

Abstract
Toll-like-receptor mediated signaling is finely regulated by a complex intracellular protein network including the interleukin-1 receptor associate kinases (IRAKs). IRAK-4, 1, and 2 may positively regulate innate immunity signaling through the activation of various downstream kinases such as MAPKs. In contrast, IRAK-M plays an inhibitory role through unknown mechanism. In this report, we show that IRAK-M is ubiquitously present in the cell, and becomes exclusively cytoplasmic upon bacterial lipoprotein Pam3CSK4 challenge. Furthermore, using bone marrow derived macrophages (BMDM) from wild type, IRAK1−/−, and IRAK-M−/− mice, we have herein demonstrated that IRAK-M selectively attenuates bacterial lipopeptide Pam3CSK4-induced p38 activation, but not ERK or JNK. IRAK1−/− and IRAK-M−/− BMDM display distinct activation profile of various MAP kinases upon Pam3CSK4 challenge, indicating that IRAK-M exerts its inhibitory effect through an IRAK1 independent pathway. Pam3CSK4 challenge leads to rapid decrease of MKP-1 protein level in IRAK-M−/− BMDM as well as THP-1 cells with decreased IRAK-M expression through siRNA interference. Our findings indicate that IRAK-M selectively attenuates p38 activation and inhibits innate immunity through stabilizing MKP-1.
Keywords: Innate immunity, signaling, kinase, phosphatase, IRAK-M, cellular distribution
Introduction
Human innate immunity is critical for host defense as well as other cellular and tissue metabolisms. Toll-like-receptors (TLRs)-mediated signaling processes can recognize diverse substances from microbial as well as non-microbial sources, and relay signals downstream to activate the expression of various pro- and anti- inflammatory mediators (1). TLR signaling is finely regulated through a series of intra-cellular proteins including the interleukin-1 receptor associated kinases (IRAKs). There are four distinct human genes coding for four IRAK proteins (1, 2, M, and 4) (2), among which IRAK-4 is critical for activating transcription factor NFκB (3), and IRAK1 is mostly involved in STAT and IRF activation (4;5;6). IRAK-M, in contrast, may counteract these effects elicited by IRAK-4 (7;8).
IRAK-M was originally cloned through a computer-based EST database search for IRAK homologues (9). It was designated as IRAK-M since it was mainly found in cells of monomyeloic origin. Initial studies using over expression of IRAK-M showed that IRAK-M can activate NFκB reporter gene activity (9). However, a later study using IRAK-M deficient mice indicate that IRAK-M inhibits NFκB activation and negatively regulates TLR signaling (7). IRAK-M−/− macrophages exhibit enhanced MAP kinase activation, elevated TNFα production, and increased inflammatory response upon challenges with bacteria or various TLR ligands (7). Furthermore, IRAK-M deficient mice spontaneously develop osteoporosis due to elevated differentiation and activation of osteoclasts (8).
Despite the significant role IRAK-M plays in innate immunity and cellular differentiation, the biochemical regulation of IRAK-M and the mechanism for its inhibitory function is not clearly understood. In this report, we have performed systematic analyses of IRAK-M protein expression and sub-cellular localization upon Pam3CSK4 challenge using primary murine macrophages, cultured human THP-1 cells, as well as primary human peripheral blood mononuclear cells. Furthermore, the mechanism for IRAK-M mediated MAP kinase downregulation was studied. We found that IRAK-M selectively modulated Pam3CSK4 mediated p38 activation through regulating MKP-1 stability.
Materials and methods
Reagents
E. coli 0114 Lipopolysaccharide (LPS) and anti-IRAK-M antibody were obtained from Sigma (St Lois, MO). Pam3CSK4 was from EMC Microcollections (Tubingen, Germany). Anti-MKP-1, anti-IRAK1, anti-phospho-Erk1/2(Thr185/Tyr187) and anti-phospho-JNK (Thr183/Tyr185, Thr221/Tyr223) antibodies were from Upstate Biotechnology (Lake Placid, NY). The antibody against phosphorylated p38 was from Santa Cruz Biotechnology (Santa Cruz, CA).
Mice
C57BL/6 wild type mice were purchased from the Charles River laboratory. IRAK1-deficient mice were kindly provided by Dr. James Thomas from the University of Texas Southwestern Medical School. IRAK-M-deficient mice were kindly provided by Dr. Richard A. Flavell at Yale University School of Medicine. These mice were bred and maintained in the animal facility at Virginia Tech with the approved Animal Care and Use Committee protocol. All mice were 7–10 weeks of age when experiments were initiated.
Cell Culture and transfection
THP-1 cells, an undifferentiated human promonocytic cell line, were obtained from the American Type Culture Collection (ATCC, Rockville, MD), and were maintained at 37°C with 5% CO2 in RPMI 1640 medium supplemented with 2 mM glutamine (Sigma), 100 units/ml penicillin, 100 mg/ml streptomycin (ICN, Aurora, OH), and 10% fetal bovine serum (Sigma). MAT-2 cells, obtained from Dr. Fabio Re, were Hela cells stably transfected with TLR2 (10). MAT-2 cells were maintained in DMEM with 2 mM glutamine (Sigma), 100 units/ml penicillin, 100 mg/ml streptomycin (ICN, Aurora, OH), and 10% fetal bovine serum (Sigma). pcDNA empty vector or pflag-IRAK-M plasmid (9) were transiently transfected into MAT-2 cells using the lipofectamine reagent as described by the manufacturer (Invitrogen).
Isolation and culture of murine bone marrow-derived macrophages
Bone marrow from tibia and femur was obtained by flushing with DMEM. Bone marrow cells were cultured in a 125×50mm Lab-Tek non-tissue-culture-treated dish with 50ml DMEM medium containing 30% L929 cell supernatant, 1mM Sodium Pyruvate, 50μM 2-Mercaptoethanol and 2 mM glutamine. On the third day of culture, the cells were fed with additional 20ml fresh medium and cultured for additional three days. Cells were harvested and washed with PBS, resuspended in DMEM supplemented with 10% FBS, and allowed to rest overnight before further treatment.
Isolation of human primary blood mononuclear cells (PBMC)
Human bloods were obtained from healthy donors with consent and in compliance with the protocol of Institutional Review Board. PBMC were isolated from heparinized blood as previously described (11) by Isolymph sedimentation (Gallard-Schlesinger Industries, Carle Place Inc., NY) followed by centrifugation for 5 min at 200×g. The pellet was resuspended at a final concentration of 5×106 cells/ml in complete RPMI 1640 medium supplemented with 10% FBS.
Isolation of Cytoplasmic and Nuclear Extracts and Western Blot
Cell lyses, isolation of total, cytoplasmic, and nuclear extracts were as described previously (4). Briefly, various cells (5×106/ml) were washed in 10 mM HEPES, pH 7.9, and subsequently lysed on ice in the lysis buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM EDTA, 0.5 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml pepstatin). After centrifugation for 10min at 3,500 rpm, the supernatant cytoplasmic fractions were transferred and saved as cytoplasmic extracts. Pellets containing intact nuclei were lysed and solubilized with the high salt buffer (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 0.4 M NaCl, 0.2 mM EDTA, 0.5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride) for 30 min and yielded the nuclear extracts. Western detection of corresponding proteins were performed as described (12).
Immunohistochemistry and Immunofluorescence Microscopy
Bone marrow macrophages were grown on chamber slides and subsequently treated with Pam3CSK4 for one hour. Treated cells were fixed in 4% paraformaldehyde, blocked with 1%BSA in PBS and incubated with IRAK-M antibody. The slides were then stained with cy-2 -conjugated anti-Rabbit IgG antibody (10 μg/ml), and double stained with 0.1μg/ml DAPI. The slides were visualized under Zeiss Axioplan 2 fluorescent microscope. Cy2-stained IRAK-M and the DAPI-stained nuclei were visualized at excitation wavelengths of 490 and 410 nm, respectively.
Transfection of Small Interfering RNAs (siRNAs)
siRNA against IRAK-M and scrambled control siRNA were obtained from Qiagen ( San Diego, CA), and used to transfect THP-1 cells with Amaxa nucleofector kit V as previously described (11). Transfected cells were incubated in RPMI with 10% fetal bovine serum for 48-h, and subsequently used for further studies as described in the text.
RESULTS
IRAK-M is constitutively expressed in human mononuclear cells
We systematically examined the IRAK-M protein levels in various cells. First, human promonocytic THP-1 cells were challenged with Pam3CSK4 for various time periods. Total cell extracts were prepared and equal amounts of cellular extracts were separated on SDS-PAGE. The IRAK-M levels were determined through Western Blot using anti-IRAK-M antibody. As shown in figure 1A, IRAK-M protein was detected through Western Blot as a 68KD band and constitutively present in resting THP-1 cells. There was no apparent induction of IRAK-M protein within 8 hr of Pam3CSK4 treatment. Intriguingly, there was a quick induction of an upper band (∼80KD) upon 15 min of Pam3CSK4 treatment. The nature of that upper band is not clear and may be covalently modified IRAK-M. In order to confirm that the major 68KD band we detected was indeed IRAK-M, we performed siRNA knock down study. THP-1 cells were transfected with either control scrambled siRNA or IRAK-M specific siRNA, and the transfected cells were subsequently challenged with either LPS or Pam3CSK4. As shown in figure 1B, the IRAK-M specific siRNA but not the control siRNA caused significant reduction of the 68 KD band, indicating the band we detected was indeed IRAK-M.
Figure 1. IRAK-M is constitutively expressed in human mononuclear cells.
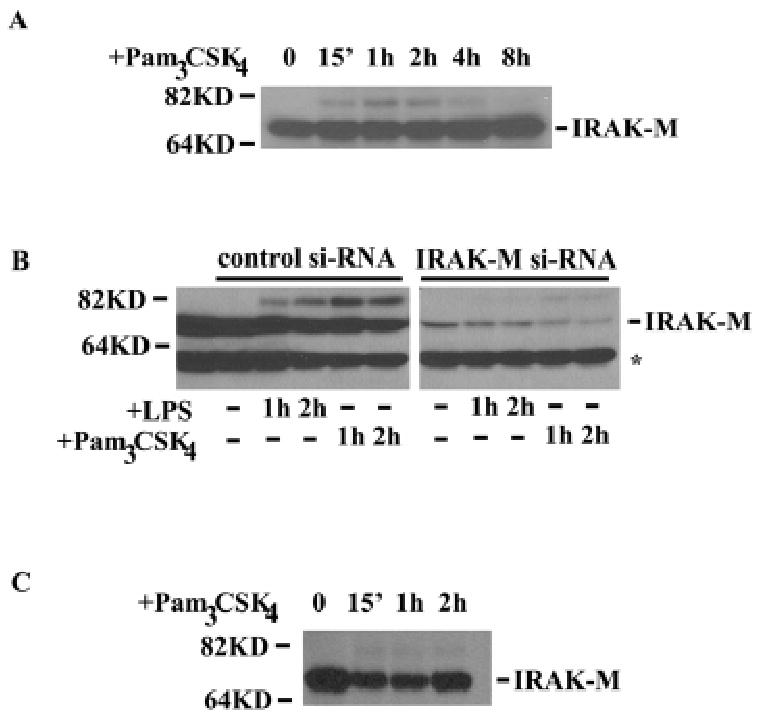
A), THP-1 cells were treated with 100 ng/ml Pam3CSK4 for various time periods. Whole cell lysates were collected, and immunoblot analyses were performed using anti-IRAK-M antibody. B), THP-1 cells were transfected with either scrambled control siRNA or siRNA against IRAK-M. After 48 hours, transfected cells were treated with either LPS or Pam3CSK4 for the indicated time periods. Cellular extracts were harvested and separated by SDS-PAGE. Immunoblots were performed using anti-IRAK-M antibody. *, non-specific protein band. C, human PBMC were isolated as described in the Experimental Procedures. Isolated cells were treated with 100 ng/ml Pam3CSK4 for various time periods. Subsequent analyses were performed as described in A and B.
We then studied the IRAK-M levels in human peripheral blood mononuclear cells (PBMC). As shown in figure 1C, IRKA-M was constitutively present and not acutely induced by Pam3CSK4 challenge. Similarly, an upper band was quickly induced upon Pam3CSK4 treatment. Overnight treatment of human PBMC did lead to slight increase of IRAK-M levels (data not shown). This is in agreement with previous studies showing IRAK-M is a late inducing protein by TLR ligands and its induction is not apparent within 8 hr challenge(7;13;14). A previous study using human PBMC showed that the IRAK-M level even decreased slightly upon short period of LPS challenge, and only started to increase after 24 hr LPS treatment (13).
IRAK-M undergoes nuclear export upon challenge
There has been no previous report regarding the subcellular localization of IRAK-M. We therefore examined the IRAK-M distribution in THP-1 cells upon challenge. Cytoplasmic and nuclear extracts harvested from THP-1 cells treated with Pam3CSK4 were separated on SDS-PAGE and blotted with IRAK-M antibody. As shown in figure 2A, IRAK-M was present in both the cytoplamic and nuclear fractions in resting THP-1 cells. However, Pam3CSK4 challenge caused significant reduction of nuclear IRAK-M levels, indicating that IRAK-M may undergo nuclear export upon challenge. In order to confirm that IRAK-M indeed underwent shuttling from nucleus to cytoplasm, we incubated the THP1- cells with leptomycin-B, an inhibitor for nuclear export. As shown in figure 2A, compared with non-treated cells, leptomycin pre-treated cells exhibited significant increase in nuclear IRAK-M levels upon subsequent Pam3CSK4 challenge.
Figure 2. Pam3CSK4 induces IRAK-M nuclear export.
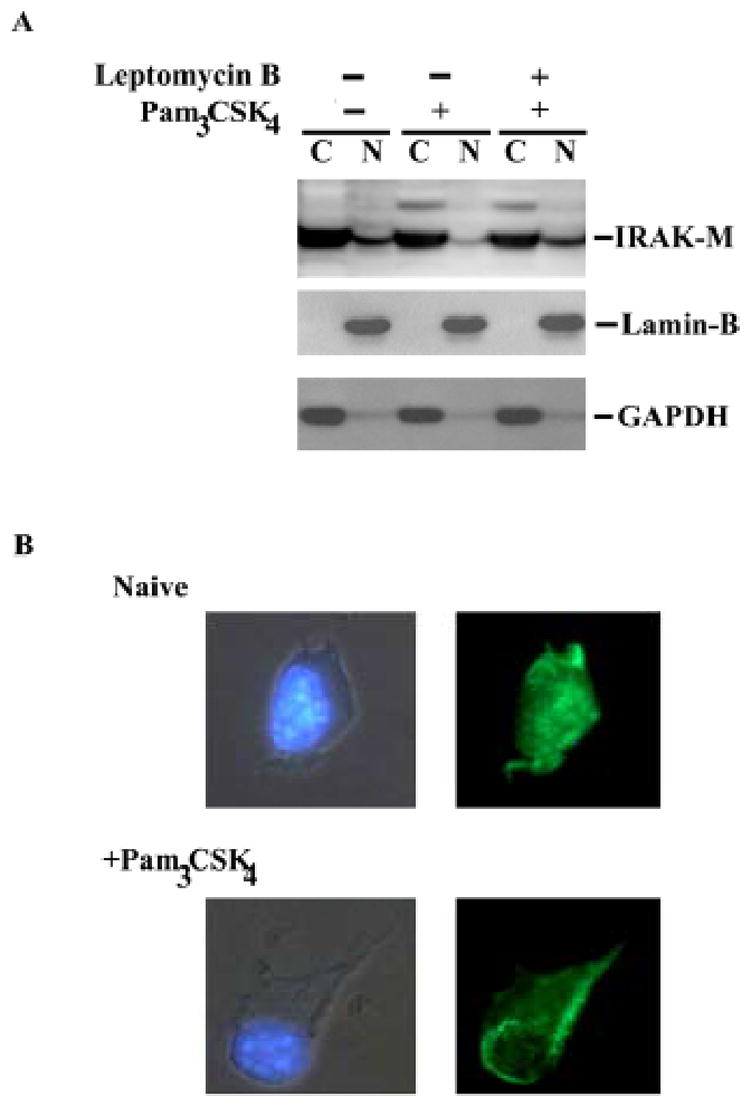
A), Naïve or leptomycin B-pretreated THP-1 cells were subsequently challenged with 100 ng/ml Pam3CSK4 for one hour. Cytoplasmic (C) and nuclear (N) extracts were harvested, separated on SDS-PAGE and blotted with IRAK-M, GAPDH or Lamin-B antibody. GAPDH and Lamin-B are cytoplasmic and nuclear markers, respectively. B), primary murine bone marrow derived macrophages (BMDM) were challenged with 100 ng/ml Pam3CSK4 for one hour. Treated or non-treated cells were fixed, incubated with IRAK-M antibody, and then stained with cy-2 -conjugated anti-Rabbit IgG (right panel). The cells were double stained with DAPI to visualize the nuclei (left panel).
IRAK-M subcellular distribution was further studied using immunohistochemistry. Wild type murine BMDM were treated with 100ng/ml Pam3CSK4 for 1 hr. Treated cells were subsequently fixed with formaldehyde and stained with anti-IRAK-M antibody as well as control IgG, followed by staining with cy-2 fluorescence-conjugated secondary antibody. Consistent with our above results, IRAK-M staining pattern was permissive in resting BMDM. Upon Pam3CSK4 challenge, IRAK-M staining was exclusively cytoplasmic, further indicating that IRAK-M underwent nuclear export upon challenge (Fig. 2B). Control antibody did not yield any noticeable staining (data not shown).
IRAK-M and IRAK1 differentially regulate MAP kinase activation
BMDM from IRAK-M−/− mice were consistently shown to exhibit elevated MAP kinase phosphorylation upon various treatments (7;8). The mechanism for IRAK-M mediated MAPK inhibition is not clearly understood. Overexpression of IRAK-M can prevent dissociation of IRAK1 and IRAK-4 from MyD88, suggesting that IRAK-M may exert its inhibitory function through IRAK1/4. However, studies employing the over expression approach may not truly reflect endogenous condition. In order to determine whether IRAK-M may functionally interact with IRAK1, we therefore tested activation of various MAP kinases in IRAK-M as well as IRAK1 deficient murine macrophages. We consistently observed that the activation patterns of various MAP kinases were distinct among BMDM from wild type, IRAK1−/−, and IRAK-M−/− mice (figure 3). Induction of p38 phosphorylation by Pam3CSK4 was comparable in wild type and IRAK1−/− BMDM, and was much prolonged in IRAK-M−/− BMDM (figure 3). In contrast, inductions of ERK and JNK phosphorylations were similar in wild type and IRAK-M−/− BMDM, and greatly attenuated in IRAK1−/− BMDM (figure 3). Our finding indicates IRAK-M specifically attenuates p38, but not ERK or JNK activation. In contrast, IRAK1 selectively contributes to the activations of ERK and JNK, instead of p38 upon Pam3CSK4 challenge.
Figure 3. IRAK-M specifically attenuates p38, but not ERK or JNK activation.
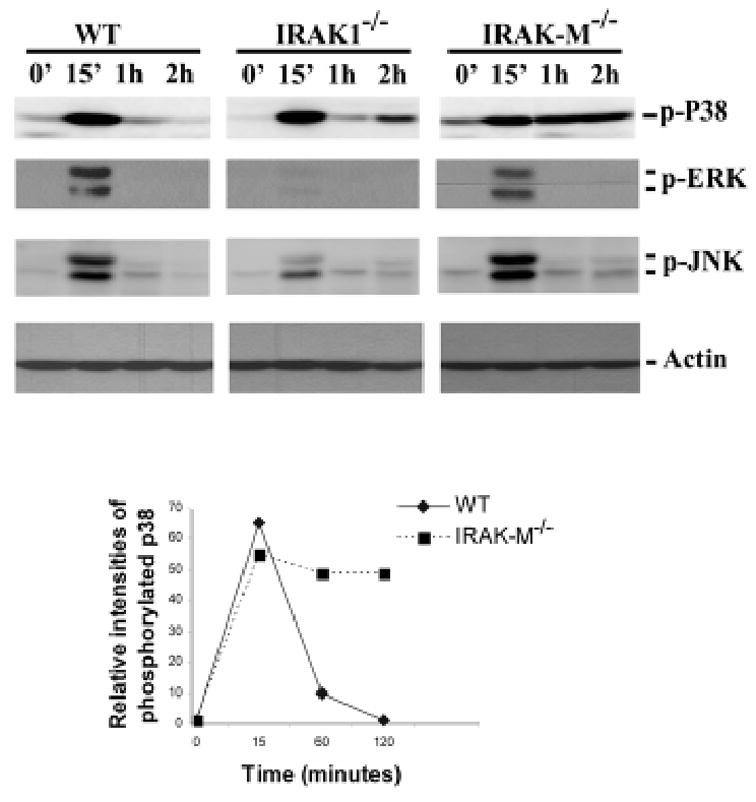
Wild type, IRAK1−/− and IRAK-M−/− murine BMDM were treated with 100 ng/ml Pam3CSK4 for the indicated time periods. Whole cell lysates were collected, and immunoblot analyses were performed using p–p38, p-ERK or p-JNK antibody. The relative p38 phosphorylation levels in wild type and IRAK-M−/− BMDM following Pam3CSK4 challenge from three independent experiments were plotted in the graph below. The relative intensities of MKP-1 protein bands in wild type and IRAK-M−/− BMDM following Pam3CSK4 challenge from three independent experiments were plotted in the graph below.
IRAK-M regulates p-38 activity via MKP-1
MKP-1/CL100 has been shown to be primarily responsible for negatively regulating p38 phosphoryation (15)., MKP-1 was shown to be a labile protein undergoing constant synthesis and turnover through proteosome-mediated degradation. Since IRAK-M selectively attenuates Pam3CSK4-induced p38 phosphorylation, we examined the MKP-1 protein levels in Pam3CSK4-stimulated BMDM from IRAK1−/−, IRAK-M−/− and wild-type mice. As shown in figure 4, MKP-1 protein levels remained constant upon 1 hr of Pam3CSK4 challenge in wild type and IRAK1−/− BMDM. In contrast, Pam3CSK4 treatment led to a rapid and dramatic decrease in MKP-1 protein levels in IRAK-M−/− BMDM (figure 4). Proteosome inhibitor MG132 prevented Pam3CSK4-induced MKP-1 level decrease in IRAK-M−/− BMDM, indicating that protein degradation accounted for MKP-1 level decrease in Pam3CSK4–treated IRAK-M−/− BMDM (figure 4).
Figure 4. IRAK-M contributes to MKP-1 stability in murine macrophages.
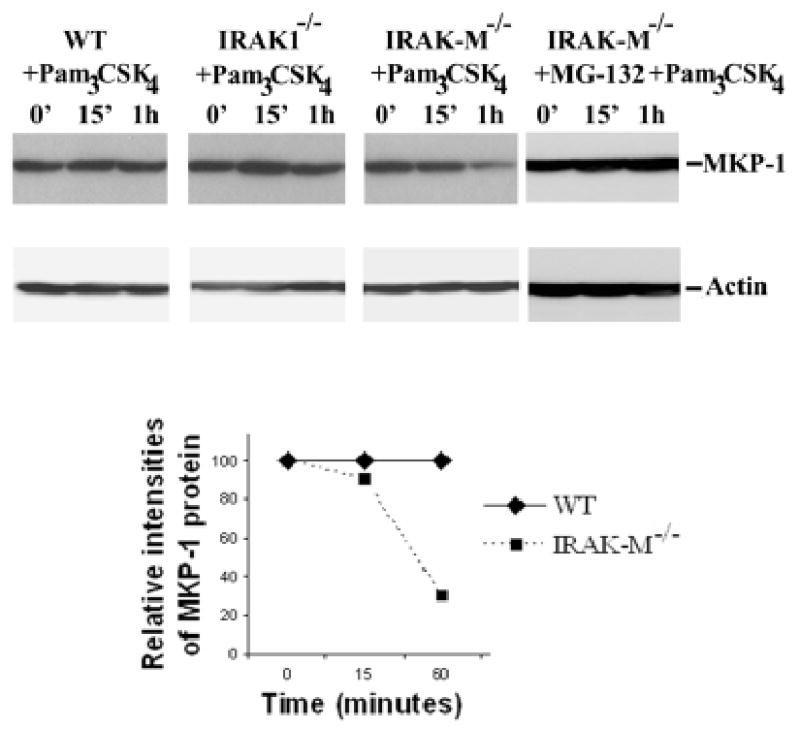
Wild type, IRAK1−/− and IRAK-M−/− murine BMDM were treated with 100 ng/ml Pam3CSK4 for the indicated time periods. Equal amounts of cellular extracts were separated on SDS-PAGE. MKP-1 levels were determined through western blot using anti-MKP-1 antibody. Bottom panel, IRAK-M−/− murine BMDM were pretreated with MG-132 for 1 hr, and subsequently challenged with 100 ng/ml Pam3CSK4 for various time periods. Results were representative of three independent experiments.
We further examined the contribution of IRAK-M to MKP-1 stability in human cells. IRAK-M expression was knocked down in THP-1 cells using siRNA approach (figure 5). Compared with cells transfected with control siRNA, cells transfected with siRNA against IRAK-M exhibited rapid MKP-1 degradation upon Pam3CSK4 challenge. Correspondingly, Pam3CSK4 induced-p38 phosphorylation was significantly higher in cells with decreased IRAK-M expression.
Figure 5. IRAK-M regulates MKP-1 stability in THP-1 cells.
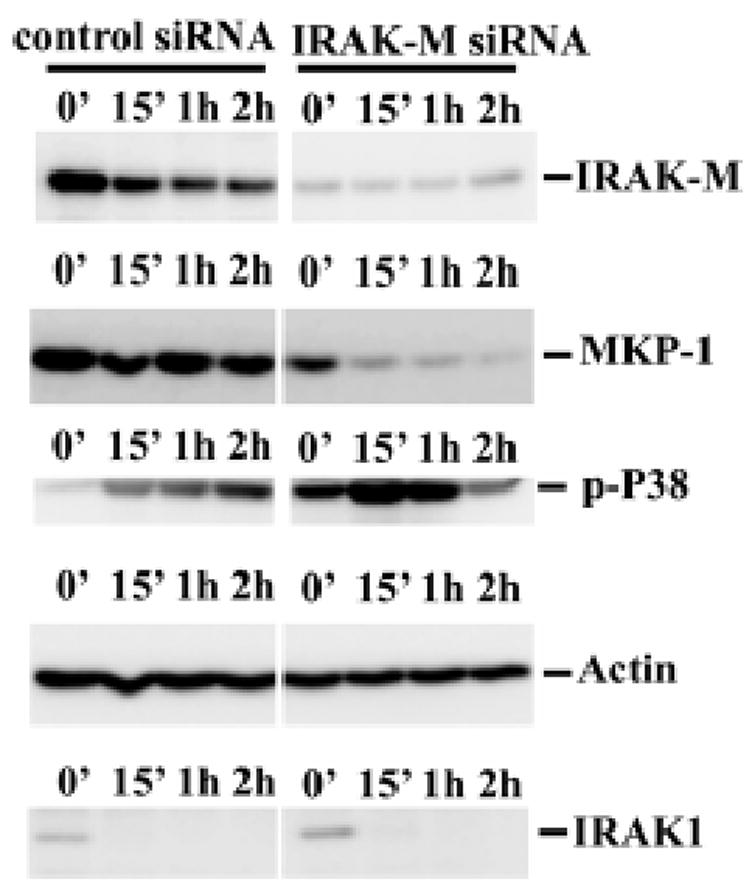
A), THP-1 cells were transfected with control or IRAK-M specific siRNA as described in the materials and methods. Transfected cells were subsequently challenged with 100 ng/ml Pam3CSK4 for various time periods. Equal amounts of total cell lysates were separated on SDS-PAGE, and MKP-1 protein levels were determined through Western blot using anti-MKP-1 antibody. B), Hela-TLR2 (MAT-2) cells were transfected with either empty pcDNA vector or pflag-IRAK-M as described in the Experimental Procedures. Transfected cells were subsequently challenged with 100 ng/ml Pam3CSK4 for various time periods. Equal amounts of cell lysates were separated on SDS-PAGE, and Western blot analyses were performed using anti-MKP-1 antibody. Results were representative of three independent experiments.
The contribution of IRAK-M to MKP-1 stability was also studied by over-expressing IRAK-M in Hela-TLR2 cells (10). As shown in figure 6B, overexpression of IRAK-M led to increased MKP-1 protein levels upon Pam3CSK4 challenge. Furthermore, we observed that IRAK-M knock-down did not affect Pam3CSK4-induced IRAK1 degradation, a hallmark for ligand-induced IRRAK1 activation (figure 4). Taken together, our results indicate that IRAK-M was functionally distinct from IRAK1, and selectively attenuate p38 activation via stabilizing MKP-1.
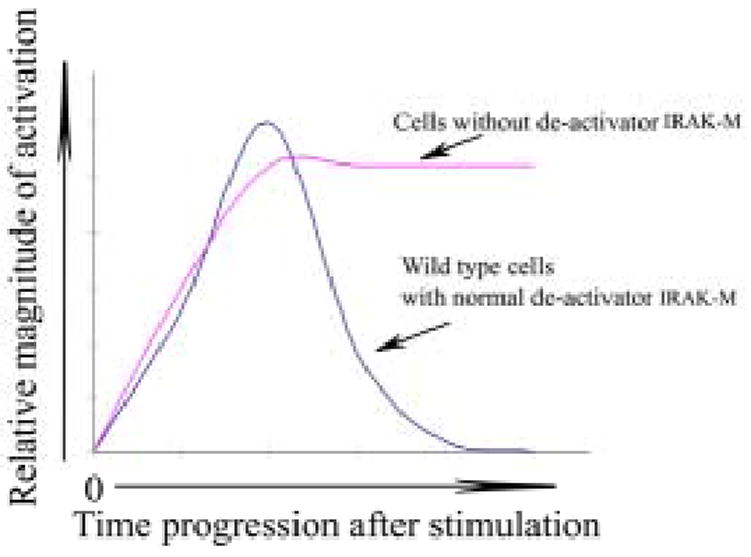
Figure 6. Schematic illustration of IRAK-M function in de-activating innate immunity signaling.
DISCUSSION
In this report, we demonstrate that IRAK-M selectively attenuates Pam3CSK4-induced p38 activation through an IRAK1 independent and MKP-1 dependent pathway. p38 activation is comparable in wild type and IRAK1 deficient cells. In contrast, IRAK-M deficient cells exhibit enhanced p38 phosphorylation upon challenge. Deletion of IRAK-M has no effect on Pam3CSK4-induced ERK and JNK phosphorylations, which are primarily affected by IRAK1 deletion. MKP-1 is the critical phosphatase responsible for inactivating p38. Deficiency in IRAK-M, but not IRAK1, leads to accelerated MKP-1 degradation upon Pam3CSK4 challenge. On the other hand, overexpression of IRAK-M contributes to increased MKP-1 protein level.
Although IRAK family proteins were initially thought to play redundant and/or overlapping function, increasing evidence has pointed to unique roles for these proteins in innate immunity. IRAK4 is the critical kinase essential for NFκB activation. In contrast, IRAK1 is primarily involved in the activation of STATs as well as IRFs (4–6). IRAK-M on the other hand seems to attenuate TLR signaling as evidenced by increased MAP kinase phosphorylation in IRAK-M−/− cells upon various challenges (7). Our data presented herein support this concept and further indicate that endogenous IRAK-M functions independently of IRAK1. We demonstrate that Pam3CSK4-induced activation profile of various MAP kinases are distinct among macrophages isolated from wild type, IRAK1−/−, and IRAK-M−/− mice. p38 is comparably activated in wild type and IRAK1−/− BMDM, and dramatically prolonged in IRAK-M−/− BMDM. The modulatory effect of IRAK-M on p38 activation is also observed in human monocytic THP-1 cells. In contrast, Pam3CSK4-induced ERK and JNK activation is comparable in wild type and IRAK-M−/− BMDM, and greatly attenuated in IRAK1−/− BMDM. Our data indicate that IRAK1 is selectively involved in Pam3CSK4-induced ERK and JNK activation, and is not related to p38 activation. In contrast, IRAK-M is primarily responsible for de-activating Pam3CSK4-induced activation of p38, but not ERK or JNK. We also demonstrate that IRAK-M is NOT contributing to IRAK1 regulation and function. Upon challenges with various TLR ligands, IRAK1 undergoes rapid activation followed by degradation (12;16;17). We observe that IRAK1 in IRAK-M knocked-down cells undergoes similar Pam3CSK4-induced degradation (figure 4), a hallmark for ligand-induced IRAK1 activation. Furthermore, our previous work has shown that IRAK1 enters cell nucleus upon challenge (4). In contrast, we show in this report that IRAK-M translocates from nucleus to cytoplasm upon Pam3CSK4 treatment. The biological implication for IRAK-M translocation is not clear and warrant further investigation. Taken together, it is evident that IRAK1 and IRAK-M play distinct and unrelated roles in mediating innate immunity.
It is worth note that IRAK-M is not simply an inhibitor of innate immunity signaling. As evident from our findings, the initial wave of p38 activation following Pam3CSK4 challenge is similar between wild type and IRAK-M-/- macrophages. Rather, activated p38 is quickly dephosphorylated and returns back to resting state in wild type cells. In contrast, IRAK-M macrophages experience prolonged p38 activation. We therefore classify IRAK-M as a deactivator instead of inhibitor for innate immunity signaling process. Such de-activator is not involved in inhibiting and/or dampening the initial activation signal. Rather, it primarily serves as a switch to help turning off the activated events, ensuring a tightly-controlled activation. This will allows rapid activation of host innate immune response, and subsequently helps preventing excessive inflammatory complications (figure 6). Indeed, studies using IRAK-M deficient mice indicate that IRAK-M deficiency contributes to the pathogenesis of osteoporosis and sepsis (8, 22).
The regulation and function of IRAK-M may be stimulus and cell type dependent. For example, it was reported that IRAK-M disruption causes increased activation of all MAP kinases including ERK, JNK, and p38 by TLR9 ligand CpG DNA (7). In contrast, our unpublished data as well as previous report indicate that disruption of IRAK-M does not significantly affect LPS-mediated MAP kinase activation (7). Our data presented in this report show that IRAK-M deficiency selectively contributes to enhanced and prolonged p38 activation upon TLR2 ligand Pam3CSK4 challenge. Correspondingly, with regard to MKP-1 levels, we demonstrate that IRAK-M deficiency leads to decrease in MKP-1 levels in cells treated with Pam3CSK4. In contrast, our unpublished observation indicates that treatment with the TLR4 ligand LPS increases MKP-1 levels in both wild-type and IRAK-M−/ − cells, indicating that TLR2- and TLR4-mediated intracellular signaling pathways may differentially utilize IRAK-M. Furthermore, both wild-type and IRAK-M−/ − cells treated with either LPS or Pam3CSK4 for extended time periods (∼16 hours) exhibit increased MKP-1 expression (data not shown). These findings indicate that the effect of IRAK-M-mediated MKP-1 protein stability is transient and TLR2 ligand-specific. On the other hand, MKP-1 gene expression at the transcriptional level may be likely controlled by an IRAK-M-independent pathway and may account for increased MKP-1 protein levels upon prolonged stimulation. This is yet another reflection of multiple control mechanisms innate immune cells utilize to regulate one of the key negative modulators. These findings implicate selective and complex regulation of different TLR downstream signaling processes. Further studies are needed to determine the pathways leading to the activation of distinct MAP kinases from various TLRs.
The de-activation of p38 is achieved through its dephosphorylation by MAPK phosphatase 1 (MKP1). MKP-1, originally discovered to be able to dephosphorylate ERK, has since been shown by many groups to have a higher potency dephosphorylating p38 in human and murine cells (15;18;19). Two recent studies using MKP-1−/− macrophages indicate that MKP-1 deletion selectively causes elevated p38 phosphorylation, and has no effect on ERK or JNK activation upon LPS challenge (15;20). Our findings are consistent with these studies and provide a mechanism for IRAK-M mediated p38 attenuation. MKP-1 is a labile protein undergoing constant synthesis and turnover through the proteosome pathway (21). MKP-1 levels remain constant in wild type or IRAK1−/− macrophages upon Pam3CSK4 challenge, reflecting stable equilibrium of induced protein expression and degradation. Strikingly, we observe a dramatic decrease of MKP-1 levels in IRAK-M−/− macrophages upon Pam3CSK4 treatment. This is due to elevated protein degradation since proteosome inhibitor MG-132 prevents Pam3CSK4-induced MKP-1 level decrease in IRAK-M−/− macrophages. Our finding indicates that Pam3CSK4 induces a rapid MKP-1 degradation in the absence of IRAK-M, which in turn contributes to enhanced and prolonged p38 activation in IRAK-M deficient cells. It is worth note that although MKP-1 is known to be a labile protein, the exact mechanism for stimuli-induced MKP-1 degradation is not fully understood. Given the significance of MKP-1 regulation in diverse physiological processes including innate immunity regulation, further systematic and mechanistic studies are needed to address molecular machinery required for the regulation of MKP-1 stability and degradation.
Taken together, our present study indicates that IRAK1 and IRAK-M differentially regulate activation of various MAP kinases. IRAK-M selectively attenuates Pam3CSK4-induced p38 activation through stabilizing MKP-1. Our findings reveal the divergence of intracellular signaling pathways controlling innate immunity, and warrant future studies regarding the selectivity and specificity of innate immunity regulation.
Acknowledgments
This work is supported in part by NIH grants to LL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain
References
- 1.Li L. Curr Drug Targets Inflamm Allergy. 2004;3:81–86. doi: 10.2174/1568010043483863. [DOI] [PubMed] [Google Scholar]
- 2.Janssens S, Beyaert R. Mol Cell. 2003;11:293–302. doi: 10.1016/s1097-2765(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 3.Li S, Strelow A, Fontana EJ, Wesche H. Proc Natl Acad Sci USA. 2002;99:5567–5572. doi: 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang Y, Li T, Sane DC, Li L. J Biol Chem. 2004;279:51697–51703. doi: 10.1074/jbc.M410369200. [DOI] [PubMed] [Google Scholar]
- 5.Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, Fitzgerald KA, Golenbock DT. J Biol Chem. 2005;280:17005–17012. doi: 10.1074/jbc.M412584200. [DOI] [PubMed] [Google Scholar]
- 6.Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. J Exp Med. 2005;201:915–923. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Cuartas E, Cui W, Choi Y, Crawford TD, Ke HZ, Kobayashi KS, Flavell RA, Vignery A. J Exp Med. 2005;201:1169–1177. doi: 10.1084/jem.20041444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wesche H, Gao X, Li X, Kirschning CJ, Stark GR, Cao Z. J Biol Chem. 1999;274:19403–19410. doi: 10.1074/jbc.274.27.19403. [DOI] [PubMed] [Google Scholar]
- 10.Re F, Strominger JL. J Biol Chem. 2001;276:37692–37699. doi: 10.1074/jbc.M105927200. [DOI] [PubMed] [Google Scholar]
- 11.Li T, Hu J, Li L. Mol Immunol. 2004;41:85–92. doi: 10.1016/j.molimm.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 12.Li L, Cousart S, Hu J, McCall CE. J Biol Chem. 2000;275:23340–23345. doi: 10.1074/jbc.M001950200. [DOI] [PubMed] [Google Scholar]
- 13.Escoll P, del Fresno C, Garcia L, Valles G, Lendinez MJ, Arnalich F, Lopez-Collazo E. Biochem Biophys Res Commun. 2003;311:465–472. doi: 10.1016/j.bbrc.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 14.Nakayama K, Okugawa S, Yanagimoto S, Kitazawa T, Tsukada K, Kawada M, Kimura S, Hirai K, Takagaki Y, Ota Y. J Biol Chem. 2004;279:6629–6634. doi: 10.1074/jbc.M308620200. [DOI] [PubMed] [Google Scholar]
- 15.Hammer M, Mages J, Dietrich H, Servatius A, Howells N, Cato AC, Lang R. J Exp Med. 2005;203:15–20. doi: 10.1084/jem.20051753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siedlar M, Frankenberger M, Benkhart E, Espevik T, Quirling M, Brand K, Zembala M, Ziegler-Heitbrock L. J Immunol. 2004;173:2736–2745. doi: 10.4049/jimmunol.173.4.2736. [DOI] [PubMed] [Google Scholar]
- 17.Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, Kurtz S, Coffield VM, Accavitti-Loper MA, Su L, Vogel SN, Braunstein M, Ting JP. J Biol Chem. 2005;280:39914–39924. doi: 10.1074/jbc.M502820200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franklin CC, Kraft AS. J Biol Chem. 1997;272:16917–16923. doi: 10.1074/jbc.272.27.16917. [DOI] [PubMed] [Google Scholar]
- 19.Wu JJ, Bennett AM. J Biol Chem. 2005;280:16461–16466. doi: 10.1074/jbc.M501762200. [DOI] [PubMed] [Google Scholar]
- 20.Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T. J Immunol. 2006;176:1899–1907. doi: 10.4049/jimmunol.176.3.1899. [DOI] [PubMed] [Google Scholar]
- 21.Brondello JM, Pouyssegur J, McKenzie FR. Science. 1999;286:2514–2517. doi: 10.1126/science.286.5449.2514. [DOI] [PubMed] [Google Scholar]
- 22.Deng JC, Cheng G, Newstead MW, Zeng X, Kobayashi K, Flavell RA, Standiford TJ. J Clin Invest. 2006;116:2532–2542. doi: 10.1172/JCI28054. [DOI] [PMC free article] [PubMed] [Google Scholar]