10.1021/bi0013905. [DOI] [PubMed] [Google Scholar]
- 71.Horton JR, Sawada K, Nishibori M, Zhang X, Cheng X. Structure. 2001;9:837–849. doi: 10.1016/s0969-2126(01)00643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klein FA, Atkinson RA, Potier N, Moras D, Cavarelli J. J.Biol.Chem. 2005;280:5682–5692. doi: 10.1074/jbc.M411697200. [DOI] [PubMed] [Google Scholar]
- 73.Bourdoncle A, Labesse G, Margueron R, Castet A, Cavailles V, Royer CA. J.Mol.Biol. 2005;347:921–934. doi: 10.1016/j.jmb.2005.01.048. [DOI] [PubMed] [Google Scholar]