

Abstract
Aggregation and fibrillization of the alpha-synuclein protein, which is the main component of Lewy bodies, may represent important processes in the pathogenesis of Parkinson’s disease (PD). Several in vivo and in vitro studies suggest that beta-synuclein may be a natural negative regulator of alpha-synuclein aggregation and fibrillization. The goal of the present study was to investigate the association of two polymorphisms (rs35035889 and rs1352303) in the beta-synuclein (SNCB) gene with PD. Our case-control study included a total of 370 case-unaffected sibling pairs and 168 case-unrelated control pairs (538 pairs total). The subjects were recruited from an ongoing study of the molecular epidemiology of PD in the Upper Midwest (USA). We employed a liberalization of the sibling transmission disequilibrium test to study the main effects of the gene variants for subjects overall and for strata defined by age at study, gender, ethnicity, clinical diagnostic certainty, dementia, and family history of PD (adjusted for age at study and gender as appropriate). The analyses were conducted for each SNCB variant separately, and also for two-locus haplotypes using score tests. Neither of the SNCB SNPs examined were associated with PD overall or in strata, and haplotype analyses were negative as well. However, one of the two SNPs (rs1352303) was associated with a delayed age at onset of PD in women. The results of this preliminary study suggest that the SNCB locus, though not a susceptibility gene for PD, might modify the age at onset of PD.
Keywords: Beta-synuclein gene, Polymorphism, Case-control study, Parkinson’s disease
Introduction
Aggregation and fibrillization of the alpha-synuclein protein, which is the main component of Lewy bodies, represent key events in the pathogenesis of Parkinson’s disease (PD) [22]. Alpha-synuclein (encoded by the SNCA gene mapped to chromosome 4q21) belongs to a family of homologous presynaptic proteins well conserved across species, which also includes beta-synuclein (encoded by the SNCB gene mapped to chromosome 5q35) and gamma-synuclein (encoded by the SNCG gene mapped to chromosome 10q23). Members of the synuclein protein family show different propensities to form soluble oligomers and fibrils, with alpha-synuclein being the most prone to fibrillate, followed by gamma-synuclein. Beta-synuclein doesn’t form fibrils [2].
Although no causal mutations have been found thus far in the SNCB gene in individuals or families with Parkinson’s disease [11], a number of in vivo and in vitro studies suggest that beta-synuclein may be a natural negative regulator of alpha-synuclein aggregation. Mice doubly transgenic for human alpha- and beta-synuclein showed decreased accumulation of alpha-synuclein-immunoreactive neuronal inclusions and less severe neurodegenerative alterations and phenotype compared to mice singly transgenic for human alpha-synuclein [10]. Two independent studies demonstrated that beta-synuclein can inhibit the process of alpha-synuclein aggregation and fibril formation in vitro [21, 17].
Materials and Methods
These findings suggest that SNCB is a biologically plausible susceptibility gene for PD. In particular, the discovery of protective alleles among at-risk individuals would guide the development of therapeutics [16]. Thus, we performed an association study of two SNPs in the SNCB gene in case-unaffected sibling and case-unrelated control pairs. The subjects were recruited as part of an ongoing study of the molecular epidemiology of PD in the Upper Midwest (USA). Case subjects were PD patients sequentially referred to the Department of Neurology at Mayo Clinic in Rochester, MN, after June 1996, and were residents of Minnesota or of one of the surroundings four states (Wisconsin, Iowa, South Dakota, or North Dakota). Diagnosis of PD was made by a movement disorder specialist using previously reported criteria [4]. Controls were free of PD at the time of the study and included unaffected siblings of probands or unrelated controls when there were no siblings available. The unrelated controls included spouses of cases and unrelated population controls. Potential control subjects were screened for parkinsonism via a validated telephone instrument [18]. Only potential controls who screened negative for PD, or who were confirmed not to have PD via clinical assessment (despite having screened positive by telephone interview), were included in the study.
The Mayo Clinic Investigational Review Board approved all study methods. All DNA samples were obtained after a written informed consent. Venous blood specimens were collected directly from the subjects examined or via mail-in blood kits (for controls who screened negative). Genomic DNA was extracted from leukocytes via the Puregene procedure (Gentra Systems, Minneapolis, MN). Two SNCB single nucleotide polymorphisms (SNPs), rs35035889 and rs1352303, were amplified from genomic DNA template by PCR using ABI Taqman® PCR Mastermix. All products were analyzed on an ABI7900 automated sequencer with SDS 2.02 allelic discrimination software (Applied Biosystems). Additional details regarding the genotyping assays are available upon request. Allele frequencies were calculated by the allele counting method. Goodness of fit of the genotype frequencies to Hardy–Weinberg expected proportions in unrelated control subjects was examined.
To study the genetic association of the SNCB gene variants with PD, we first matched cases to a single unaffected sibling of the same sex (when possible) and then of closest age at study. For cases without an available sibling, we matched an unrelated control for sex and age (±2 years). A liberalization of the sibling transmission disequilibrium test was employed to compare cases and control subjects (case-unaffected sibling and case-unrelated control pairs) for the association of genotypes defined by single SNPs with PD [19]. We performed analyses assuming autosomal dominant, autosomal recessive, log additive, and unrestricted models of inheritance. All analyses were adjusted for age and sex as appropriate. These genotype analyses were performed for subjects overall and stratified by family history of PD (defined as at least one first-degree relative with PD), age at study, and gender. We also performed analyses restricted to non-demented PD cases or to clinically definite PD cases (i.e., absence of mild or late dementia or dysautonomia) and their matched controls, or to subjects of European origin. We calculated odds ratios (ORs), 95% confidence intervals (CIs), and p values (two-tailed tests, α = 0.05).
The association of inferred two-locus haplotypes with PD was studied using score tests, as previously described [20]. The pair-wise linkage disequilibrium (LD) between markers was estimated. Furthermore, we constructed a LD map under the Malecot model using LDMAP [12] for the region surrounding the SNCB locus with SNP data from 231 unaffected siblings of PD cases that were included both in this study and in a published whole-genome association study of PD [14].
The correlation between SNCB SNP genotypes and age at onset was analyzed by Cox regression analysis. Survival free of PD was represented using Kaplan-Meier plots. We defined age at onset as the age when a subject first noticed or was first observed to have a cardinal motor symptom or sign of parkinsonism (i.e. tremor, bradykinesia, rigidity, postural instability) according to self-report, by proxy report, or by medical records review (when available).
Results
The study included 538 case-control pairs (370 case-unaffected sibling pairs and 168 case-unrelated control pairs). The demographic characteristics of the cases and controls are reported in Table 1. Genotype frequencies were in Hardy Weinberg Equilibrium in unrelated control subjects (p > 0.05). The two SNCB SNPs were in LD with each other within our sample of unrelated controls (data not shown). The LD map derived from unaffected sibling controls (Figure 1) suggested that LD is not very extensive around the SNCB gene. Flat areas on the map indicate regions of stability, whereas sloping areas indicate regions of instability. Although the two SNCB SNPs investigated by this study bound a small flat region, the areas on either side had some slope. This suggests that we did cover the region of the SNCB gene bounded by the two SNPs but possibly not the gene as a whole (particularly at the 3′ end).
Table 1.
Demographic characteristics of Parkinson’s disease cases and controls (unaffected siblings or unrelated controls)
| PD Cases – Unaffected Sibling Pairs
|
PD Cases-Unrelated Control Pairs
|
|||
|---|---|---|---|---|
| General characteristics | PD Cases n (%) | Sibling Controls n (%) | PD Cases n (%) | Unrelated Controls n (%) |
| Total sample | 370 (100) | 370 (100) | 168 (100) | 168 (100) |
| Men | 236 (63.8) | 176 (47.6) | 67 (39.9) | 67 (39.9) |
| Women | 134 (36.2) | 194 (52.4) | 101 (60.1) | 101 (60.1) |
| Age at onset of PD, median (range) | 61 (30–87) | -- | 66 (36–84) | -- |
| Age at exam (cases) or at blood draw (controls), median (range) | 67 (38–91) | 66 (41–89) | 72 (39–90) | 72 (39–91) |
| Region of origin of parentsa | ||||
| Both parents of European origin | 317 (85.7) | 304 (82.2) | 131 (78.0) | 133 (79.2) |
| Both parents Northern Europeanb | 89 (28.1) | 84 (27.6) | 38 (29.0) | 45 (33.8) |
| Both parents Central Europeanc | 121 (38.2) | 121 (39.8) | 51 (38.9) | 39 (29.3) |
| Both parents Southern Europeand | 3 (0.9) | 3 (1.0) | 1 (0.8) | 2 (1.5) |
| Both parents European, mixed region | 104 (32.8) | 96 (31.6) | 41 (31.3) | 47 (35.3) |
| Only one parent of European origine | 31 (8.4) | 35 (9.5) | 23 (13.7) | 18 (10.7) |
| One parent declared “American”f | 15 (4.1) | 14 (3.8) | 9 (5.4) | 13 (7.7) |
| Both parents declared “American”f | 17 (4.6) | 19 (5.1) | 8 (4.8) | 11 (6.5) |
| Both parents Asian | 2 (0.5) | 3 (0.8) | 2 (1.2) | 1 (0.6) |
| Both parents Mexican | 1 (0.3) | 1 (0.3) | 0 (0.0) | 0 (0.0) |
| Both parents African | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.6) |
| Unknown | 2 (0.5) | 7 (1.9) | 3 (1.8) | 2 (1.2) |
Region of origin of parents was self-reported by subjects. Note that Parkinson’s disease cases and their siblings were not always in agreement [5].
“ Northern European” includes Scandinavian, Swedish, Norwegian, Finnish, Danish, Irish, or British origin.
“Central European” includes French, Belgian, Dutch, Swiss, Luxemburgian, German, Austrian, Hungarian, Polish, Czechoslovakian, or Russian origins.
“Southern European” includes Italian, Spanish, Portuguese, Greek, or Yugoslavian origins.
Includes subjects for whom origin of one parent is unknown.
These subjects were all Caucasians and not Native Americans.
Fig. 1.
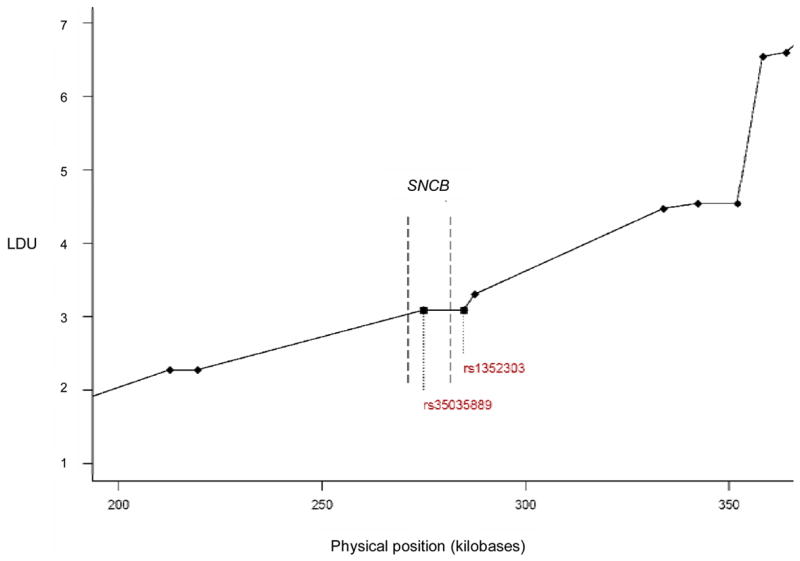
Close-up of Linkage Disequilibrium Unit (LDU) map for the SNCB region, constructed using a sample of 231 unaffected sibling controls included in this study and in a previous whole-genome association study [14]. The two SNCB SNPs genotyped as part of this study are indicated by squares and labeled by rs numbers on the plot. Diamonds indicate SNPs genotyped as part of a whole-genome association study, and the dashed lines indicate the bounds of the SNCB gene. The 3′ region of the gene is to the left and the 5′ region is to the right.
The results of our liberalized sTDT analyses for each of the two SNPs are summarized in Tables 2–3. Neither SNP was significantly associated with PD overall or in strata. The frequency of the inferred two-locus haplotypes was not significantly different in cases and controls (global score statistics, p > 0.05). Finally, in analyses restricted to PD cases (case-case comparisons), one of the two SNPs (rs1352303) was associated with a delayed age at onset in women only, when assuming a dominant model (HR 0.59, 95% CI 0.42 to 0.82, p = 0.0025) or an unrestricted model of inheritance (for the heterozygous vs the homozygous wild genotype, HR 0.56, 95% CI = 0.39 to 0.8; for the homozygous variant vs the homozygous wild genotype, HR 0.65, 95% CI = 0.43 to 0.96; p = 0.0076). The median ages at onset were 59.13 years for women with zero copies of the rs1352303 minor allele versus 65.08 years for women with one or two copies of the rs1352303 minor allele (dominant model). By contrast, median ages at onset were not significantly modified by the rs1352303 SNP variant in men (Figure 2).
Table 2.
Results of Case-control Analyses for SNCB SNP rs1352303.
| Genotype n (%) | Dominant Modela | |||||
|---|---|---|---|---|---|---|
| Sample or Stratumb | n | 1/1 | 1/2 | 2/2 | OR (95% CI) | p-value |
| Cases, all | 538 | 151 (28.1) | 258 (48.0) | 129 (24.0) | 0.98 (0.70–1.36) | 0.89 |
| Controls, all | 538 | 147 (27.3) | 262 (48.7) | 129 (24.0) | ||
| Cases, women | 197 | 48 (24.4) | 99 (50.3) | 50 (25.4) | 1.09 (0.66–1.81) | 0.73 |
| Controls, women | 197 | 51 (24.4) | 93 (47.2) | 53 (26.9) | ||
| Cases, men | 205 | 61 (29.8) | 96 (46.8) | 48 (23.4) | 1.00 (0.59–1.69) | 0.99 |
| Controls, men | 205 | 60 (29.3) | 103 (50.2) | 42 (20.5) | ||
| Cases, age ≤ 68.19 c | 269 | 87 (32.3) | 119 (44.2) | 63 (23.4) | 0.92 (0.57–1.48) | 0.72 |
| Controls, age ≤ 68.19 c | 269 | 84 (31.2) | 127 (47.2) | 58 (21.6) | ||
| Cases, age > 68.19 c | 269 | 64 (23.8) | 139 (51.7) | 66 (24.5) | 1.02 (0.64–1.61) | 0.94 |
| Controls, age > 68.19 c | 269 | 63 (23.4) | 135 (50.2) | 71 (26.4) | ||
| Cases, family historyd | 100 | 28 (28.0) | 50 (50.0) | 22 (22.0) | 1.01 (0.46–2.25) | 0.97 |
| Controls, family historyd | 100 | 28 (28.0) | 47 (47.0) | 25 (25.0) | ||
| Cases, no family historyd | 428 | 117 (27.3) | 205 (47.9) | 106 (24.8) | 0.99 (0.69–1.44) | 0.97 |
| Controls, no family historyd | 428 | 115 (26.9) | 212 (49.5) | 101 (23.6) | ||
Adjusted for gender and age at study, as appropriate. Results for additional models (trend, autosomal recessive, and unrestricted) were all non-significant (data not shown).
Additional analyses were stratified by age at study of PD cases (youngest quartile versus other three quartiles combined). Other analyses were restricted to non-demented cases or to cases with clinically definite PD (excluding clinically probable cases) or to subjects of European origin only. As those results were all non-significant, the data are not shown.
Based upon PD cases; median age at study = 68.19 years
Based upon PD cases; family history was defined as at least one first-degree relative affected.
Table 3.
Results of Case-control Analyses for SNCB SNP rs35035889
| Genotype n (%)
|
Dominant Modela |
|||||
|---|---|---|---|---|---|---|
| Sample or Stratumb | n | 1/1 | 1/2 | 2/2 | OR (95% CI) | p-value |
| Cases, all | 538 | 353 (65.6) | 168 (31.2) | 17 (3.2) | 0.99 (0.72–1.36) | 0.93 |
| Controls, all | 538 | 360 (66.9) | 161 (29.9) | 17 (3.2) | ||
| Cases, women | 197 | 129 (65.5) | 66 (33.5) | 2 (1.0) | 1.14 (0.71–1.82) | 0.60 |
| Controls, women | 197 | 133 (67.5) | 59 (30.0) | 5 (2.5) | ||
| Cases, men | 205 | 134 (65.4) | 61 (29.8) | 10 (4.9) | 1.02 (0.61–1.69) | 0.94 |
| Controls, men | 205 | 136 (66.3) | 62 (30.2) | 7 (3.4) | ||
| Cases, age ≤ 68.19 c | 269 | 179 (66.5) | 81 (30.1) | 9 (3.4) | 0.93 (0.58–1.50) | 0.77 |
| Controls, age ≤ 68.19 c | 269 | 180 (66.9) | 82 (30.5) | 7 (2.6) | ||
| Cases, age > 68.19 c | 269 | 174 (64.7) | 87 (32.3) | 8 (3.0) | 1.02 (0.66–1.59) | 0.91 |
| Controls, age > 68.19 c | 269 | 180 (66.9) | 79 (29.4) | 10 (3.7) | ||
| Cases, family historyd | 100 | 65 (65.0) | 31 (31.0) | 4 (4.0) | 1.45 (0.73–2.86) | 0.28 |
| Controls, family historyd | 100 | 74 (74.0) | 24 (24.0) | 2 (2.0) | ||
| Cases, no family historyd | 428 | 281 (65.7) | 134 (31.3) | 13 (3.0) | 0.86 (0.59–1.24) | 0.42 |
| Controls, no family historyd | 428 | 278 (65.0) | 135 (31.5) | 15 (3.5) | ||
Adjusted for gender and age at study, as appropriate. Results for additional models (trend, autosomal recessive, and unrestricted) were all non-significant (data not shown).
Additional analyses were stratified by age at study of PD cases (youngest quartile versus other three tertiles combined). Other analyses were restricted to non-demented cases or to cases with clinically definite PD (excluding clinically probable cases) or to subjects of European origin only. As those results were all non-significant, the data are not shown.
Based upon PD cases; median age at study = 68.19 years
Based upon PD cases; family history was defined as at least one first-degree relative affected.
Fig. 2.
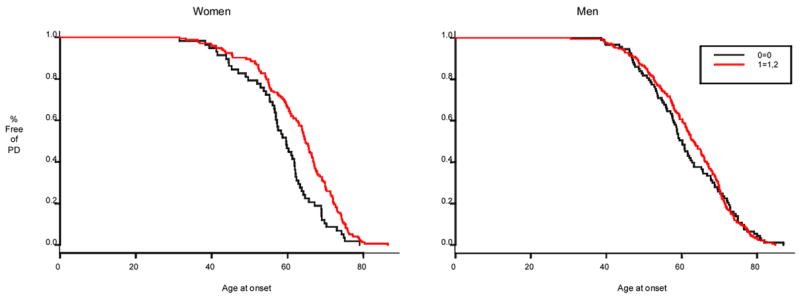
Kaplan-Meier plot showing percent surviving free of PD by age, for PD patients with zero (black line) and one or two (red line) copies of the rs1352303 minor allele. P = 0.003 in females and p = 0.29 in males.
Discussion
Our results suggest that, despite the strong biological plausibility, the SNCB gene is not a susceptibility locus for PD. Our negative findings are also consistent with findings from two recent genome-wide association studies. Those studies included respectively one SNCB SNP (ss46558233) [14] or two SNCB SNPs (rs744307 and rs4242202) [9]. The SNPs selected for those studies were preferentially haplotype-tagging. None of those SNPs were significantly associated with PD (p-values > 0.30).
However, our findings for the SNCB gene and susceptibility to PD could be falsely negative because our selection of the two SNCB SNPs preceded the availability of haplotype maps. The SNCB gene is a small gene that contains 5 coding exons and at least one 5′ untranslated exon. The rs35035889 polymorphism is located at the 3′ region of the gene and is intronic; the rs1352303 maps upstream at the 5′ region of the gene. We chose the two SNPs because they were positioned at 3′ and 5′ ends of the genes. Unfortunately, based on present knowledge, the area of LD mapped by the two SNPs did not span the entire length of the SNCB gene, particularly at its 3′ end. Using data from another study including 231 subjects [14], we constructed an LD map for the SNCB gene locus. This map will guide SNP selection for future studies of the gene. The newly launched website PDGene (http://www.pdgene.org/), which systematically reviews genetic association studies of PD, confirms that as of March 2007 information regarding the SNCB gene and its association with PD is lacking.
Variability in the length of a dinucleotide repeat sequence (REP1) within the promoter of the closely related SNCA gene has been reported to confer susceptibility to sporadic PD [15], presumably via a mechanism of gene overexpression and protein aggregation [6,7]. Because the beta-synuclein protein negatively regulates the alpha-synuclein protein, it is possible that interactions between SNCA REP 1 and SNCB genotypes might be related to PD susceptibility. However, the effect size of SNCA REP1 has been shown to be small. For example, using a scored test of genotypes according to allele length, the observed OR was 1.19 [15]. Detecting an OR of that size with 80% power would require 924 case-unrelated control pairs or 1,849 case-sib pairs and a substantially larger sample size would be required to detect interactions (beyond the scope of this preliminary study).
Nevertheless, our study has some strengths. The comparison subjects were primarily unaffected siblings to limit the possible confounding effect of population stratification. We adjusted our analyses for possible confounders (gender and age at study). The sample size employed and the minor allele frequency for the SNPs selected provided sufficient statistical power for the main effect analyses. If all 538 pairs included in our study were cases matched to unaffected siblings, we would have had 80% power to detect an OR of 0.6 or smaller (or 1.7 or greater) for the rs13523903 SNP and an OR of 0.6 or smaller (or 1.6 or larger) for the rs35035889 SNP (dominant model). Because we included a mixture of case-unaffected siblings and case-unrelated control pairs, these are conservative estimates of the detectable ORs (smallest deviation from 1.0). We also considered multiple genetic models and stratified our sample for multiple variables to explore possible effect modifications.
Furthermore, age at onset analyses revealed an association between one of the two SNPs and delayed age at onset restricted to women with PD. This may be a spurious finding; alternatively it may be real and reflect the unequal role of genetic and environmental factors in the pathogenesis of PD in men and women [4, 13]. Moreover, since the associated SNP maps upstream (5′) of the gene, a differential regulation of SNCB transcription upon interaction with estrogens might account for the association of the SNP variant with age at onset in women only. Indeed estrogen response elements are located at the 5′ promoter region of the genes [1]. This finding awaits replication and it remains possible that SNCB polymorphism might also modify outcomes in PD (to be studied).
Acknowledgments
The authors acknowledge funding support from the NIH grant R01 ES10751, R01 NS33978, and P50 NS40256.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Behl C. Oestrogen as a neuroprotective hormone. Nat Rev Neurosci. 2002;3:433–42. doi: 10.1038/nrn846. [DOI] [PubMed] [Google Scholar]
- 2.Biere AL, Wood SJ, Wypych J, Steavenson S, Jiang Y, Anafi D, Jacobsen FW, Jarosinski MA, Wu GM, Louis JC, Martin F, Narhi LO, Citron M. Parkinson’s disease-associated alpha-synuclein is more fibrillogenic than beta- and gamma-synuclein and cannot cross-seed its homologs. J Biol Chem. 2000;275:34574–9. doi: 10.1074/jbc.M005514200. [DOI] [PubMed] [Google Scholar]
- 3.Bonsch D, Lederer T, Reulbach U, Hothorn T, Kornhuber J, Bleich S. Joint analysis of the NACP-REP1 marker within the alpha synuclein gene concludes association with alcohol dependence. Hum Mol Genet. 2005;14:967–71. doi: 10.1093/hmg/ddi090. [DOI] [PubMed] [Google Scholar]
- 4.Bower JH, Maraganore DM, McDonnell SK, Rocca WA. Incidence and distribution of parkinsonism in Olmsted County, Minnesota, 1976–1990. Neurology. 1999;52:1214–20. doi: 10.1212/wnl.52.6.1214. [DOI] [PubMed] [Google Scholar]
- 5.Burnett MS, Strain KJ, Lesnick TG, de Andrade M, Rocca WA, Maraganore DM. Reliability of self-reported ancestry among siblings: implications for genetic association studies. Am J Epidemiol. 2006;163:486–92. doi: 10.1093/aje/kwj057. [DOI] [PubMed] [Google Scholar]
- 6.Chiba-Falek O, Nussbaum RL. Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum Mol Genet. 2001;10:3101–3109. doi: 10.1093/hmg/10.26.3101. [DOI] [PubMed] [Google Scholar]
- 7.Chiba-Falek O, Touchman JW, Nussbaum RL. Functional analysis of intra-allelic variation at NACP-Rep1 in the alpha-synuclein gene. Hum Genet. 2003;113:426–31. doi: 10.1007/s00439-003-1002-9. [DOI] [PubMed] [Google Scholar]
- 8.Farrer M, Maraganore DM, Lockhart P, Singleton A, Lesnick TG, de Andrade M, West A, de Silva R, Hardy J, Hernandez D. Alpha-Synuclein gene haplotypes are associated with Parkinson’s disease. Hum Mol Genet. 2001;10:1847–51. doi: 10.1093/hmg/10.17.1847. [DOI] [PubMed] [Google Scholar]
- 9.Fung HC, Scholz S, Matarin M, Simon-Sanchez J, Hernandez D, Britton A, Gibbs JR, Langefeld C, Stiegert ML, Schymick J, Okun MS, Mandel RJ, Fernandez HH, Foote KD, Rodriguez RL, Peckham E, De Vrieze FW, Gwinn-Hardy K, Hardy JA, Singleton A. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006;5:911–6. doi: 10.1016/S1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- 10.Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. Beta-synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron. 2001;32:213–23. doi: 10.1016/s0896-6273(01)00462-7. [DOI] [PubMed] [Google Scholar]
- 11.Lincoln S, Crook R, Chartier-Harlin MC, Gwinn-Hardy K, Baker M, Mouroux V, Richard F, Becquet E, Amouyel P, Destee A, Hardy J, Farrer M. No pathogenic mutations in the beta-synuclein gene in Parkinson’s disease. Neurosci Lett. 1999;269:107–9. doi: 10.1016/s0304-3940(99)00420-6. [DOI] [PubMed] [Google Scholar]
- 12.Maniatis N, Collins A, Xu CF, McCarthy LC, Hewett DR, Tappe W, Ennis S, Ke X, Morton NE. The first linkage disequilibrium (LD) maps: Delineation of hot and cold blocks by diplotype analysis. PNAS. 2002;99:2228–2233. doi: 10.1073/pnas.042680999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maraganore DM, de Andrade M, Lesnick TG, Farrer MJ, Bower JH, Hardy JA, Rocca WA. Complex interactions in Parkinson’s disease: a two-phased approach. Mov Disord. 2003;18:631–636. doi: 10.1002/mds.10431. [DOI] [PubMed] [Google Scholar]
- 14.Maraganore DM, de Andrade M, Lesnick TG, Strain KJ, Farrer MJ, Rocca WA, Pant PV, Frazer KA, Cox DR, Ballinger DG. High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet. 2005;77:685–93. doi: 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Kruger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan EK, Van Broeckhoven C. Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium, Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296:661–70. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- 16.Nadeau JH, Topol EJ. The genetics of Health. Nature Genet. 2006;38:1095– 1098. doi: 10.1038/ng1006-1095. [DOI] [PubMed] [Google Scholar]
- 17.Park JY, Lansbury PT., Jr Beta-synuclein inhibits formation of alpha-synuclein protofibrils: a possible therapeutic strategy against Parkinson’s disease. Biochemistry. 2003;42:3696–700. doi: 10.1021/bi020604a. [DOI] [PubMed] [Google Scholar]
- 18.Rocca WA, Maraganore DM, McDonnell SK, Schaid DJ. Validation of a telephone questionnaire for Parkinson’s disease. J Clin Epidemiol. 1998;51:517–23. doi: 10.1016/s0895-4356(98)00017-1. [DOI] [PubMed] [Google Scholar]
- 19.Schaid DJ, Rowland C. Use of parents, sibs, and unrelated controls for detection of associations between genetic markers and disease. Am J Hum Genet. 1998;63:1492–1506. doi: 10.1086/302094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70:425–34. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, Goedert M, Fink AL AL. Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem. 2002;277:11970–8. doi: 10.1074/jbc.M109541200. [DOI] [PubMed] [Google Scholar]
- 22.Volles MJ, Lansbury PT., Jr Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson’s disease. Biochemistry. 2003;42:7871–8. doi: 10.1021/bi030086j. [DOI] [PubMed] [Google Scholar]