

Abstract
Adiponectin is an adipokine with potent anti-inflammatory properties. However, the mechanisms by which adiponectin suppresses macrophage function are not well understood. Treatment of RAW264.7 macrophages with adiponectin for 18 h decreased lipopolysaccharide (LPS)-stimulated tumor necrosis factor-α (TNF-α) production. Here we demonstrate that globular adiponectin (gAcrp) initially increased TNF-α expression in RAW264.7 macrophages; this TNF-α then contributed to increased expression of interleukin-10, which in turn was required for the development of tolerance to subsequent LPS exposure. gAcrp-mediated increases in TNF-α mRNA accumulation were associated with increased TNF-α promoter activity. gAcrp increased the DNA binding activity of both Egr-1 and NFκB; mutation of either the Egr-1 or NFκB binding sites in the TNF-α promoter decreased gAcrp-stimulated promoter activity. Further, co-transfection with either dominant negative Egr-1 or the IκB super-repressor prevented gAcrp-stimulated TNF-α promoter activity. gAcrp also increased Egr-1 promoter activity, mRNA accumulation, and DNA binding activity. Inhibition of ERK1/2 with U0126 potently suppressed gAcrp-stimulated Egr-1 promoter activity, as well as TNF-α promoter activity. In summary, these data demonstrate that adiponectin initially increases TNF-α production by macrophages via ERK1/2→Egr-1 and NFκB-dependent mechanisms; these increases in TNF-α in turn lead to increased expression of interleukin-10 and an eventual dampening of LPS-mediated cytokine production in macrophages.
Adiponectin (a 30-kDa peptide also known as adipocyte complement-related protein; Acrp30)2 is an adipokine secreted by adipose tissue (1, 2). Adiponectin concentration is decreased in the blood during obesity, insulin resistance, and type 2 diabetes, and insulin sensitivity can be restored in obese and diabetic animals by treatment with adiponectin (3, 4). Treatment of mice with adiponectin increases fatty acid oxidation in liver and glucose utilization in muscle (4). Adiponectin acts on target tissues via the activation of adiponectin receptors 1 and 2 (AdipoR1 and AdipoR2) (5). The liver primarily expresses AdipoR2, whereas skeletal muscle expresses predominantly AdipoR1 (5).
In addition to regulating glucose and lipid metabolism, adiponectin influences the activity of the innate immune system. Both AdipoR1 and AdipoR2 are expressed in monocytes and macrophages (6). Accumulating evidence indicates that AdipoR1 is critical to the anti-inflammatory effects of adiponectin (7). Adiponectin is hypothesized to dampen the early phases of macrophage inflammatory responses (8), acting to inhibit the growth of myelomonocytic progenitor cells (8) and decrease the ability of mature macrophages to respond to activation (8, 9). Adiponectin suppresses phagocytic activity, as well as lipopolysaccharide (LPS)-stimulated cytokine production in macrophages (8-10). This anti-inflammatory response to adiponectin has been implicated as a potential mechanism for the anti-atherogenic properties of adiponectin (11), as well as the potential therapeutic effects of adiponectin in alcoholic and non-alcoholic liver injury (10, 12, 13).
In response to either the globular or full-length forms of adiponectin, macrophages exhibit decreased sensitivity to LPS-stimulated signaling. In particular, LPS-stimulated NFκB and ERK1/2 activation have been found to be sensitive to the inhibitory effects of adiponectin (7, 9, 10). Suppression of these toll-like receptor 4-mediated signaling pathways is thought to contribute to decreased activation of key transcription factors (nuclear factor κB (NFκB) and early growth response protein 1 (Egr-1)) that regulate cytokine expression in macrophages (7, 10). The mechanisms for the profound inhibitory effects of adiponectin on macrophage function are not well understood. The phenotypic characteristics of adiponectin-treated macrophages are similar to that observed in response to repeated/ prolonged exposure to LPS/endotoxin, or what is often called “endotoxin tolerance” (14). One recent report demonstrates that short-term treatment of macrophages with adiponectin first increases the expression of inflammatory cytokines, such as TNF-α and IL-6 (15); continued exposure to adiponectin then promotes the development of tolerance to subsequent pro-inflammatory signals, such as the toll-like receptor 4 ligand LPS or the toll-like receptor 3 ligand poly (I-C) (15). Wolf et al. have demonstrated that adiponectin induces the expression of anti-inflammatory mediators, such as IL-10 (16); however, it is not yet known whether IL-10 is essential for the inhibitory effects of adiponectin on LPS-stimulated signaling in macrophages.
To better understand the mechanisms by which adiponectin leads to the development of tolerance to pro-inflammatory signals, here we have investigated in detail the short-term responses of RAW 264.7 macrophages to adiponectin. In addition to the activation of NFκB by adiponectin (15), we found that adiponectin also activates the transcription factor Egr-1 via an ERK1/2-dependent pathway. In combination, NFκB and Egr-1, two potent transcription factors that regulate pro-inflammatory gene expression in macrophages, then contributed to increased TNF-α expression. This rapid increase in TNF-α expression then led to increased IL-10 expression; IL-10 in turn mediated the development of tolerance to subsequent stimulation of RAW 264.7 macrophages by LPS after a longer period of exposure to adiponectin.
EXPERIMENTAL PROCEDURES
Materials
LPS from Escherichia coli serotype 026:B6 (tissue culture-tested, L-2654) was purchased from Sigma; all experiments were carried out with a single lot of LPS (Lot 064K4077). All cell culture reagents were from Invitrogen. Antibodies were from the following sources: phospho-ERK1/2, Egr-1, p50, and p65 (Santa Cruz Biotechnology, Santa Cruz, CA) and total ERK1/2 (Upstate Biotechnology, Lake Placid, NY). Anti-rabbit and anti-mouse IgG peroxidase were purchased from Chemicon (Indianapolis, IN). Recombinant human globular adiponectin (gAcrp) expressed in E. coli was purchased from Peprotech, Inc. (Rocky Hill, NJ). Endotoxin contamination of all reagents used to treat RAW264.7 macrophages was routinely monitored in the laboratory using a kinetic chromogenic test based on the Limulus amebocyte lysate assay (Kinetic-QCL; BioWhittaker, Walkersville, MD).
Endotoxin-free plasmid preparation kits were from Qiagen (Valencia, CA). Luciferase reporter constructs have been previously described: pTNF-α-LUC (containing −615 to +1 nucleotide TNF-α promoter and mutated constructs in a luciferase reporter) (17) and the Egr-1 LUC (containing 1.2 kb of the Egr-1 promoter) (18) were from N. Mackman, University of California San Diego. The plasmid containing the dominant negative Egr-1 (pCMVETTL) was a gift from D. L. Wong and V. Sukhatme (19), and the IκB super-repressor (IκB-AA (pRc/CMV IκB S32A/S36A) was a gift from J. Didonato (20). Silencer-validated siRNA for TNFR SF1A (TNF-RI) or scrambled siRNA were from Ambion (Austin, TX). Enzyme-linked immunosorbent assay kits for TNF-α and IL-10 were from BIO-SOURCE (Camarillo, CA); neutralizing antibodies to TNF-α and IL-10 were from R&D Systems (Minneapolis, MN) and BD Biosciences, respectively.
Culture of RAW 264.7 Macrophages and Luciferase Assays
The murine RAW 264.7 macrophage-like cell line was routinely cultured in Dulbecco's modified Eagle medium with 10% fetal bovine serum and penicillin-streptomycin at 37°C and 5% CO2 (21). To test the role of secreted TNF-α or IL-10 in mediating the effects of gAcrp, in some experiments cells were co-cultured with increasing concentrations of neutralizing antibodies to TNF-α or IL-10 or the appropriate normal IgG controls during gAcrp treatment.
For luciferase reporter assays, RAW 264.7 macrophages were grown in 6-well plates to 60–70% confluency and then transiently transfected with control and expression vectors using Superfect transfection reagent (for DNA-only transfections) or TransMessenger transfection reagent (for siRNA transfections) (Qiagen) according to the manufacturer's instructions. Cells were co-transfected with pTK-RL (Promega, Madison, WI), an expression vector for Renilla luciferase under the control of the thymidine kinase promoter, as a control for transfection efficiency. Transfected cells were subcultured and seeded at 10.2 × 104/cm2 in 96-well plates. After 24 h (for DNA-only transfections) or 4 h (for siRNA transfections), medium was removed and cells stimulated or not with gAcrp or LPS in Dulbecco's modified Eagle's medium/fetal bovine serum. Cells were then extracted in lysis buffer and luciferase activities measured using the Dual Luciferase assay system (Promega). In some experiments, RAW264.7 macrophages were preincubated with 50 [H9262]g/ml polymyxin B.
Western Blot Analysis of ERK1/2 Phosphorylation
RAW264.7 macrophages were stimulated with or without 1 μg/ml gAcrp for 0–60 min. Cells were then moved to ice, washed with 2 ml of ice-cold phosphate-buffered saline buffer containing 1 mm sodium orthovanadate, and lysed in Laemmli sample buffer containing 1 mm sodium orthovanadate. Samples were separated by SDS-polyacrylamide gel electrophoresis and probed by Western blot for phospho-ERK1/2 and total ERK1/2.
Isolation of Nuclear Extracts and Electrophoretic Mobility Shift Assays
RAW264.7 macrophages were treated with or without 1 μg/ml gAcrp for 0–4 h. In some experiments, cells were pretreated with 10 μg/ml polymixin B for 60 min. Nuclei were isolated and nuclear proteins extracted using the Nuclei EZ Prep kit from Sigma. Nuclear extracts (5 μg of protein) were then used to assess DNA binding activity by electrophoretic mobility shift assay using an oligonucleotide probe for the sequence for the Egr-1 binding site in the TNF-α promoter (Integrated DNA Technologies, Inc., Coralville, IA) or for the consensus sequence for the NFκB binding site (Santa Cruz Bio-technology) as previously described (22).
Real-time PCR
Total RNA was isolated from RAW264.7 macrophages using the RNeasy Micro kit (Qiagen), with on-column DNA digestion using the RNase-free DNase set (Qiagen) according to the manufacturer's instructions. Two hundred to three hundred nanograms of total RNA were reverse transcribed using the RETROscript kit (Ambion, Austin, TX) with random decamers as primers. Real-time polymerase chain reaction (PCR) amplification was performed in an MX3000P apparatus (Stratagene, La Jolla, CA), using the Brilliant SYBR Green QPCR Master Mix (Applied Biosystems, Foster City, CA). The relative amount of target mRNA was determined using the comparative threshold (Ct) method by normalizing target mRNA Ct values to those for β-actin (ΔCt). The primer sequences are as follows: TNF-α, F 5′-CCC TCA CAC TCA GAT CAT CTT CT-3′, R5′-GCT ACG ACG TGG GCT ACA G-3′; IL-10, F 5′-CCA AGC CTT ATC GGA AAT GA-3′, R 5′TTT TCA CAG GGG AGA AAT CG-3′; Egr-1, F 5′-AGC GAA CAA CCC TAT GAG CAC-3′, R 5′-TCG TTT GGC TGG GAT AAC TCG-3′; β-actin, F 5′-CTT TGC AGC TCC TTC GTT GC-3′, R 5′-ACG ATG GAG GGG AAT ACA GC-3′. All primers used for real-time PCR analysis were synthesized by Integrated DNA Technologies, Inc. Statistical analyses of real-time PCR data were performed using ΔCt values.
Statistical Analysis
Values reported are means + S.E. Data were analyzed by the general linear models procedure followed by least square means analysis of differences between groups (SAS, Carey, IN).
RESULTS
Adiponectin has potent anti-inflammatory effects on macrophages. We have previously reported that exposure of primary cultures of rat Kupffer cells to globular or full-length adiponectin for 18 h suppresses subsequent LPS-stimulated TNF-α production (10). Here we report a similar effect of an 18-h treatment with adiponectin on LPS-stimulated TNF-α protein secretion in RAW 264.7 macrophages (Fig. 1A). Treatment of RAW264.7 macrophages with increasing concentrations of gAcrp or full-length adiponectin dose dependently decreased LPS-stimulated TNF-α production (Fig. 1A). TNF-α concentration in the medium of cells not treated with LPS was undetectable (data not shown). RAW 264.7 macrophages were more sensitive to gAcrp compared with full-length adiponectin (Fig. 1A). Therefore, all subsequent experiments in this report were carried out with gAcrp at a concentration of 1 μg/ml. Decreased LPS-stimulated TNF-α production after overnight treatment with gAcrp was associated with decreased accumulation of TNF-α mRNA accumulation in response to LPS treatment (Fig. 1B).
FIGURE 1. Role of IL-10 in mediating the suppression of LPS-stimulated TNF-α expression by overnight exposure to adiponectin in RAW 264.7 macrophages.
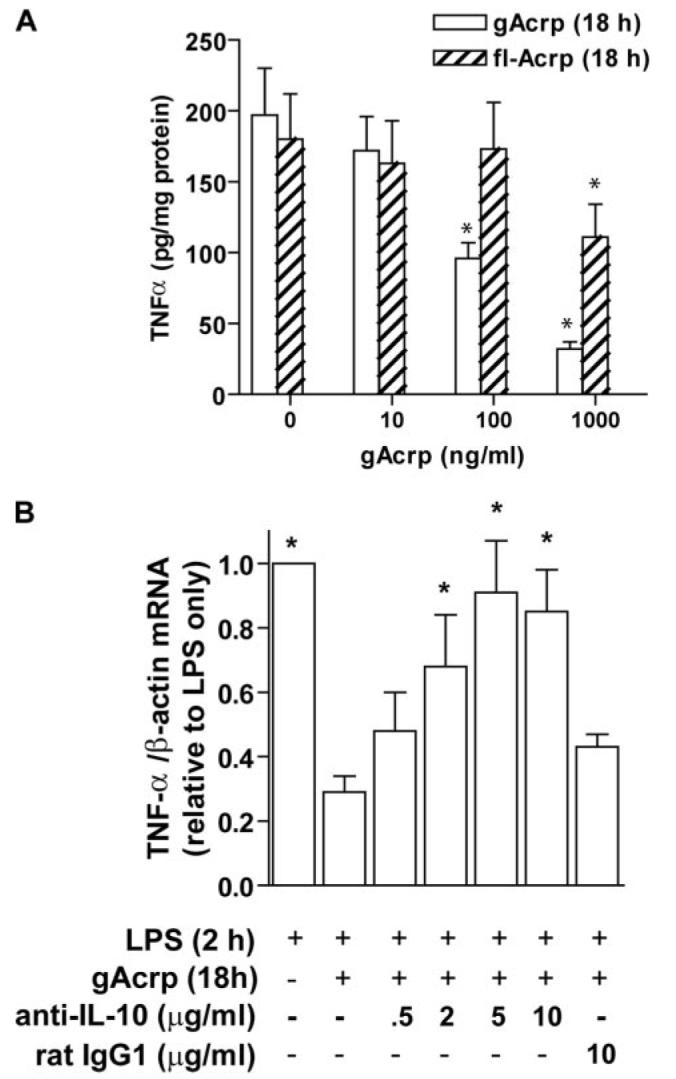
A, RAW 264.7 cells were cultured for 18 h in the absence or presence of globular (gAcrp) or full-length (flAcrp) adiponectin. Cells were then stimulated with 100 ng/ml LPS for 4 h and TNF-α concentration in the medium measured by enzyme-linked immunosorbent assay. Values represent means + S.E., n = 8, *, p <0.05 compared with cells not treated with adiponectin. B, RAW264.7 macrophages were treated with or without 1 μg/ml gAcrp for 18 h. Cells were co-cultured with neutralizing antibody to IL-10 or IgG controls. Cells were then stimulated with 100 ng/ml LPS for 2 h, and accumulation of TNF-α mRNA was measured by real-time PCR. Values represent TNF-α mRNA normalized to β-actin mRNA, means ± S.E., n = 4–11; *, p <0.05 compared with cells treated with gAcrp overnight without neutralizing antibody.
The characteristics of macrophages after overnight exposure to Acrp, i.e. decreased toll-like receptor 4-stimulated signal transduction and cytokine expression (7, 10), are reminiscent of the development of tolerance to LPS (14, 23). Because tolerance to LPS can be mediated by the anti-inflammatory cytokine IL-10 (24), we next asked whether the desensitization of LPS-stimulated TNF-α production by gAcrp was mediated by IL-10. gAcrp increased IL-10 mRNA accumulation, with a peak accumulation at5hof exposure to gAcrp (Fig. 2A), similar to the increase in IL-10 mRNA observed in human leukocytes after treatment with adiponectin (16, 23). IL-10 peptide accumulated in the medium during gAcrp treatment (Fig. 2B). Co-culture of RAW264.7 macrophages with neutralizing antibodies against IL-10 during 18 h of exposure to gAcrp prevented the gAcrp-mediated desensitization of LPS-stimulated TNF-α mRNA accumulation (Fig. 1B).
FIGURE 2. Short-term treatment of RAW264.7 macrophages with globular adiponectin (gAcrp) increased IL-10 production.
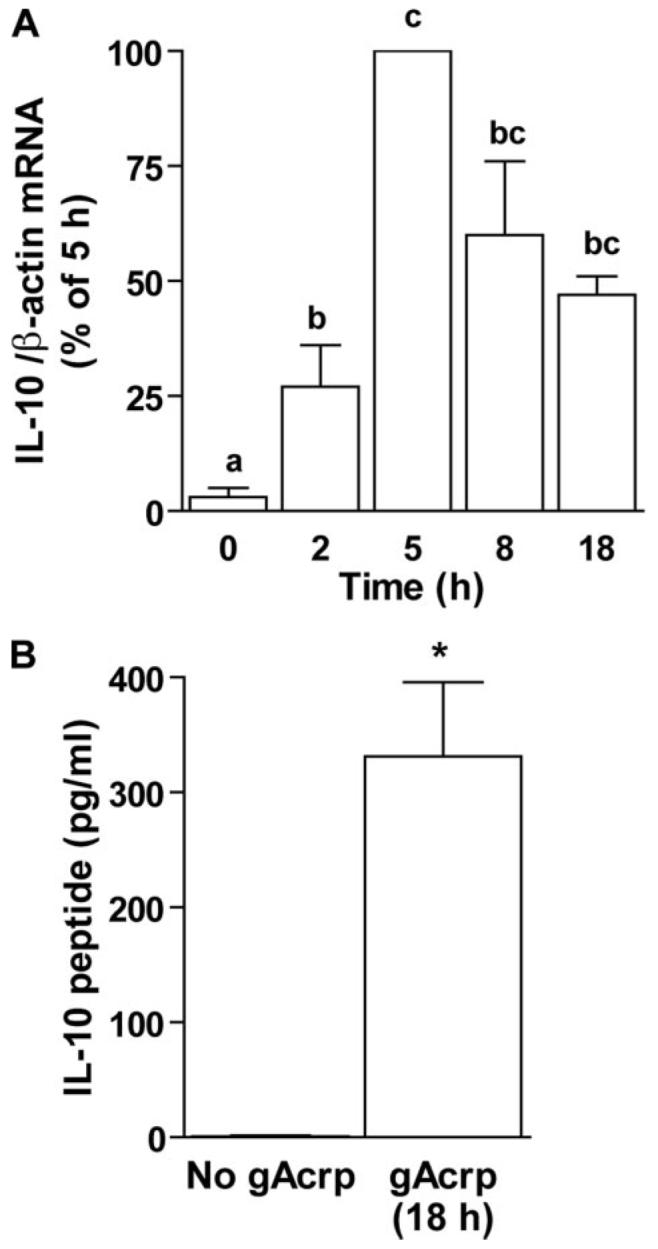
A, RAW264.7 macrophages were treated with or without 1 μg/ml gAcrp for 0–18 h, and accumulation of IL-10 mRNA was measured by real-time PCR. Values represent IL-10 mRNA normalized to β-actin mRNA, means ± S.E., n = 4–8. Values with different superscripts are significantly different from each other; p <0.05. B, RAW264.7 macrophages were treated with or without 1 μg/ml gAcrp for 18 h and accumulation of IL-10 secreted into the medium was measured by enzyme-linked immunosorbent assay. Values represent means ± S.E., n = 6. *, p <0.05.
Because gAcrp-stimulated IL-10 expression was required for long-term desensitization of LPS-stimulated TNF-α expression by gAcrp, we next investigated the mechanisms by which gAcrp increased IL-10 expression. IL-10 expression is induced by a number of pro-inflammatory mediators, including viruses, LPS, and TNF-α (25); LPS induction of IL-10 expression is dependent on TNF-α production (26). Therefore, we hypothesized that short-term treatment of RAW 264.7 macrophages with gAcrp would rapidly increase the expression of TNF-α and that this increase in TNF-α expression would contribute to gAcrp-stimulated IL-10 expression. Exposure of RAW 264.7 macrophages to 1 μg/ml gAcrp rapidly increased the accumulation of TNF-α mRNA with an apparent peak at 2 h (Fig. 3A); this increase preceded the peak in IL-10 mRNA accumulation at 5 h (Fig. 2A). Secretion of TNF-α peptide by RAW264.7 macrophages was also increased in response to gAcrp treatment, with TNF-α detected in the medium within1hof exposure to gAcrp (Fig. 3B). Increased accumulation of TNF-α mRNA was associated with an increase in the expression of a luciferase reporter construct driven by the murine TNF-α promoter (Fig. 3C). To rule out any effect of contaminating endotoxin in the gAcrp preparation in mediating this increase in TNF-α promoter activity, RAW 264.7 macrophages were pretreated with polymyxin B, an antibiotic that binds to LPS, prior to stimulation with either 100 ng/ml LPS or 1 μg/ml gAcrp. Although polymyxin B decreased LPS-stimulated TNF-α promoter activity, it had no effect on the response to gAcrp (Fig. 3D).
FIGURE 3. Short-term treatment of RAW264.7 macrophages with globular adiponectin (gAcrp) increased TNF-α promoter activity and mRNA accumulation.
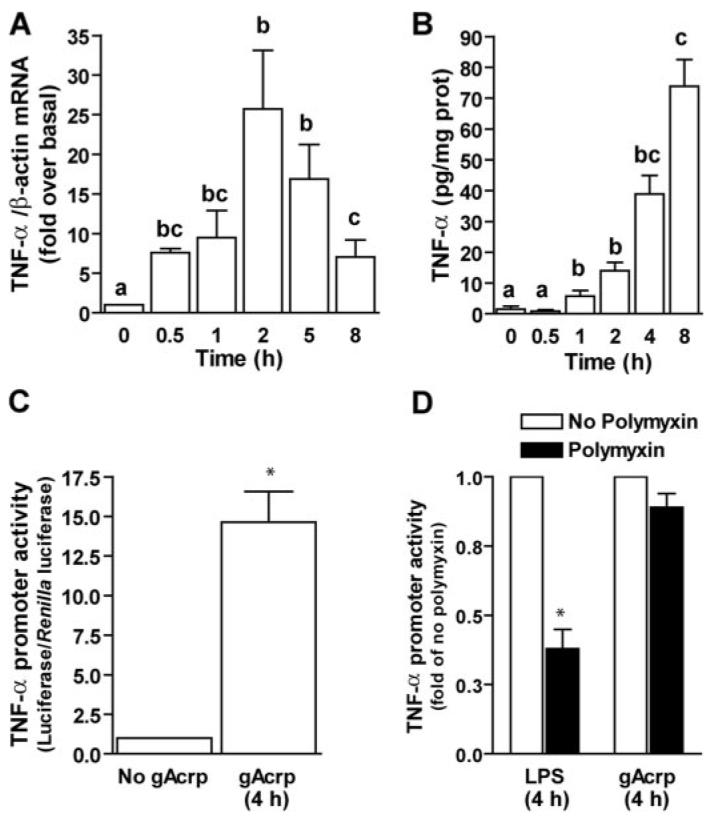
A, RAW264.7 macrophages were treated with or without 1 μg/ml gAcrp for 0–8 h min, and accumulation of TNF-α mRNA was measured by real-time PCR. Values represent TNF-α mRNA normalized to β-actin mRNA, means ± S.E., n = 4–8. Values with different superscripts are different from each other; *, p <0.05. B, RAW264.7 macrophages were treated with 1 μg/ml gAcrp for 0–8 h, and accumulation of TNF-α peptide in the cell culture medium was measured by enzyme-linked immunosorbent assay. Values represent means ± S.E., n = 4. Values with different superscripts are different from each other; *, p <0.05. C, TNF-α promoter activity was measured in RAW264.7 macrophages transiently transfected with a full-length TNF-α promoter-luciferase reporter construct. RAW264.7 macrophages were co-transfected with the TNF-α promoter-luciferase reporter and a Renilla luciferase control and then, after 24 h, treated with or without 1 μg/ml gAcrp for 4 h. Values represent relative luciferase activity (corrected for Renilla luciferase activity), means ± S.E., n=8; *, p <0.05. D, pretreatment with polymyxin B, an LPS-binding protein, prevents LPS-stimulated TNF-α promoter activity but not gAcrp-mediated responses. RAW 264.7 macrophages were co-transfected with the TNF-α promoter-luciferase reporter and a Renilla luciferase control and then, after 24 h, pretreated with 50 μg/ml polymyxin B. After 30 min cells were treated with 100 ng/ml LPS or 1 μg/ml gAcrp for 4 h. Values represent fold suppression of relative luciferase activity (corrected for Renilla luciferase activity) compared with cells not treated with polymyxin B, means ± S.E., n = 4. *, p <0.05 compared with cells not treated with polymyxin.
Two strategies were taken to test the role of gAcrp-stimulated TNF-α production in mediating increased IL-10 production during gAcrp treatment. First, RAW 264.7 macrophages were co-cultured with a neutralizing antibody to TNF-α during exposure to gAcrp. Antibodies against TNF-α decreased gAcrp-stimulated increases in IL-10 mRNA, whereas control antibodies had no effect (Fig. 4A). A second approach was to knock down TNF-RI with siRNA strategies. We hypothesized that if TNF-α were required for gAcrp-stimulated IL-10 expression, then knock down of TNF-RI expression should prevent this response. RAW264.7 macrophages were transiently transfected with siRNA against TNF-RI or scrambled siRNA, as a control. Cells were co-transfected with a luciferase reporter plasmid containing the full-length IL-10 promoter. gAcrp increased IL-10 promoter-driven luciferase activity in cells cotransfected with scrambled siRNA by 48-fold over basal (Fig. 4B). This response was reduced to 11-fold in cells co-transfected with siRNA against TNF-RI (Fig. 4B). As a further control for the efficacy of the siRNA knock down of the TNF-RI, TNF-α-stimulated expression of an NFκB-cis reporter plasmid was also found to be suppressed in RAW264.7 macrophages transfected with siRNA against TNF-RI compared with cells transfected with scrambled siRNA (data not shown). Taken together, these experiments demonstrate that gAcrp-stimulated TNF-α expression contributed to the gAcrp-stimulated production of IL-10 and therefore to the anti-inflammatory effects of gAcrp.
FIGURE 4. TNF-α-neutralizing antibodies or transfection with siRNA against TNF-RI suppressed globular adiponectin (gAcrp)-stimulated IL-10 production.
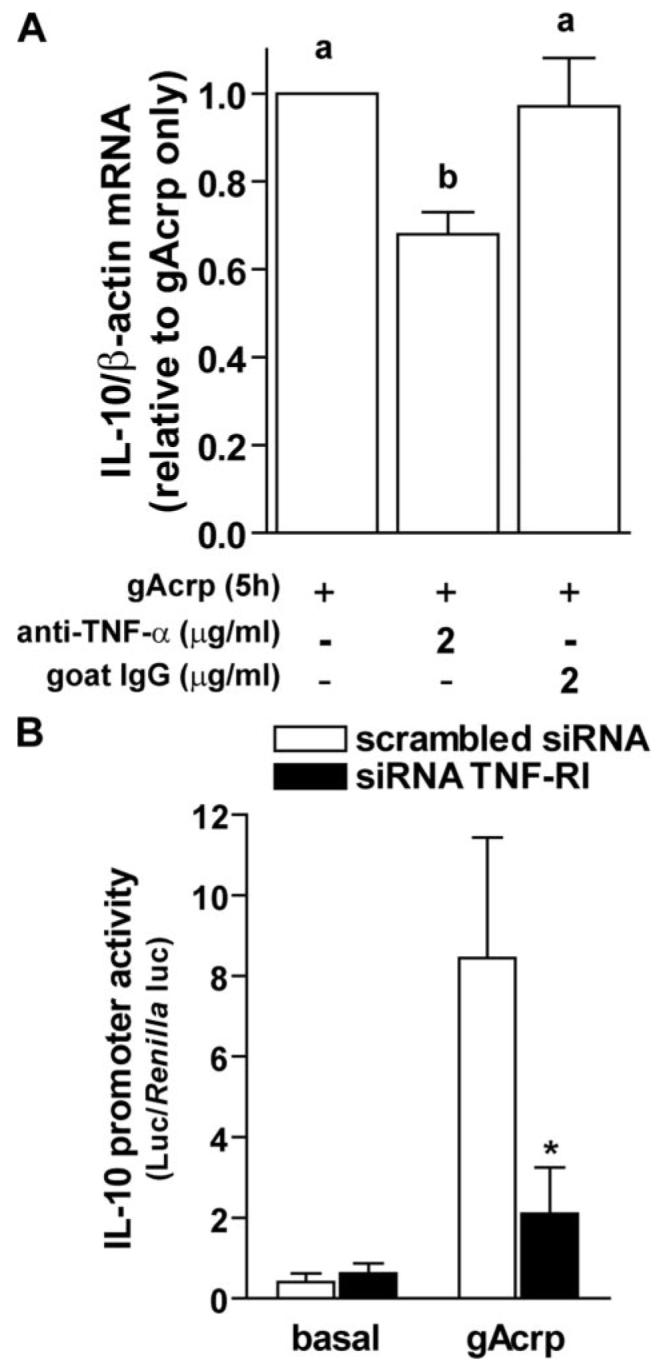
A, RAW264.7 macrophages were treated with or without 1 μg/ml gAcrp for 5 h, and IL-10 mRNA was measured by real-time PCR. Cells were co-cultured with neutralizing antibody to TNF-α or IgG controls. Values represent IL-10 mRNA normalized to β-actin mRNA, means ± S.E., n = 9 for anti-TNF-α and n = 3 for IgG controls. Values with different superscripts are significantly different from each other; p <0.05. B, RAW264.7 macrophages were transiently transfected with siRNA against TNF-RI or a scrambled siRNA, along with the IL-10 promoter-luciferase reporter and a Renilla luciferase control. Four hours post-transfection, cells were treated with or without 1 μg/ml gAcrp for 18 h. Values represent relative luciferase activity (corrected for Renilla luciferase activity), means ± S.E., n=3; *, p <0.05 compared with cells transfected with scrambled siRNA.
Transcriptional activation of TNF-α can involve the activation of distinct sets of transcription factors binding to at least two regions of the TNF-α promoter that include NFκB, Egr-1, and AP-1 binding sites (17, 27). Although the exact array of transcription factors interacting with the TNF-α promoter is to some extent cell- and species-specific (28), recruitment of NFκB and Egr-1 appears to be required for full activation of TNF-α expression in most types of macrophages (17, 27). Treatment of RAW 264.7 macrophages with 1 μg/ml gAcrp activated the DNA binding activity of both Egr-1 and NFκB (Fig. 5A). Sixty minutes after treatment with gAcrp, nuclear extracts exhibited increased DNA binding activity of Egr-1, as well as both the p50 and p65 subunits of NFκB (Fig. 5A). The identity of each DNA-binding protein was confirmed in super-shift assays using specific antibodies against each transcription factor protein (Fig. 5A).
FIGURE 5. Short-term treatment of RAW264.7 macrophages with globular adiponectin (gAcrp) stimulated DNA binding activity of Egr-1 and NFκB and increased TNF-α promoter activity.

A, RAW264.7 macrophages were treated with and without 1 μg/ml gAcrp for 60 min. Nuclear extracts were then isolated and used to measure the binding of nuclear proteins to oligonucleotides specific for Egr-1 and NFκB DNA binding sites. Extracts were also incubated with normal IgG or antibodies specific for each transcription factor for supershift assays. Supershifted bands are indicated with an arrowhead to the right of the supershifted band. Images are representative of at least four independent experiments. B, gAcrp-stimulated TNF-α promoter activity was measured using a series of luciferase reporter constructs containing either the full-length TNF-α promoter or constructs mutated at the κB3 or Egr-1 binding sites. RAW264.7 macrophages were co-transfected with TNF-α promoter luciferase and Renilla luciferase reporters and then stimulated with 100 ng/ml LPS or 1 μg/ml gAcrp. Values represent relative luciferase activity (corrected for Renilla luciferase activity), means ± S.E., n = 6; *, p <0.05 compared with full-length promoter.
To test the functional contribution of Egr-1 and NFκB in mediating gAcrp-stimulated TNF-α expression, RAW 264.7 macrophages were transfected with a full-length TNF-α promoter-luciferase reporter construct or constructs in which the κB-3 site or Egr-1 site was mutated. Both 100 ng/ml LPS and 1 μg/ml gAcrp stimulated TNF-α promoter-driven luciferase expression to an equivalent level. Mutation of the Egr-1 or κB-3 sites reduced both LPS- and gAcrp-stimulated luciferase expression by 60–70 and 90%, respectively (Fig. 5B). To further validate the contribution of these two transcription factors, RAW 264.7 macrophages were co-transfected with the TNF-α promoter-luciferase reporter and a dominant negative Egr-1 construct or the IκB super-repressor. In both conditions, gAcrp-stimulated TNF-α promoter activity was reduced (Fig. 6). Taken together, these data indicate that both Egr-1 and NFκB were involved in gAcrp-stimulated TNF-α promoter activation.
FIGURE 6. Overexpression of dominant negative Egr-1 or IκB super-repressor decreased globular adiponectin-stimulated TNF-α promoter activity.
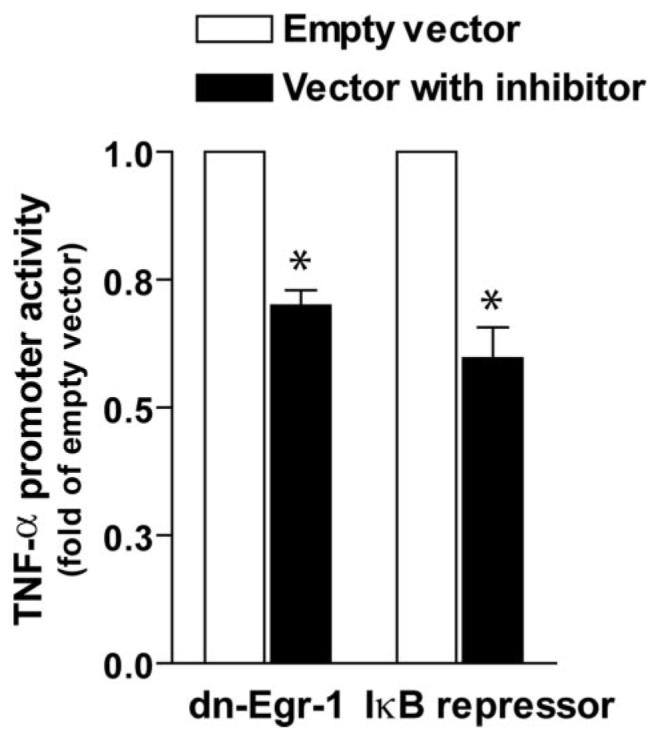
RAW264.7 macrophages were co-transfected with full-length TNF-α promoter luciferase reporter and Renilla luciferase, along with a plasmid containing dominant negative Egr-1 (pCMV-ETTL), the IκB super-repressor, or empty vector. Cells were then stimulated with 1 μg/ml gAcrp for 4 h. Values represent relative luciferase activity (corrected for Renilla luciferase activity), means ± S.E., n = 5; *, p <0.05 compared with cells transfected with empty vector.
Because a previous report also found that gAcrp can activate NFκB in monocytes (15), here we have further investigated the novel observation that gAcrp increases the activity of Egr-1. Egr-1 is an immediate early gene involved in regulating the expression of a number of pro-inflammatory cytokines and chemokines (29). gAcrp rapidly and transiently increased Egr-1 mRNA (Fig. 7A) and DNA binding activity (Fig. 7B).
FIGURE 7. Globular adiponectin (gAcrp) transiently increased Egr-1 expression and DNA binding activity.
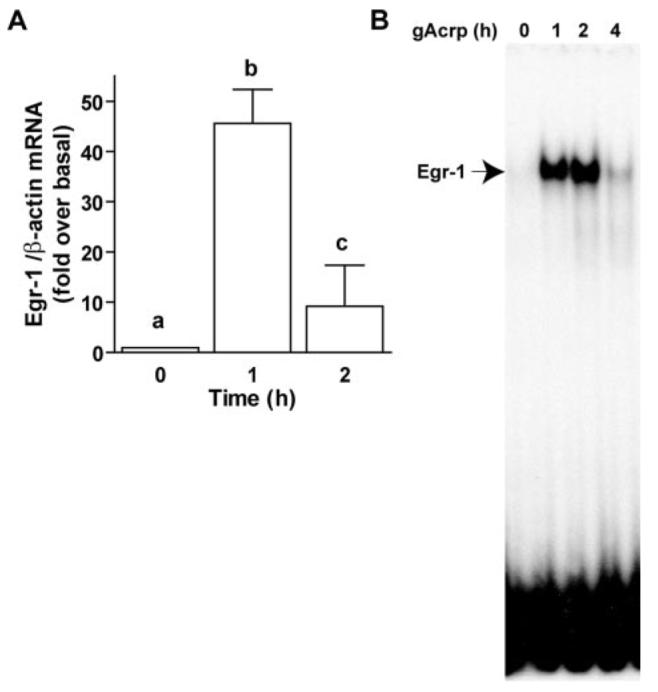
A, RAW264.7 macrophages were stimulated with 1 μg/ml gAcrp for 0–2 h, and Egr-1 mRNA was measured by real-time PCR. Values represent means ± S.E., n = 3. Values with different superscripts are significantly different from each other; p <0.05. B, RAW264.7 macrophages were treated with 1 μg/ml gAcrp for 0–4 h. Nuclear extracts were prepared and used to measure the binding of nuclear proteins to an oligonucleotide specific for Egr-1 binding. Images are representative of four independent experiments.
Increased expression of Egr-1 in response to LPS or tissue factor requires the activation of the MAPK family member ERK1/2 (30, 31). Therefore, we asked whether inhibition of ERK1/2, p38, or JNK would prevent gAcrp-stimulated Egr-1 mRNA accumulation. Pretreatment of RAW264.7 macrophages with the MEK1/2 inhibitor U0126 completely inhibited gAcrp-stimulated Egr-1 mRNA accumulation, whereas pretreatment with SB203580 to inhibit p38, or SP600125 to inhibit JNK, had no effect on gAcrp-stimulated Egr-1 mRNA accumulation (Fig. 8A). Similarly, pretreatment of RAW264.7 macrophages with U0126 suppressed both gAcrp-stimulated Egr-1 and TNF-α promoter-luciferase activity (Fig. 8B); the extent of inhibition of TNF-α promoter activation by U0126 (Fig. 8B) was similar to that observed using the TNF-α promoter-luciferase reporter construct in which the Egr-1 binding site was mutated (Fig. 4B). gAcrp rapidly increased phosphorylation of ERK1/2 (Fig. 8C). Polymyxin B pretreatment had no effect on basal or 30-min gAcrp-stimulated phospho-ERK but inhibited LPS-stimulated phosphorylation of ERK1/2 (data not shown). Taken together, these data point to an essential role for gAcrp-stimulated ERK1/2 phosphorylation and subsequently increased expression of Egr-1 in mediating the loss of LPS-stimulated TNF-α production after long-term exposure of RAW264.7 macrophages to gAcrp.
FIGURE 8. Globular adiponectin (gAcrp) increased ERK1/2 phosphorylation, and inhibition of ERK1/2 decreased gAcrp-stimulated expression of Egr-1 and TNF-α.
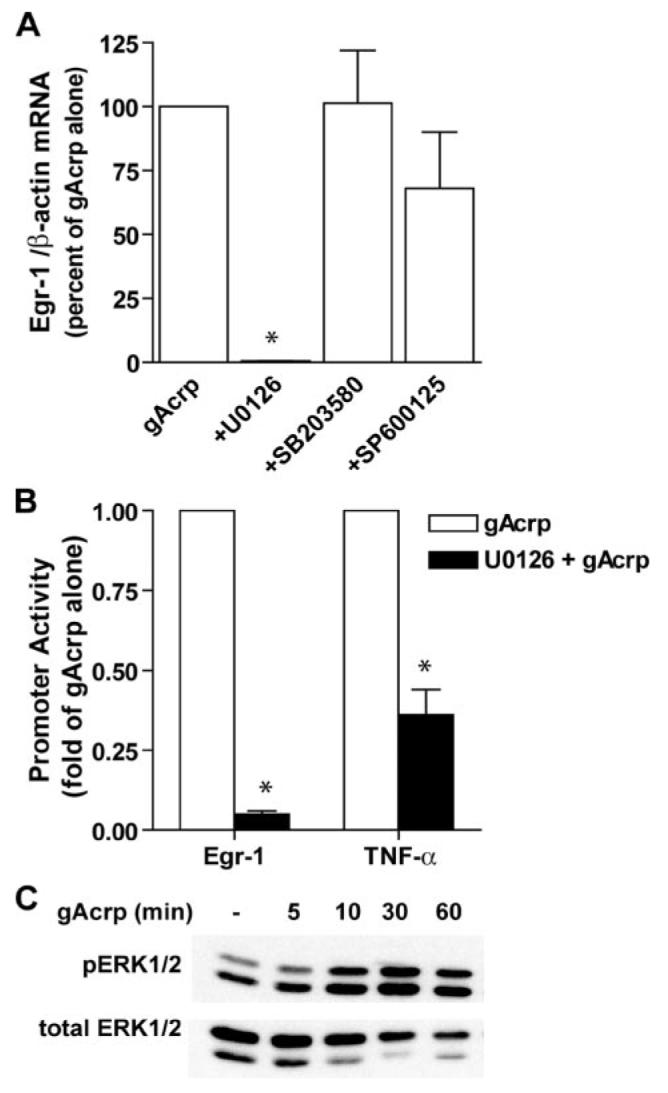
A, RAW264.7 macrophages were pretreated with MAPK inhibitors for 60 min and then stimulated for 1 h with 1 μg/ml gAcrp. Egr-1 mRNA was then measured by real-time PCR. Values represent Egr-1 mRNA/β-actin mRNA normalized to cells not treated with inhibitors, means ± S.E., n = 3. *, p <0.05 compared with cells not treated with inhibitors. B, RAW264.7 macrophages were co-transfected with Egr-1 promoter lucifer-ase or TNF-α promoter luciferase reporters along with Renilla luciferase. Cells were then preincubated with U0126 for 60 min prior to stimulation with 1 μg/ml gAcrp for 4 h. Values represent relative luciferase activity (corrected for Renilla luciferase activity), means ± S.E., n = 4 for Egr-1 and 6 for TNF-α; *, p < 0.05 compared with cells not treated with inhibitors. C, RAW264.7 macrophages were stimulated or not with 1 μg/ml gAcrp for 0 – 60 min, and phosphorylation of ERK1/2 was assessed by Western blot. Images are representative of three independent experiments.
DISCUSSION
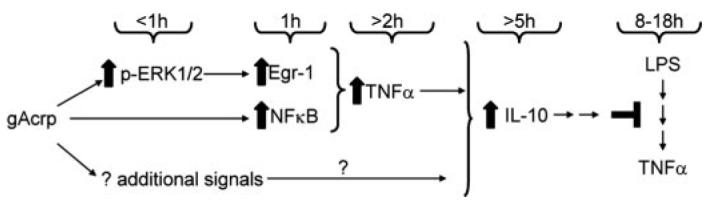
Adiponectin has potent anti-inflammatory effects both in vivo and in isolated macrophage cultures. Continued exposure to adiponectin in vivo likely results in a tonic suppression of pro-inflammatory signals. This anti-inflammatory effect of adiponectin can be modeled in cultured macrophages during an overnight treatment with adiponectin. Here we have shown that in RAW264.7 macrophages overnight exposure to 1 μg/ml gAcrp or full-length adiponectin suppressed LPS-stimulated TNF-α production, associated with a decrease in LPS-stimulated TNF-α mRNA accumulation. Desensitization of LPS-stimulated responses was mediated via increased IL-10 expression during gAcrp exposure. In turn, gAcrp-stimulated IL-10 expression was dependent on a rapid increase in TNF-α expression. Thus, in contrast to the well documented long-term/tonic anti-inflammatory effects of adiponectin (7, 10), here we have demonstrated that short-term exposure with gAcrp increased TNF-α promoter activity and mRNA accumulation. gAcrp-stimulated TNF-α production was dependent on the activation of both NFκB and Egr-1. Working together, these two potent transcriptional activators of TNF-α expression thus contributed to the ability of gAcrp to eventually suppress the responsivity of macrophages to LPS (Fig. 9).
FIGURE 9. Schematic diagram illustrating the proposed time course of globular adiponectin (gAcrp)-mediated events leading to desensitization of LPS-stimulated responses in RAW264. 7 macrophages.
Two adiponectin receptors have been identified, AdipoR1 and AdipoR2 (5, 32). Although not completely understood, the available data suggest that gAcrp may have a higher affinity for AdipoR1 receptors, whereas full-length adiponectin has a higher affinity to AdipoR2 (32). Differential distribution of AdipoR1 and AdipoR2 is thought to contribute, at least in part, to the differential efficacy of gAcrp compared with full-length adiponectin in different tissues (33). Macrophages express varying proportions of AdipoR1 and AdipoR2 (6, 7, 10), and antibodies against AdipoR1, but not AdipoR2, are effective at blocking the development of LPS tolerance in RAW264.7 macrophages upon exposure to gAcrp (7). Here we have demonstrated that gAcrp is more potent in inhibiting LPS-stimulated TNF-α production in RAW264.7 macrophages (Fig. 1A). Interestingly, whereas circulating concentrations of gAcrp are undetectable, macrophages secrete a protease capable of cleaving full-length adiponectin to gAcrp (34). Taken together, the available data are consistent with the hypothesis for a predominant role of gAcrp and AdipoR1 in mediating the anti-inflammatory effects of adiponectin.
The anti-inflammatory effects of adiponectin are mediated by a number of mechanisms. Adiponectin inhibits the proliferation of myelomonocytic progenitor cells, as well as inhibiting phagocytosis (8). We and others (7, 9, 10) have found that treatment of macrophages with adiponectin for 6–24 h suppresses LPS-stimulated signal transduction and pro-inflammatory cytokine expression in rat Kupffer cells, porcine macrophages, and RAW264.7 macrophages. In rat Kupffer cells and porcine macrophages, this tolerance to LPS is characterized by a suppression of LPS-stimulated NFκB activation, as well as a decrease in LPS-stimulated ERK1/2 phosphorylation and subsequent Egr-1 expression (9, 10). In contrast, exposure of macrophages and dendritic cells to Acrp increases the expression of the anti-inflammatory mediators IL-10 and IL-10 receptor antagonist (9, 16).
Similar anti-inflammatory effects of long-term/continuous exposure to adiponectin have been observed in vivo. For example, increasing adiponectin expression in mice using recombinant adenoviral constructs suppresses the development of atherosclerosis in apolipoprotein E-deficient mice, at least in part by decreasing the production of TNF-α (11). Restoration of circulating adiponectin concentrations in mice chronically exposed to ethanol inhibited ethanol-induced increases in TNF-α production, contributing to a protection from ethanol-induced liver injury (35). Similarly, pretreatment of mice with adiponectin protects from LPS-induced liver injury in KK-Ay obese mice by suppressing TNF-α expression (36). In contrast, adiponectin-deficient mice exhibit increased sensitivity to galactosamine/LPS-induced liver injury (37). Taken together, these studies in isolated cells and in vivo studies demonstrate the critical anti-inflammatory effects of adiponectin on macrophage function.
Despite this well characterized effect of Acrp on modulating LPS sensitivity of macrophages, very little is known about the mechanisms for desensitization of LPS-mediated responses. Here we have identified the increase in IL-10 production during gAcrp treatment as a critical mediator for the anti-inflammatory effects of gAcrp. This essential role for IL-10 in mediating the actions of gAcrp on macrophages is consistent with the potent anti-inflammatory effects of IL-10 (25, 38).
gAcrp also rapidly induced the expression of TNF-α in RAW264.7 macrophages (15) (Fig. 3). Further, we have demonstrated for the first time that the secretion of TNF-α contributed to the subsequent increased expression of IL-10 during exposure to gAcrp. siRNA knock down of the TNF-RI receptor reduced gAcrp-stimulated IL-10 promoter activity by 75% (Fig. 4B). Although TNF-α-neutralizing antibodies did not completely inhibit gAcrp-stimulated IL-10 mRNA accumulation, this partial inhibition is similar to the effect of TNF-α-neutralizing antibodies in suppressing LPS-stimulated IL-10 production in other cell types (26) and may be due to incomplete neutralization of locally acting TNF-α and/or a contribution of additional paracrine factors in mediating the ability of gAcrp to increase IL-10 expression.
In addition to the previously proposed role for NFκB in mediating this short-term response to gAcrp (15), here we have demonstrated that gAcrp also increased ERK1/2 phosphorylation, which subsequently increased expression of Egr-1. Importantly, we found that both NFκB and Egr-1 were required for maximally increased gAcrp-stimulated TNF-α promoter activation. Egr-1 (also known as nerve growth factor-induced-A (NGFI-A), Krox-24, ZIF268, ETR103, and TIS8) is a zinc finger transcription factor discovered for its role in regulation of cell growth and proliferation (39). Synthesis of Egr-1, an immediate early gene, is rapidly and transiently induced in response to a variety of stimuli, including cytokines and growth factors, as well as environmental stress, including ischemic stress and tissue damage (40). Egr-1 has been characterized as a transcription factor that coordinates cellular responses to environmental stress, mediating increased expression of a number of target genes, including transforming growth factor β, platelet-derived growth factor, chemokines, and adhesion molecules (41). These gene products are required for progression of tissue repair, involving the acute inflammatory response and the release of cytokines and growth factors, neutrophil migration, collagen synthesis, and extracellular matrix remodeling (40). Here we identified for the first time a role of Egr-1 in mediating macrophage responses to gAcrp, thus adding to the list of the critical functions mediated by Egr-1 in coordinating the response of the organism to stress.
Transcription of Egr-1 is the primary mediator for increased Egr-1 DNA binding activity (39). The promoter sequence of the Egr-1 gene contains five serum response elements, as well as two AP-1 sites (31). We and others (21, 22, 31) have found that Egr-1 expression is dependent on the activation of ERK1/2 in Kupffer cells and macrophages. Importantly, here we have shown that gAcrp also potently stimulated Egr-1 transcription in an ERK1/2-dependent mechanism (Fig. 8A). Further, we found that gAcrp-stimulated ERK1/2 and subsequent increases in Egr-1 expression are upstream of gAcrp-stimulated TNF-α promoter activity, because inhibition of ERK1/2 activation with the MEK1/2 inhibitor U1026, as well as inhibition of Egr-1 transactivating activity, using a dominant negative Egr-1 construct, ameliorated the effects of gAcrp on TNF-α promoter activity. These data thus demonstrate that gAcrp has the ability to activate multiple transcription factors involved in increasing pro-inflammatory cytokine expression.
Taken together, the data presented here are consistent with the hypothesis that maintenance of a high circulating concentration of adiponectin has a tonic, anti-inflammatory effect on macrophages. In pathophysiological situations characterized by decreased concentrations of adiponectin, such as chronic ethanol exposure (12) or obesity (35, 42), the normal/“tonic” suppression of macrophage activity, usually maintained as a consequence of high circulating concentrations of adiponectin, would likely be alleviated. Decreased adiponectin would thus lead to enhanced sensitivity of macrophages to pro-inflammatory mediators, like LPS, and contribute to the development of the chronic inflammatory states observed in response to chronic ethanol exposure or obesity.
Footnotes
The abbreviations used are: Acrp, adipocyte complement-related protein; gAcrp, globular Acrp; AdipoR, adiponectin receptor; LPS, lipopolysaccha-ride; IL, interleukin; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; JNK, c-Jun N-terminal kinase; siRNA, small interfering RNA; MEK, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase.
This work was supported by United States Public Health Service Grants AA011975 and AA013868.
REFERENCES
- 1.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. J. Biol. Chem. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 2.Berg AH, Combs TP, Scherer PE. Trends Endocrinol. Metab. 2002;13:84–89. doi: 10.1016/s1043-2760(01)00524-0. [DOI] [PubMed] [Google Scholar]
- 3.Hu E, Liang P, Spiegelman BM. J. Biol. Chem. 1996;271:10697–10703. doi: 10.1074/jbc.271.18.10697. [DOI] [PubMed] [Google Scholar]
- 4.Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. Nat. Med. 2001;7:947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- 5.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 6.Chinetti G, Zawadski C, Fruchart JC, Staels B. Biochem. Biophys. Res. Commun. 2004;314:151–158. doi: 10.1016/j.bbrc.2003.12.058. [DOI] [PubMed] [Google Scholar]
- 7.Yamaguchi N, Argueta JG, Masuhiro Y, Kagishita M, Nonaka K, Saito T, Hanazawa S, Yamashita Y. FEBS Lett. 2005;579:6821–6826. doi: 10.1016/j.febslet.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 8.Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N, Kihara S, Funahashi T, Tenner AJ, Tomiyama Y, Matsuzawa Y. Blood. 2000;96:1723–1732. [PubMed] [Google Scholar]
- 9.Wulster-Radcliffe MC, Ajuwon KM, Wang J, Christian JA, Spurlock ME. Biochem. Biophys. Res. Commun. 2004;316:924–929. doi: 10.1016/j.bbrc.2004.02.130. [DOI] [PubMed] [Google Scholar]
- 10.Thakur V, Pritchard MT, McMullen MR, Nagy LE. Am. J. Physiol. 2006;290:G998–G1007. doi: 10.1152/ajpgi.00553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okamoto Y, Kihara S, Ouchi N, Nishida M, Arita Y, Kumada M, Ohashi K, Sakai N, Shimomura I, Kobayashi H, Terasaka N, Inaba T, Funahashi T, Matsuzawa Y. Circulation. 2002;106:2767–2770. doi: 10.1161/01.cir.0000042707.50032.19. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Lu G, Wong WP, Vliegenthart JF, Gerwig GJ, Lam KS, Cooper GJ, Xu A. Proteomics. 2004;4:3933–3942. doi: 10.1002/pmic.200400826. [DOI] [PubMed] [Google Scholar]
- 13.Furukawa K, Hori M, Ouchi N, Kihara S, Funahashi T, Matsuzawa Y, Miyazaki A, Nakayama H, Horiuchi S. Biochem. Biophys. Res. Commun. 2004;317:831–836. doi: 10.1016/j.bbrc.2004.03.123. [DOI] [PubMed] [Google Scholar]
- 14.Dobrovolskaia MA, Vogel SN. Microbes Infect. 2002;4:903–914. doi: 10.1016/s1286-4579(02)01613-1. [DOI] [PubMed] [Google Scholar]
- 15.Tsatsanis C, Zacharioudaki V, Androulidaki A, Dermitzaki E, Charalampopoulos I, Minas V, Gravanis A, Margioris AN. Biochem. Biophys. Res. Commun. 2005;335:1254–1263. doi: 10.1016/j.bbrc.2005.07.197. [DOI] [PubMed] [Google Scholar]
- 16.Wolf AM, Wolf D, Rumpold H, Enrich B, Tilg H. Biochem. Biophys. Res. Commun. 2004;323:630–635. doi: 10.1016/j.bbrc.2004.08.145. [DOI] [PubMed] [Google Scholar]
- 17.Yao J, Mackman N, Edgington TS, Fan ST. J. Biol. Chem. 1997;272:17795–17801. doi: 10.1074/jbc.272.28.17795. [DOI] [PubMed] [Google Scholar]
- 18.Calandra T, Bucala R. Crit. Rev. Immunol. 1997;17:77–88. doi: 10.1615/critrevimmunol.v17.i1.30. [DOI] [PubMed] [Google Scholar]
- 19.Papanikolaou NA, Sabban EL. J. Biol. Chem. 2000;275:26683–26689. doi: 10.1074/jbc.M000049200. [DOI] [PubMed] [Google Scholar]
- 20.DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mol. Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi L, Kishore R, McMullen M, Nagy LE. J. Biol. Chem. 2002;277:14777–14785. doi: 10.1074/jbc.M108967200. [DOI] [PubMed] [Google Scholar]
- 22.Kishore R, Hill JR, McMullen MR, Frenkel J, Nagy LE. Am. J. Physiol. 2002;282:G6–G15. doi: 10.1152/ajpgi.00328.2001. [DOI] [PubMed] [Google Scholar]
- 23.Fan H, Cook JA. J. Endotoxin. Res. 2004;10:71–84. doi: 10.1179/096805104225003997. [DOI] [PubMed] [Google Scholar]
- 24.Grutz G. J. Leukocyte Biol. 2005;77:3–15. doi: 10.1189/jlb.0904484. [DOI] [PubMed] [Google Scholar]
- 25.Moore MJ, Rosbash M. Science. 2001;294:1841–1842. doi: 10.1126/science.1067676. [DOI] [PubMed] [Google Scholar]
- 26.Sato TA, Keelan JA, Mitchell MD. J. Immunol. 2003;170:158–166. doi: 10.4049/jimmunol.170.1.158. [DOI] [PubMed] [Google Scholar]
- 27.Tsai EY, Falvo JV, Tsytsykova AV, Barczak AK, Reimold AM, Glimcher LH, Fenton MJ, Gordon DC, Dunn IF, Goldfeld AE. Mol. Cell Biol. 2000;20:6084–6094. doi: 10.1128/mcb.20.16.6084-6094.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Means TK, Pavlovich RP, Roca D, Vermeulen MW, Fenton MJ. J. Leukocyte Biol. 2000;67:885–893. doi: 10.1002/jlb.67.6.885. [DOI] [PubMed] [Google Scholar]
- 29.Guha M, Mackman N. Cell. Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 30.Shi L, Kishore R, McMullen M, Nagy LE. Am. J. Physiol. 2002;282:C1205–C1211. doi: 10.1152/ajpcell.00511.2001. [DOI] [PubMed] [Google Scholar]
- 31.Guha M, O'Connell MA, Pawlinski R, Hollis A, McGovern P, Yan SF, Stern D, Mackman N. Blood. 2001;98:1429–1439. doi: 10.1182/blood.v98.5.1429. [DOI] [PubMed] [Google Scholar]
- 32.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Nat. Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 33.Goldstein BJ, Scalia R. J. Clin. Endocrinol. Metab. 2004;89:2563–2568. doi: 10.1210/jc.2004-0518. [DOI] [PubMed] [Google Scholar]
- 34.Waki H, Yamauchi T, Kamon J, Kita S, Ito Y, Hada Y, Uchida S, Tsuchida A, Takekawa S, Kadowaki T. Endocrinology. 2005;146:790–796. doi: 10.1210/en.2004-1096. [DOI] [PubMed] [Google Scholar]
- 35.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. J. Clin. Investig. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masaki T, Chiba S, Tatsukawa H, Yasuda T, Noguchi H, Seike M, Yoshimatsu H. Hepatology. 2004;40:177–184. doi: 10.1002/hep.20282. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto H, Tamura S, Kamada Y, Kiso S, Fukushima J, Wada A, Maeda N, Kihara S, Funahashi T, Matsuzawa Y, Shimomura I, Hayashi N. World J. Gastroenterol. 2006;12:3352–3358. doi: 10.3748/wjg.v12.i21.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamilton TA, Ohmori Y, Tebo J. Immunol. Res. 2002;25:229–245. doi: 10.1385/IR:25:3:229. [DOI] [PubMed] [Google Scholar]
- 39.Gashler A, Sukhatme VP. Prog. Nucleic Acids Res. Mol. Biol. 1995;50:191–224. doi: 10.1016/s0079-6603(08)60815-6. [DOI] [PubMed] [Google Scholar]
- 40.Braddock M. Ann. Med. 2001;33:313–318. doi: 10.3109/07853890109002083. [DOI] [PubMed] [Google Scholar]
- 41.Yan SF, Fujita T, Lu J, Okada K, Zou YS, Mackman N, Pinsky DJ, Stern DM. Nature Med. 2000;6:1355–1361. doi: 10.1038/82168. [DOI] [PubMed] [Google Scholar]
- 42.Tilg H, Hotamisligil GS. Gastroenterology. 2006;131:934–945. doi: 10.1053/j.gastro.2006.05.054. [DOI] [PubMed] [Google Scholar]